Redox-Mediated Mechanism of Chemoresistance in Cancer Cells


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Redox Homeostasis in Tumorigenesis
2.1. ROS Generation
2.1.1. ATP Synthesis in Mitochondria
2.1.2. Endoplasmic Reticulum (ER)
2.1.3. NADPH Oxidases (NOXs)
2.2. ROS Elimination
2.2.1. SODs
2.2.2. Catalase
2.2.3. Prxs
2.2.4. Nrf2
2.3. Redox Homeostasis of Chemoresistance
2.3.1. Oxaliplatin Resistance
2.3.2. 5-Fluorouracil (5-FU) Resistance
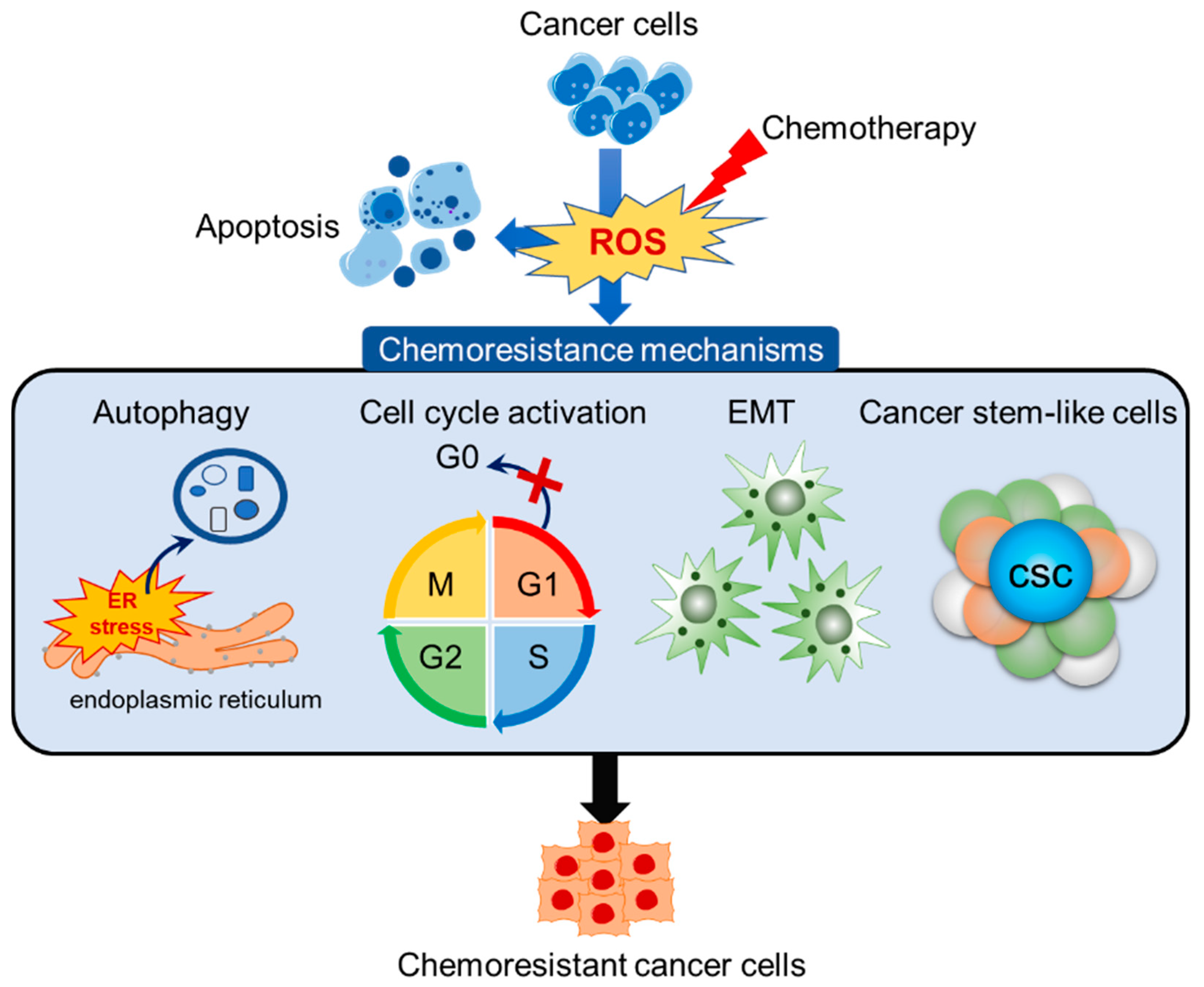
3. Redox-Mediated Mechanism of Chemoresistance
3.1. ER Stress-Mediated Autophagy
3.1.1. Oxaliplatin-Resistance
3.1.2. 5-FU-Resistance
3.2. Overcoming Cell Cycle Arrest
3.2.1. Oxaliplatin-Resistance
3.2.2. 5-FU-Resistance
3.3. Epithelial-Mesenchymal Transition (EMT) and Cancer Stem-Like Cells (CSC)
3.3.1. Oxaliplatin-Resistance
3.3.2. 5-FU-Resistance
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.B.; Wan, M.Y.; Wang, P.Y.; Zhang, C.X.; Xu, D.Y.; Liao, X.; Sun, H.J. Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFkappaB/mTOR/P70S6K signaling cascade. Redox Biol. 2018, 14, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Nordzieke, D.E.; Medrano-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H.; Lee, I.K.; Kim, G.W.; Kim, B.U.; Han, Y.H.; Yu, D.Y.; Park, H.S.; Kim, K.Y.; Lee, J.S.; Choi, C.; et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature 2005, 435, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Dharmaraja, A.T. Role of Reactive Oxygen Species (ROS) in Therapeutics and Drug Resistance in Cancer and Bacteria. J. Med. Chem. 2017, 60, 3221–3240. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Balibrea, E.; Martinez-Cardus, A.; Gines, A.; Ruiz de Porras, V.; Moutinho, C.; Layos, L.; Manzano, J.L.; Bugés, C.; Bystrup, S.; Esteller, M.; et al. Tumor-Related Molecular Mechanisms of Oxaliplatin Resistance. Mol. Cancer Ther. 2015, 14, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Nobel, S.; Abrahmsen, L.; Oppermann, U. Metabolic conversion as a pre-receptor control mechanism for lipophilic hormones. Eur. J. Biochem. 2001, 268, 4113–4125. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.-H.; Chen, Z.-S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Porporato, P.E.; Payen, V.L.; Baselet, B.; Sonveaux, P. Metabolic changes associated with tumor metastasis, part 2: Mitochondria, lipid and amino acid metabolism. Cell. Mol. Life Sci. 2016, 73, 1349–1363. [Google Scholar] [CrossRef]
- Oliva, C.R.; Markert, T.; Ross, L.J.; White, E.L.; Rasmussen, L.; Zhang, W.; Everts, M.; Moellering, D.R.; Bailey, S.M.; Suto, M.J.; et al. Identification of Small Molecule Inhibitors of Human Cytochrome c Oxidase That Target Chemoresistant Glioma Cells. J. Biol. Chem. 2016, 291, 24188–24199. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.W.; Kuo, C.Y.; Fan, C.C.; Fang, W.C.; Jiang, S.S.; Lo, Y.K.; Kao, M.C.; Lee, A.Y.L. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell Death Dis. 2013, 4, e681. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2009, 3, 281–290. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Chemotherapy Resistance Explained through Endoplasmic Reticulum Stress-Dependent Signaling. Cancers 2019, 11, 338. