Glycerolic Licorice Extracts as Active Cosmeceutical Ingredients: Extraction Optimization, Chemical Characterization, and Biological Activity




Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plant Material
2.3. Preparation of the Extracts
2.4. Spectrophotometric Determination of Total Phenol Content
2.5. Spectrophotometric Determination of Total Flavonoid Content
2.6. RP-HPLC-DAD Determinations of Glycyrrhizin, Glabridin and Isoliquiritigenin
2.7. Extraction Optimization
2.8. Radical Scavenging Activity
2.9. Fe2+ Chelating Activity
2.10. Antioxidant Activity in β-Carotene-Linoleic Acid Assay
2.11. Tyrosinase Inhibitory Activity
2.12. Elastase Inhibitory Activity
2.13. Anti-Inflammatory Activity
2.14. Statistical Analysis
3. Results and Discussion
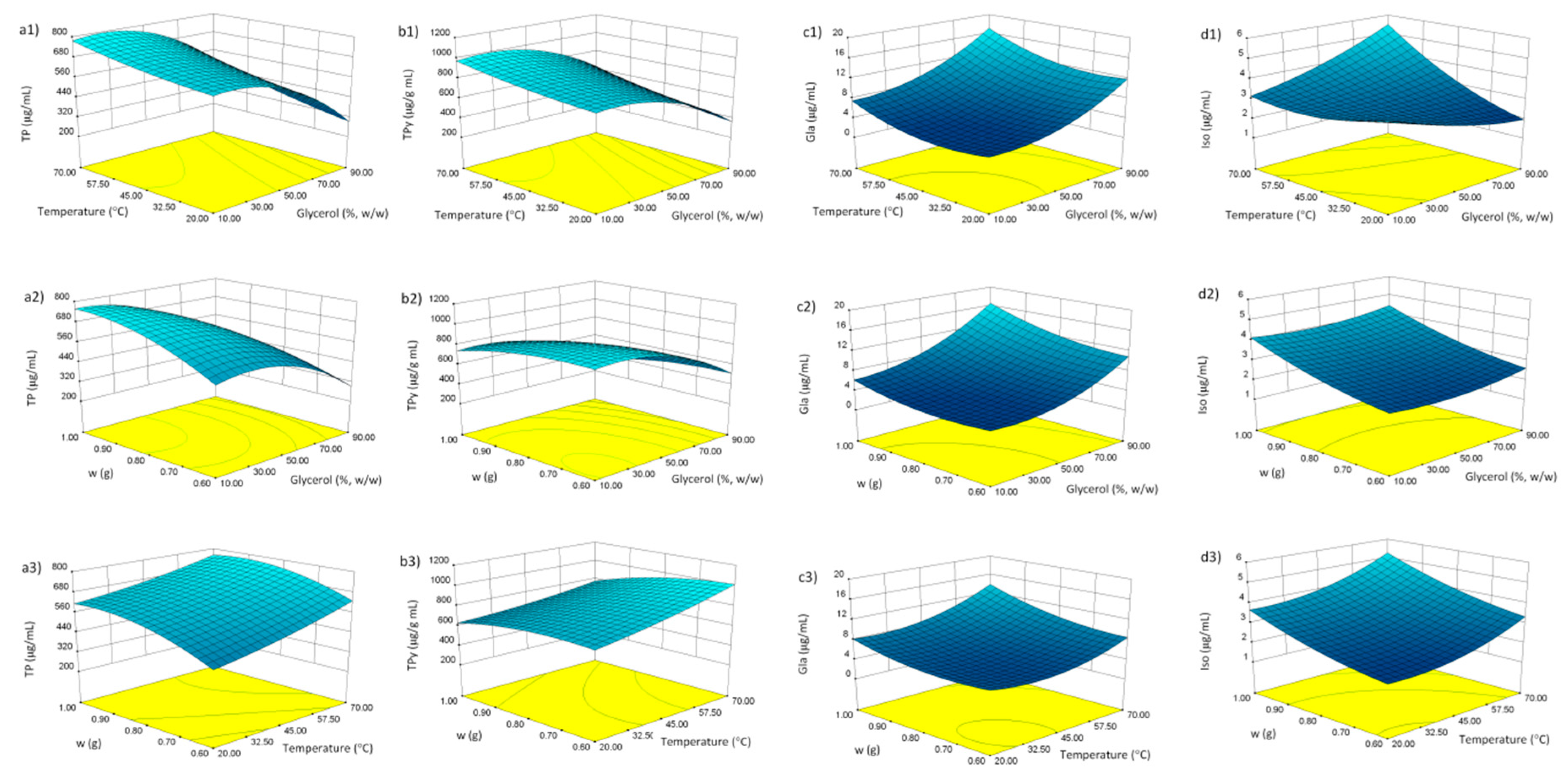
3.1. Response Surface Methodology
3.2. Fitting the Model
3.3. Model Analysis
3.4. Validation of Optimal Extraction Conditions
3.5. Chemical Composition of the Optimized Extracts
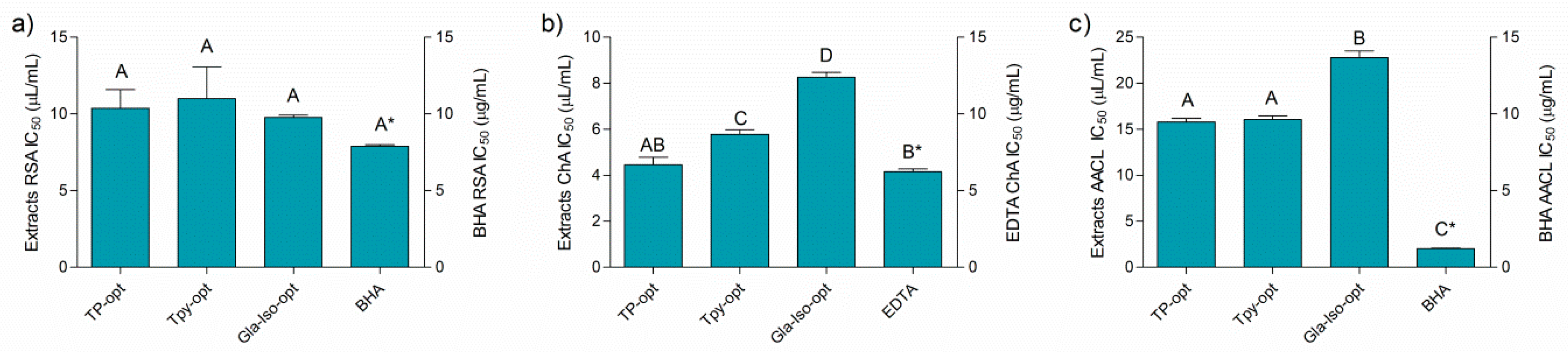
3.6. Antioxidant Activity of the Optimized Extracts
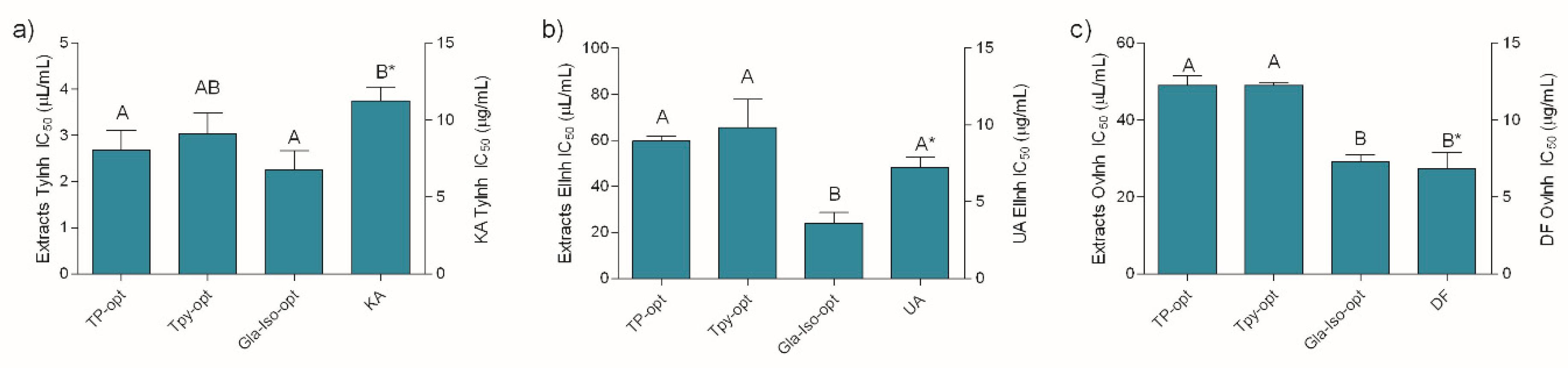
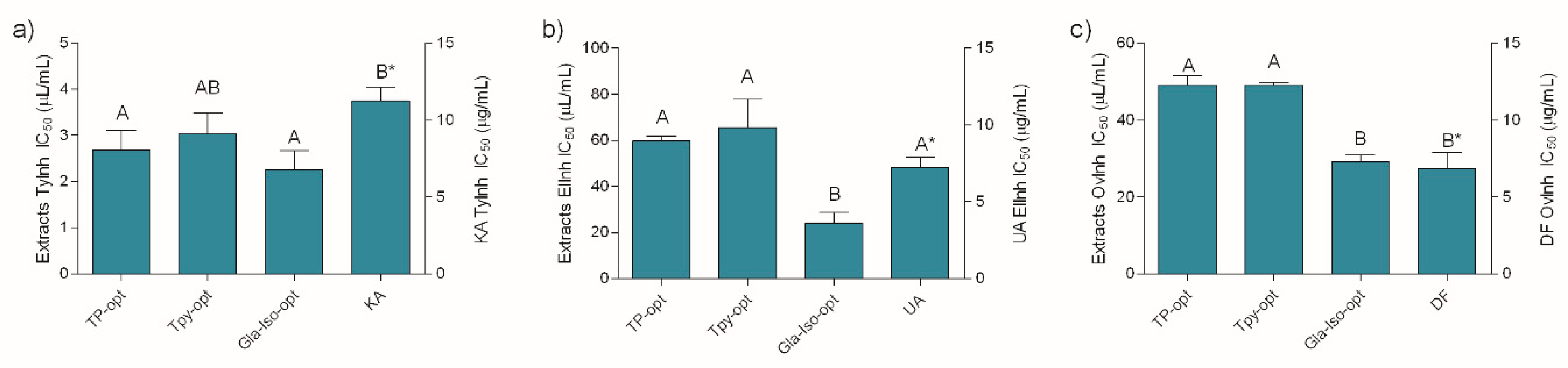
3.7. Enzyme Inhibiting and Anti-inflammatory Activity of the Optimized Extracts
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Yahya, N.A.; Attan, N.; Wahab, R.A. An overview of cosmeceutically relevant plant extracts and strategies for extraction of plant-based bioactive compounds. Food Bioprod. Process. 2018, 112, 69–85. [Google Scholar] [CrossRef]
- Costa, R.; Santos, L. Delivery systems for cosmetics—From manufacturing to the skin of natural antioxidants. Powder Technol. 2017, 322, 402–416. [Google Scholar] [CrossRef]
- Chemat, F.; Vian, M.A.; Cravotto, G. Green extraction of natural products: Concept and principles. Int. J. Mol. Sci. 2012, 13, 8615–8627. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, A.; Dlugy, C.; Shotland, Y. Glycerol as a green solvent for high product yields and selectivities. Environ. Chem. Lett. 2007, 5, 67–71. [Google Scholar] [CrossRef]
- Shehata, E.; Grigorakis, S.; Loupassaki, S.; Makris, D.P. Extraction optimisation using water/glycerol for the efficient recovery of polyphenolic antioxidants from two Artemisia species. Sep. Purif. Technol. 2015, 149, 462–469. [Google Scholar] [CrossRef]
- El Kantar, S.; Rajha, H.N.; Boussetta, N.; Vorobiev, E.; Maroun, R.G.; Louka, N. Green extraction of polyphenols from grapefruit peels using high voltage electrical discharges, deep eutectic solvents and aqueous glycerol. Food Chem. 2019, 295, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Apostolakis, A.; Grigorakis, S.; Makris, D.P. Optimisation and comparative kinetics study of polyphenol extraction from olive leaves (Olea europaea) using heated water/glycerol mixtures. Sep. Purif. Technol. 2014, 128, 89–95. [Google Scholar] [CrossRef]
- Rouhani, M. Modeling and optimization of ultrasound-assisted green extraction and rapid HPTLC analysis of stevioside from Stevia rebaudiana. Ind. Crop. Prod. 2019, 132, 226–235. [Google Scholar] [CrossRef]
- Glória, M.B.A. SWEETENERS|Others. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Caballero, B., Ed.; Academic Press: Oxford, MA, USA, 2003; pp. 5695–5702. [Google Scholar]
- Castangia, I.; Caddeo, C.; Manca, M.L.; Casu, L.; Latorre, A.C.; Díez-Sales, O.; Ruiz-Saurí, A.; Bacchetta, G.; Fadda, A.M.; Manconi, M. Delivery of liquorice extract by liposomes and hyalurosomes to protect the skin against oxidative stress injuries. Carbohydr. Polym. 2015, 134, 657–663. [Google Scholar] [CrossRef]
- Mostafa, D.M.; Ammar, N.M.; Abd El-Alim, S.H.; El-anssary, A.A. Transdermal microemulsions of Glycyrrhiza glabra L.: Characterization, stability and evaluation of antioxidant potential. Drug Deliv. 2014, 21, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Kotian, S.; Bhat, K.; Pai, S.; Nayak, J.; Souza, A.; Gourisheti, K.; Padma, D. The role of natural medicines on wound healing: A biomechanical, histological, biochemical and molecular study. Ethiop. J. Health Sci. 2018, 28, 759–770. [Google Scholar] [PubMed]