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Wang, W.; Thirumalai, D. Protein folding guides disulfide bond formation. Proc. Natl. Acad. Sci. USA 2015, 112, 11241–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Iemura, S.; Kamiya, Y.; Ron, D.; Kato, K.; Natsume, T.; Nagata, K. Ero1-alpha and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 2013, 202, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Enyedi, B.; Varnai, P.; Geiszt, M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal. 2010, 13, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Weng, M.S.; Chang, J.H.; Hung, W.Y.; Yang, Y.C.; Chien, M.H. The interplay of reactive oxygen species and the epidermal growth factor receptor in tumor progression and drug resistance. J. Exp. Clin. Cancer Res. 2018, 37, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, Y.; Matsuo, T.; Sagara, Y.; Ohba, K.; Ohyama, K.; Sakai, H. A Mini-Review of Reactive Oxygen Species in Urological Cancer: Correlation with NADPH Oxidases, Angiogenesis, and Apoptosis. Int. J. Mol. Sci. 2017, 18, 2214. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Watt, J.M.; Fortunato, T.M.; Mellor, H.; Burgess, M.; Wicks, K.; Mace, K.; Reeksting, S.; Lubben, A.; Wheeler-Jones, C.P.D.; et al. Direct Activation of NADPH Oxidase 2 by 2-Deoxyribose-1-Phosphate Triggers Nuclear Factor Kappa B-Dependent Angiogenesis. Antioxid. Redox Signal. 2018, 28, 110–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef] [PubMed]
- Ameziane-El-Hassani, R.; Schlumberger, M.; Dupuy, C. NADPH oxidases: New actors in thyroid cancer? Nat. Rev. Endocrinol. 2016, 12, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, F.R.; Araujo, T.L.; Abrahao, T.B. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef]
- Fulton, D.J. Nox5 and the regulation of cellular function. Antioxid. Redox Signal. 2009, 11, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.V.; Hines, M.C. Alpha, beta, and gamma crystallins in the ocular lens of rabbits: Preparation and partial characterization. Investig. Ophthalmol. 1966, 5, 601–609. [Google Scholar]
- Holl, M.; Koziel, R.; Schafer, G.; Pircher, H.; Pauck, A.; Hermann, M.; Klocker, H.; Jansen-Dürr, P.; Sampson, N. ROS signaling by NADPH oxidase 5 modulates the proliferation and survival of prostate carcinoma cells. Mol. Carcinog. 2016, 55, 27–39. [Google Scholar] [CrossRef]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef]
- Wang, P.; Shi, Q.; Deng, W.H.; Yu, J.; Zuo, T.; Mei, F.C.; Wang, W.X. Relationship between expression of NADPH oxidase 2 and invasion and prognosis of human gastric cancer. World J. Gastroenterol. 2015, 21, 6271–6279. [Google Scholar] [CrossRef] [PubMed]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases: Insights into Selected Functions and Mechanisms of Action in Cancer and Stem Cells. Oxid Med. Cell. Longev. 2017, 2017, 9420539. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Sun, Y.C.; Lu, W.H.; Huang, P.; Hu, Y. Selective killing of K-ras-transformed pancreatic cancer cells by targeting NAD(P)H oxidase. Chin. J. Cancer 2015, 34, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Redon, C.E.; Parekh, P.R.; Dupuy, C.; Bonner, W.M. NADPH Oxidases NOXs and DUOXs as putative targets for cancer therapy. Anti-Cancer Agents Med. Chem. 2013, 13, 502–514. [Google Scholar]
- Che, M.; Wang, R.; Li, X.; Wang, H.Y.; Zheng, X.F.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef]