- Yu, H.; Li, H.; Li, Y.; Li, M.; Chen, G. Effect of isoliquiritigenin for the treatment of atopic dermatitis-like skin lesions in mice. Arch. Dermatol. Res. 2017, 309, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Simmler, C.; Pauli, G.F.; Chen, S.N. Phytochemistry and biological properties of glabridin. Fitoterapia 2013, 90, 160–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorino, G.; Cornara, L.; Soares, S.; Rodrigues, F.; Oliveira, M.B.P.P. Liquorice (Glycyrrhiza glabra): A phytochemical and pharmacological review. Phytother. Res. 2018, 32, 2323–2339. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Shiba, M.; Nakamura, R.; Morota, T.; Shoyama, Y. Constituent properties of licorices derived from Glycyrrhiza uralensis, G. glabra, or G. inflata identified by genetic information. Biol. Pharm. Bull. 2007, 30, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.C.; Lin, Y.H.; Chang, T.M.; Huang, W.Y. Identification of two licorice species, Glycyrrhiza uralensis and Glycyrrhiza glabra, based on separation and identification of their bioactive components. Food Chem. 2012, 132, 2188–2193. [Google Scholar] [CrossRef]
- European Pharmacopoeia Commission. European Pharmacopoeia, 8th ed.; Council of Europe: Strasbourg, France, 2013. [Google Scholar]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1999; Volume 299, pp. 152–178. [Google Scholar]
- Kumazawa, S.; Hamasaka, T.; Nakayama, T. Antioxidant activity of propolis of various geographic origins. Food Chem. 2004, 84, 329–339. [Google Scholar] [CrossRef]
- Kleinschmidt, G. Case Study: Validation of an HPLC-Method for Identity, Assay, and Related Impurities. In Method Validation in Pharmaceutical Analysis: A Guide to Best Practice; Ermer, J., Miller, J.H., Eds.; Wiley-VCH: Weinheim, Germany, 2005; pp. 195–212. [Google Scholar]
- Tada, A.; Ishizuki, K.; Sugimoto, N.; Yoshimatsu, K.; Kawahara, N.; Suematsu, T.; Arifuku, K.; Fukai, T.; Tamura, Y.; Ohtsuki, T.; et al. Determination of the plant origin of licorice oil extract, a natural food additive, by principal component analysis based on chemical components. J. Food Hyg. Soc. Jpn. 2015, 56, 217–227. [Google Scholar] [CrossRef]