- Dhar, S.K.; St Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef]
- Papa, L.; Hahn, M.; Marsh, E.L.; Evans, B.S.; Germain, D. SOD2 to SOD1 switch in breast cancer. J. Biol. Chem. 2014, 289, 5412–5416. [Google Scholar] [CrossRef]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 reduces experimental non-small-cell lung cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef]
- Oberley, L.W.; Buettner, G.R. Role of superoxide dismutase in cancer: A review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar] [PubMed]
- Hempel, N.; Carrico, P.M.; Melendez, J.A. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anti-Cancer Agents Med. Chem. 2011, 11, 191–201. [Google Scholar] [CrossRef]
- Hempel, N.; Melendez, J.A. Intracellular redox status controls membrane localization of pro- and anti-migratory signaling molecules. Redox Biol. 2014, 2, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, I.; Kim, T.Y.; Kim-Ha, J. Identification of Drosophila SOD3 and its protective role against phototoxic damage to cells. FEBS Lett. 2011, 585, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.B. Redox signaling across cell membranes. Antioxid. Redox Signal. 2009, 11, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Folz, R.J.; Landmesser, U.; Harrison, D.G. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc. Res. 2002, 55, 239–249. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Zamocky, M.; Sandoval, J.M.; Verrax, J.; Calderon, P.B. Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 2015, 87, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Rainis, T.; Maor, I.; Lanir, A.; Shnizer, S.; Lavy, A. Enhanced oxidative stress and leucocyte activation in neoplastic tissues of the colon. Dig. Dis. Sci. 2007, 52, 526–530. [Google Scholar] [CrossRef]
- Hwang, T.S.; Choi, H.K.; Han, H.S. Differential expression of manganese superoxide dismutase, copper/zinc superoxide dismutase, and catalase in gastric adenocarcinoma and normal gastric mucosa. Eur. J. Surg. Oncol. 2007, 33, 474–479. [Google Scholar] [CrossRef]
- Sander, C.S.; Hamm, F.; Elsner, P.; Thiele, J.J. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br. J. Dermatol. 2003, 148, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.C.; Li, S.H.; Majima, H.J.; Huang, Y.H.; Chen, C.P.; Liu, C.C.; Tu, Y.C.; Chen, C.W. Up-regulation of antioxidant enzymes and coenzyme Q(10) in a human oral cancer cell line with acquired bleomycin resistance. Free Radic. Res. 2011, 45, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Kuramitsu, Y.; Taba, K.; Ryozawa, S.; Yoshida, K.; Zhang, X.; Tanaka, T.; Maehara, S.I.; Maehara, Y.; Sakaida, I.; Nakamura, K. Identification of up- and down-regulated proteins in gemcitabine-resistant pancreatic cancer cells using two-dimensional gel electrophoresis and mass spectrometry. Anti-Cancer Res. 2010, 30, 3367–3372. [Google Scholar]
- Xu, H.; Choi, S.M.; An, C.S.; Min, Y.D.; Kim, K.C.; Kim, K.J.; Choi, C.H. Concentration-dependent collateral sensitivity of cisplatin-resistant gastric cancer cell sublines. Biochem. Biophys. Res. Commun. 2005, 328, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.H.; Gupta, V.; Singh, S.V. Characterization of a human bladder cancer cell line selected for resistance to mitomycin C. Int. J. Cancer 1994, 58, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jang, H.H. Role of Cytosolic 2-Cys Prx1 and Prx2 in Redox Signaling. Antioxidants 2019, 8, 169. [Google Scholar] [CrossRef] [PubMed]