- Jug, M.; Končić, M.Z.; Kosalec, I. Modulation of antioxidant, chelating and antimicrobial activity of poplar chemo-type propolis by extraction procures. LWT Food Sci. Technol. 2014, 57, 530–537. [Google Scholar] [CrossRef]
- Končić, M.Z.; Barbarić, M.; Perković, I.; Zorc, B. Antiradical, chelating and antioxidant activities of hydroxamic acids and hydroxyureas. Molecules 2011, 16, 6232–6242. [Google Scholar] [CrossRef]
- Rajić, Z.; Končić, M.; Miloloža, K.; Perković, I.; Butula, I.; Bucar, F.; Zorc, B. Primaquine-NSAID twin drugs: Synthesis, radical scavenging, antioxidant and Fe2+ chelating activity. Acta Pharm. 2010, 60, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Bose, B.; Choudhury, H.; Tandon, P.; Kumaria, S. Studies on secondary metabolite profiling, anti-inflammatory potential, in vitro photoprotective and skin-aging related enzyme inhibitory activities of Malaxis acuminata, a threatened orchid of nutraceutical importance. J. Photochem. Photobiol. B 2017, 173, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Chatterjee, P.; Dey, P.; Bhattacharya, S. Evaluation of in vitro anti-inflammatory activity of coffee against the denaturation of protein. Asian Pac. J. Trop. Biomed. 2012, 2, S178–S180. [Google Scholar] [CrossRef]
- Li, Y.; Cao, S.Y.; Lin, S.J.; Zhang, J.R.; Gan, R.Y.; Li, H.B. Polyphenolic profile and antioxidant capacity of extracts from Gordonia axillaris fruits. Antioxidants 2019, 8, 150. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, X.Y.; Gan, R.Y.; Zheng, J.; Li, Y.; Zhang, J.J.; Xu, D.P.; Li, H.B. Optimization of ultrasound-assisted extraction of antioxidant polyphenols from the seed coats of red sword bean (Canavalia gladiate (Jacq.) DC.). Antioxidants 2019, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Limwachiranon, J.; Jiang, L.; Huang, H.; Sun, J.; Luo, Z. Improvement of phenolic compounds extraction from high-starch lotus (Nelumbo nucifera G.) seed kernels using glycerol: New insights to amylose/amylopectin—Phenolic relationships. Food Chem. 2019, 274, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, S. Main Cosmetic Vehicles. In Handbook of Cosmetic Science and Technology; Barel, A., Paye, M., Maibach, H., Eds.; Marcel Dekker: New York, NY, USA, 2001. [Google Scholar]
- Mlakar, A.; Batna, A.; Dudda, A.; Spiteller, G. Iron (II) ions induced oxidation of ascorbic acid and glucose. Free Radic. Res. 1996, 25, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Ratz-Łyko, A.; Arct, J. Resveratrol as an active ingredient for cosmetic and dermatological applications: A review. J. Cosmet. Laser Ther. 2019, 21, 84–90. [Google Scholar] [CrossRef]
- Thiele, J.; Elsner, P. Oxidants and Antioxidants in Cutaneous Biology; Karger: Basel, Switzerland, 2001; Volume 29. [Google Scholar]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Bessada, S.M.F.; Alves, R.C.; Oliveira, M.B. Coffee silverskin: A review on potential cosmetic applications. Cosmetics 2018, 5, 5. [Google Scholar] [CrossRef]
- Akhma, Z.; Ibrahim, F.; Nor Rashid, N.; Mohamad Razif, M.; Yusof, R. The importance of some plant extracts as skin anti-aging resources: A Review. Curr. Pharm. Biotechnol. 2017, 19, 864–876. [Google Scholar]
- Imokawa, G. Recent advances in characterizing biological mechanisms underlying UV-induced wrinkles: A pivotal role of fibrobrast-derived elastase. Arch. Dermatol. Res. 2008, 300, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Penkova, R.; Goshev, I.; Gorinstein, S.; Nedkov, P. Stability of collagen during denaturation. J. Protein Chem. 1999, 18, 397–401. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Analyte | Slope (a) | Intercept (b) | r2 | LD (μg) | LQ (μg) |
|---|---|---|---|---|---|
| Glycyrrhizin | 257.96 | 1.54 | 0.99998638 | 0.006112 | 0.018522 |
| Gla | 3402.71 | 26.12 | 0.9999998 | 0.000741 | 0.002246 |
| Iso | 5079.81 | 21.21 | 0.9999931 | 0.005013 | 0.015191 |
| Run | X1 | X2 | X3 | TP | TPy | Gla | Iso |
|---|---|---|---|---|---|---|---|
| (%. w/w) | (°C) | (g) | (μg/mL) | (μg/g mL) | (μg/mL) | (μg/mL) | |
| 1 | 50 | 70 | 0.6 | 605.3 | 1008.8 | 9.12 | 3.20 |
| 2 | 10 | 45 | 0.6 | 529.6 | 882.7 | 4.37 | 2.48 |
| 3 | 50 | 70 | 1.0 | 753.9 | 753.9 | 14.11 | 5.20 |
| 4 | 50 | 45 | 0.8 | 606.3 | 757.8 | 5.29 | 3.31 |
| 5 | 10 | 70 | 0.8 | 790.6 | 988.3 | 6.56 | 3.11 |