- Sharapov, M.G.; Novoselov, V.I. Catalytic and Signaling Role of Peroxiredoxins in Carcinogenesis. Biochemistry 2019, 84, 79–100. [Google Scholar] [CrossRef]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A(2) activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.H.; Kim, S.U.; Kwon, T.H.; Kim, J.M.; Song, I.S.; Shin, H.J.; Lee, B.K.; Bang, D.H.; Lee, S.J.; Lee, D.S. Peroxiredoxin II promotes hepatic tumorigenesis through cooperation with Ras/Forkhead box M1 signaling pathway. Oncogene 2016, 35, 3503–3513. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Kang, D.H.; Choi, M.; Choi, Y.J.; Lee, J.Y.; Park, J.H.; Park, Y.J.; Lee, K.W.; Kang, S.W. Peroxiredoxin-2 represses melanoma metastasis by increasing E-Cadherin/beta-Catenin complexes in adherens junctions. Cancer Res. 2013, 73, 4744–4757. [Google Scholar] [CrossRef] [PubMed]
- Giudice, A.; Arra, C.; Turco, M.C. Review of molecular mechanisms involved in the activation of the Nrf2-ARE signaling pathway by chemopreventive agents. Methods Mol. Biol. 2010, 647, 37–74. [Google Scholar] [PubMed]
- Giudice, A.; Barbieri, A.; Bimonte, S.; Cascella, M.; Cuomo, A.; Crispo, A.; D’Arena, G.; Galdiero, M.; Della Pepa, M.E.; Botti, G.; et al. Dissecting the prevention of estrogen-dependent breast carcinogenesis through Nrf2-dependent and independent mechanisms. Onco Targets Ther. 2019, 12, 4937–4953. [Google Scholar] [CrossRef] [PubMed]
- Grewal, G.K.; Kukal, S.; Kanojia, N.; Saso, L.; Kukreti, S.; Kukreti, R. Effect of Oxidative Stress on ABC Transporters: Contribution to Epilepsy Pharmacoresistance. Molecules 2017, 22, 365. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A.M. Current Approaches and Emerging Directions in HER2-resistant Breast Cancer. Breast Cancer 2014, 8, 109–118. [Google Scholar] [CrossRef]
- Ganan-Gomez, I.; Wei, Y.; Yang, H.; Boyano-Adanez, M.C.; Garcia-Manero, G. Oncogenic functions of the transcription factor Nrf2. Free Radic. Biol. Med. 2013, 65, 750–764. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Macewan, D.J. The role of nrf2 and cytoprotection in regulating chemotherapy resistance of human leukemia cells. Cancers 2011, 3, 1605–1621. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Cives-Losada, C.; Asensio, M.; Lozano, E.; Briz, O.; Macias, R.I.R. Mechanisms of Anticancer Drug Resistance in Hepatoblastoma. Cancers 2019, 11, 407. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, L.; Shi, H.; Chen, H.; Tao, J.; Shen, R.; Wang, T. Ursolic acid enhances the therapeutic effects of oxaliplatin in colorectal cancer by inhibition of drug resistance. Cancer Sci. 2018, 109, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tang, B.; Yu, P.W.; Tang, B.; Hao, Y.X.; Lei, X.; Luo, H.X.; Zeng, D.Z. Autophagy protects against oxaliplatin-induced cell death via ER stress and ROS in Caco-2 cells. PLoS ONE 2012, 7, e51076. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.B.; Hui, B.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Gu, C.Y.; Yang, H.; Shi, G.M.; Ke, A.W.; Wang, X.Y. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin. Cancer Res. 2011, 17, 6229–6238. [Google Scholar] [CrossRef]