| 6 | 50 | 45 | 0.8 | 779.1 | 973.9 | 6.56 | 2.86 |
| 7 | 50 | 45 | 0.8 | 676.8 | 846.0 | 6.09 | 2.79 |
| 8 | 90 | 20 | 0.8 | 279.5 | 349.3 | 12.9 | 2.00 |
| 9 | 10 | 45 | 1.0 | 748.7 | 748.7 | 6.96 | 4.27 |
| 10 | 10 | 20 | 0.8 | 633.6 | 792.0 | 3.99 | 3.47 |
| 11 | 50 | 45 | 0.8 | 582.2 | 727.7 | 4.40 | 2.07 |
| 12 | 90 | 70 | 0.8 | 518.7 | 648.3 | 17.30 | 5.76 |
| 13 | 90 | 45 | 0.6 | 302.1 | 503.4 | 10.14 | 2.47 |
| 14 | 50 | 45 | 0.8 | 620.8 | 776.0 | 6.39 | 3.29 |
| 15 | 50 | 20 | 1.0 | 638.3 | 638.3 | 7.63 | 3.79 |
| 16 | 90 | 45 | 1.0 | 346.6 | 346.6 | 16.18 | 4.26 |
| 17 | 50 | 20 | 0.6 | 447.4 | 745.7 | 5.48 | 2.02 |
| Response | Unit | The Equation Coefficients: a × X12 + b × X22 + c × X32 + d × X1 × X2 + e × X1 × X3 + f × X2 × X3 + g × X1 + h × X2 + i × X3 + j | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | D | e | f | g | h | i | j | ||
| TP | mg/mL | −113.482 a | 16.020 | −57.818 b | 20.547 | −43.631 | −10.56 | −156.966 a | 83.702 a | 75.393 a | 653.045 |
| TPy | mg/g mL | −144.084 a | 22.256 | −51.862 | 25.684 | −5.703 | −36.864 | −195.495 a | 109.235 a | −81.642 a | 816.306 |
| Gla | μg/mL | 2.384 a | 2.058 a | 1.283 a | 0.459 | 0.863 | 0.708 | 4.329 a | 2.135 a | 1.972 a | 5.746 |
| Iso | μg/mL | 0.271 | 0.451 | 0.237 b | 1.032 a | −0.003 | 0.055 | 0.146 | 0.748 a | 0.919 a | 2.864 |
| TP | TPy | |||||||||
| r2 | r2 = 0.9329; rA2 = 0.8467; rP2 = 0.8027 | r2 = 0.9325; rA2 = 0.8457; rP2 = 0.8389 | ||||||||
| Source | SS | df | MS | F Value | p-value | SS | df | MS | F Value | p-value |
| Model | 379,961.7 | 9 | 42,218 | 10.82 | 0.0024 | 565,610.6 | 9 | 62,845.62 | 10.74213 | 0.0025 |
| Lack of Fit | 2607.6 | 3 | 869 | 0.14 | 0.9305 | 2337.216 | 3 | 779.072 | 0.0807 | 0.9671 |
| Pure Error | 24,713.9 | 4 | 6178 | 38,615.51 | 4 | 9653.877 | ||||
| Gla | Iso | |||||||||
| r2 | r2 = 0.9739; rA2 = 0.9403; rP2 = 0.7444 | r2 = 0.9307; rA2 = 0.8415; rP2 = 0.6810 | ||||||||
| Source | SS | df | MS | F Value | p-value | SS | df | MS | F Value | p-value |
| Model | 277.3 | 9 | 30.811 | 29.01 | <0.0001 | 17.23 | 9 | 1.915 | 10.44 | 0.0027 |