- Canta, A.; Pozzi, E.; Carozzi, V.A. Mitochondrial Dysfunction in Chemotherapy-Induced Peripheral Neuropathy (CIPN). Toxics 2015, 3, 198–223. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Jiang, Y.; Guo, C.; Reed, A.; Meng, H.; Vasko, M.R. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS ONE 2014, 9, e106485. [Google Scholar] [CrossRef]
- Plasencia, C.; Martinez-Balibrea, E.; Martinez-Cardus, A.; Quinn, D.I.; Abad, A.; Neamati, N. Expression analysis of genes involved in oxaliplatin response and development of oxaliplatin-resistant HT29 colon cancer cells. Int. J. Oncol. 2006, 29, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Pirpour Tazehkand, A.; Akbarzadeh, M.; Velaie, K.; Sadeghi, M.R.; Samadi, N. The role of Her2-Nrf2 axis in induction of oxaliplatin resistance in colon cancer cells. Biomed. Pharmacother. 2018, 103, 755–766. [Google Scholar] [CrossRef]
- Chian, S.; Li, Y.Y.; Wang, X.J.; Tang, X.W. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the Nrf2 pathway. Asian Pac. J. Cancer Prev. 2014, 15, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Mann, J.; Vasilaki, A.; Mathers, J.; Burt, A.D.; Oakley, F.; White, S.A.; Mann, D.A. Pathogenesis of FOLFOX induced sinusoidal obstruction syndrome in a murine chemotherapy model. J. Hepatol. 2013, 59, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.W.; Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Fernando, P.; Oh, M.C.; Park, J.E.; Shilinkova, K.; Na, S.Y.; Jeong, S.U.; et al. Reduced Autophagy in 5-Fluorouracil Resistant Colon Cancer Cells. Biomol. Ther. (Seoul) 2017, 25, 315–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Kang, H.K.; Chang, W.Y.; Park, I.C.; Keum, Y.S.; Surh, Y.J.; Hyun, J.W. Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon cancer cells: Involvement of TET-dependent DNA demethylation. Cell Death Dis. 2014, 5, e1183. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Ryu, Y.S.; Piao, M.J.; Shilnikova, K.; Kang, H.K.; Yi, J.M.; Boulanger, M.; Paolillo, R.; Bossis, G.; Yoon, S.Y.; et al. DUOX2-mediated production of reactive oxygen species induces epithelial mesenchymal transition in 5-fluorouracil resistant human colon cancer cells. Redox Biol. 2018, 17, 224–235. [Google Scholar] [CrossRef]
- Kim, J.K.; Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Han, X.; Fernando, P.M.; Oh, M.C.; Park, J.E.; Shilnilova, K.; Boo, S.Y.; et al. Endoplasmic reticulum stress induces 5-fluorouracil resistance in human colon cancer cells. Environ. Toxicol. Pharmacol. 2016, 44, 128–133. [Google Scholar] [CrossRef]
- Chan, J.Y.; Phoo, M.S.; Clement, M.V.; Pervaiz, S.; Lee, S.C. Resveratrol displays converse dose-related effects on 5-fluorouracil-evoked colon cancer cell apoptosis: The roles of caspase-6 and p53. Cancer Biol. Ther. 2008, 7, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef]
- Shin, Y.K.; Yoo, B.C.; Chang, H.J.; Jeon, E.; Hong, S.H.; Jung, M.S.; Lim, S.J.; Park, J.G. Down-regulation of mitochondrial F1F0-ATP synthase in human colon cancer cells with induced 5-fluorouracil resistance. Cancer Res. 2005, 65, 3162–3170. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.T.; Chung, Y.M.; Kim, J.J.; Chung, J.S.; Kim, B.S.; Kim, H.J.; Kim, J.S.; Yoo, Y.D. Drug resistance to 5-FU linked to reactive oxygen species modulator 1. Biochem. Biophys. Res. Commun. 2007, 359, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wu, L.; Chen, Z.; Yang, J.; Chen, X.; Yu, F.; Zheng, F.; Lin, X. MiR-141 Activates Nrf2-Dependent Antioxidant Pathway via Down-Regulating the Expression of Keap1 Conferring the Resistance of Hepatocellular Carcinoma Cells to 5-Fluorouracil. Cell. Physiol. Biochem. 2015, 35, 2333–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-differentiation confers multidrug resistance via noncanonical PERK-Nrf2 signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.F.; Yao, J.; Gao, S.G.; Wang, X.S.; Peng, X.Q.; Yang, Y.T.; Feng, X.S. Nrf2 overexpression predicts prognosis and 5-FU resistance in gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 5231–5235. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Kang, H.K.; Chang, W.Y.; Keum, Y.S.; Hyun, J.W. Interaction of DNA demethylase and histone methyltransferase upregulates Nrf2 in 5-fluorouracil-resistant colon cancer cells. Oncotarget 2016, 7, 40594–40620. [Google Scholar] [CrossRef] [Green Version]
- Akhdar, H.; Loyer, P.; Rauch, C.; Corlu, A.; Guillouzo, A.; Morel, F. Involvement of Nrf2 activation in resistance to 5-fluorouracil in human colon cancer HT-29 cells. Eur. J. Cancer 2009, 45, 2219–2227. [Google Scholar] [CrossRef]
- Alimbetov, D.; Askarova, S.; Umbayev, B.; Davis, T.; Kipling, D. Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells. Int. J. Mol. Sci. 2018, 19, 1690. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial-mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef]
- Shen, Y.; Yang, J.; Zhao, J.; Xiao, C.; Xu, C.; Xiang, Y. The switch from ER stress-induced apoptosis to autophagy via ROS-mediated JNK/p62 signals: A survival mechanism in methotrexate-resistant choriocarcinoma cells. Exp. Cell Res. 2015, 334, 207–218. [Google Scholar] [CrossRef]
- Dayem, A.A.; Choi, H.Y.; Kim, J.H.; Cho, S.G. Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers 2010, 2, 859–884. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Heintz, N.H. The cell cycle is a redox cycle: Linking phase-specific targets to cell fate. Free Radic. Biol. Med. 2009, 47, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhou, L.; Chen, Z.; Nice, E.C.; Huang, C. Stress management by autophagy: Implications for chemoresistance. Int. J. Cancer 2016, 139, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Dufey, E.; Sepulveda, D.; Rojas-Rivera, D.; Hetz, C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Cell Physiol. 2014, 307, C582–C594. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Healy, S.J.; Gorman, A.M.; Mousavi-Shafaei, P.; Gupta, S.; Samali, A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur. J. Pharmacol. 2009, 625, 234–246. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, S.; He, L.; Rong, Y.; Brier, L.W.; Sun, Q.; Liu, R.; Fan, W.; Chen, S.; Yue, S.; et al. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy 2017, 13, 592–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.; Park, E.; Dinesh-Kumar, S.P. Differential processing of Arabidopsis ubiquitin-like Atg8 autophagy proteins by Atg4 cysteine proteases. Proc. Natl. Acad. Sci. USA 2014, 111, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Gines, A.; Bystrup, S.; Ruiz de Porras, V.; Guardia, C.; Musulen, E.; Martinez-Cardus, A.; Manzano, J.L.; Layos, L.; Abad, A.; Martinez-Balibrea, E. PKM2 Subcellular Localization Is Involved in Oxaliplatin Resistance Acquisition in HT29 Human Colorectal Cancer Cell Lines. PLoS ONE 2015, 10, e0123830. [Google Scholar] [CrossRef]