| Lack of Fit | 4.24 | 3 | 1.412 | 1.76 | 0.2926 | 0.27 | 3 | 0.09 | 0.36 | 0.7892 |
| Pure Error | 3.2 | 4 | 0.8 | 1.01 | 4 | 0.253 | ||||
| Extract | Measured Response | X1 | X2 | X3 | Resppred | Respms | RD (%) |
|---|---|---|---|---|---|---|---|
| (%, w/w) | (°C) | (g) | |||||
| TP-opt | TP (μg/mL) | 20 | 70 | 0.93 | 830.2 | 854.6 | 2.9 |
| Tpy-opt | TP (μg/mL) | 30 | 70 | 0.7 | 734.8 | 791.6 | 7.7 |
| Gla-Iso-opt | Glabridin (μg/mL) | 85 | 70 | 1 | 20.67 | 21.89 | 5.9 |
| Gla-Iso-opt | Isoliquiritigenin (μg/mL) | 85 | 70 | 1 | 6.51 | 6.23 | −4.3 |
| Extract | TP | TF | Gla | Iso | Glycyrrhizin |
|---|---|---|---|---|---|
| (μg/mL) | (μg/mL) | (μg/mL) | (μg/mL) | (mg/mL) | |
| TP-opt | 854.6 ± 42.7 | 667.5 ± 42.7 | 9.62 ± 0.72 | 4.02 ± 0.26 | 4.31 ± 0.22 |
| Tpy-opt | 791.6± 48.0 | 521.4 ± 8.9 | 8.38 ± 0.17 | 3.51 ± 0.18 | 4.20 ± 0.17 |
| Gla-Iso-opt | 535.4 ± 32.1 | 692 ± 32.4 | 21.89 ± 1.09 | 6.23 ± 0.16 | 4.67 ± 0.34 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciganović, P.; Jakimiuk, K.; Tomczyk, M.; Zovko Končić, M. Glycerolic Licorice Extracts as Active Cosmeceutical Ingredients: Extraction Optimization, Chemical Characterization, and Biological Activity. Antioxidants 2019, 8, 445. https://doi.org/10.3390/antiox8100445
Ciganović P, Jakimiuk K, Tomczyk M, Zovko Končić M. Glycerolic Licorice Extracts as Active Cosmeceutical Ingredients: Extraction Optimization, Chemical Characterization, and Biological Activity. Antioxidants. 2019; 8(10):445. https://doi.org/10.3390/antiox8100445
Chicago/Turabian StyleCiganović, Petar, Katarzyna Jakimiuk, Michał Tomczyk, and Marijana Zovko Končić. 2019. "Glycerolic Licorice Extracts as Active Cosmeceutical Ingredients: Extraction Optimization, Chemical Characterization, and Biological Activity" Antioxidants 8, no. 10: 445. https://doi.org/10.3390/antiox8100445
APA StyleCiganović, P., Jakimiuk, K., Tomczyk, M., & Zovko Končić, M. (2019). Glycerolic Licorice Extracts as Active Cosmeceutical Ingredients: Extraction Optimization, Chemical Characterization, and Biological Activity. Antioxidants, 8(10), 445. https://doi.org/10.3390/antiox8100445