- Delgado, M.; Tesfaigzi, Y. Is BMF central for anoikis and autophagy? Autophagy 2014, 10, 168–169. [Google Scholar] [CrossRef]
- Tischner, D.; Manzl, C.; Soratroi, C.; Villunger, A.; Krumschnabel, G. Necrosis-like death can engage multiple pro-apoptotic Bcl-2 protein family members. Apoptosis 2012, 17, 1197–1209. [Google Scholar] [CrossRef] [Green Version]
- Grespi, F.; Soratroi, C.; Krumschnabel, G.; Sohm, B.; Ploner, C.; Geley, S.; Hengst, L.; Hacker, G.; Villunnger, A. BH3-only protein Bmf mediates apoptosis upon inhibition of CAP-dependent protein synthesis. Cell Death Differ. 2010, 17, 1672–1683. [Google Scholar] [CrossRef]
- Du, H.; Yang, W.; Chen, L.; Shi, M.; Seewoo, V.; Wang, J.; Lin, A.; Liu, Z.; Qui, W. Role of autophagy in resistance to oxaliplatin in hepatocellular carcinoma cells. Oncol. Rep. 2012, 27, 143–150. [Google Scholar] [PubMed]
- Das, C.K.; Linder, B.; Bonn, F.; Rothweiler, F.; Dikic, I.; Michaelis, M.; Cinatl, J.; Mandal, M.; Kogel, D. BAG3 Overexpression and Cytoprotective Autophagy Mediate Apoptosis Resistance in Chemoresistant Breast Cancer Cells. Neoplasia 2018, 20, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2007, 26, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Maziere, C.; Trecherel, E.; Ausseil, J.; Louandre, C.; Maziere, J.C. Oxidized low density lipoprotein induces cyclin A synthesis. Involvement of ERK, JNK and NFkappaB. Atherosclerosis 2011, 218, 308–313. [Google Scholar] [CrossRef]
- Tickner, J.; Fan, L.M.; Du, J.; Meijles, D.; Li, J.M. Nox2-derived ROS in PPARgamma signaling and cell-cycle progression of lung alveolar epithelial cells. Free Radic. Biol. Med. 2011, 51, 763–772. [Google Scholar] [CrossRef]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol. Cell. Biol. 2006, 26, 4701–4711. [Google Scholar] [CrossRef]
- Deshpande, S.S.; Irani, K. Oxidant signalling in carcinogenesis: A commentary. Hum. Exp. Toxicol. 2002, 21, 63–64. [Google Scholar] [CrossRef]
- Stamatakos, M.; Palla, V.; Karaiskos, I.; Xiromeritis, K.; Alexiou, I.; Pateras, I.; Kontzoglou, K. Cell cyclins: Triggering elements of cancer or not? World J. Surg. Oncol. 2010, 8, 111. [Google Scholar] [CrossRef]
- Fu, M.; Wang, C.; Li, Z.; Sakamaki, T.; Pestell, R.G. Minireview: Cyclin D1: Normal and abnormal functions. Endocrinology 2004, 145, 5439–5447. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, Y.; Hosokawa, Y.; Yoshimoto, K.; Schipani, E.; Mallya, S.; Papanikolaou, A.; Kifor, O.; Tokura, T.; Sablosky, M.; Ledgard, F.; et al. Primary hyperparathyroidism caused by parathyroid-targeted overexpression of cyclin D1 in transgenic mice. J. Clin. Investig. 2001, 107, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Yasui, M.; Yamamoto, H.; Ngan, C.Y.; Damdinsuren, B.; Sugita, Y.; Fukunaga, H.; Gu, J.; Maeda, M.; Takemasa, I.; Ikeda, M.; et al. Antisense to cyclin D1 inhibits vascular endothelial growth factor-stimulated growth of vascular endothelial cells: Implication of tumor vascularization. Clin. Cancer Res. 2006, 12, 4720–4729. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, T.; Casimiro, M.C.; Ju, X.; Quong, A.A.; Katiyar, S.; Liu, M.; Jiao, X.; Li, A.; Zhang, X.; Lu, Y.; et al. Cyclin D1 determines mitochondrial function in vivo. Mol. Cell. Biol. 2006, 26, 5449–5469. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Chen, M.C.; Baskaran, R.; Lin, Y.M.; Day, C.H.; Lin, Y.J.; Tu, C.C.; Vijaya Padma, V.; Kuo, W.W.; Huang, C.Y. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J. Cell Physiol. 2018, 233, 5458–5467. [Google Scholar] [CrossRef]
- El Khoury, F.; Corcos, L.; Durand, S.; Simon, B.; Le Jossic-Corcos, C. Acquisition of anticancer drug resistance is partially associated with cancer stemness in human colon cancer cells. Int. J. Oncol. 2016, 49, 2558–2568. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Goessl, E.; Jin, G.; Collie-Duguid, E.S.; Cassidy, J.; Wang, W.; O’Brien, W. Cell cycle perturbation and acquired 5-fluorouracil chemoresistance. Anti-Cancer Res. 2008, 28, 9–14. [Google Scholar]
- Wang, W.B.; Yang, Y.; Zhao, Y.P.; Zhang, T.P.; Liao, Q.; Shu, H. Recent studies of 5-fluorouracil resistance in pancreatic cancer. World J. Gastroenterol. 2014, 20, 15682–15690. [Google Scholar] [CrossRef]
- Wang, W.; Cassidy, J.; O’Brien, V.; Ryan, K.M.; Collie-Duguid, E. Mechanistic and predictive profiling of 5-Fluorouracil resistance in human cancer cells. Cancer Res. 2004, 64, 8167–8176. [Google Scholar] [CrossRef]
- Li, M.H.; Ito, D.; Sanada, M.; Odani, T.; Hatori, M.; Iwase, M.; Nagumo, M. Effect of 5-fluorouracil on G1 phase cell cycle regulation in oral cancer cell lines. Oral Oncol. 2004, 40, 63–70. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.; Smits, R.; Korinek, V.; Smit, L.; Kielman, M.; Fodde, R.; Clevers, H.; Pals, S.T. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am. J. Pathol. 1999, 154, 515–523. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Okumura, T.; Yamaguchi, T.; Miwa, T.; Shimada, Y.; Nagata, T. Enhanced cancer stem cell properties of a mitotically quiescent subpopulation of p75NTR-positive cells in esophageal squamous cell carcinoma. Int. J. Oncol. 2017, 51, 49–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, X.; Jiang, Z.; Li, L.; Cui, Z.; Gao, Y.; Kong, D.; Liu, X. A novel method to limit breast cancer stem cells in states of quiescence, proliferation or differentiation: Use of gel stress in combination with stem cell growth factors. Oncol. Lett. 2016, 12, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
- Jiao, L.; Li, D.D.; Yang, C.L.; Peng, R.Q.; Guo, Y.Q.; Zhang, X.S.; Zhu, X.F. Reactive oxygen species mediate oxaliplatin-induced epithelial-mesenchymal transition and invasive potential in colon cancer. Tumour Biol. 2016, 37, 8413–8423. [Google Scholar] [CrossRef]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Bu, Y.; Jiang, S.; Chang, F.; Jia, F.; Xiao, X.; Song, G.; Zhang, M.; Ning, P.; Jia, Q. CCN2-MAPK-Id-1 loop feedback amplification is involved in maintaining stemness in oxaliplatin-resistant hepatocellular carcinoma. Hepatol. Int. 2019, 13, 440–453. [Google Scholar] [CrossRef]
- Yin, X.; Tang, B.; Li, J.H.; Wang, Y.; Zhang, L.; Xie, X.Y.; Zhang, B.H.; Qiu, S.J.; Wu, W.Z.; Ren, Z.G. ID1 promotes hepatocellular carcinoma proliferation and confers chemoresistance to oxaliplatin by activating pentose phosphate pathway. J. Exp. Clin. Cancer Res. 2017, 36, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, A.; Ray, A. The CCN family proteins in carcinogenesis. Exp. Oncol. 2010, 32, 2–9. [Google Scholar] [PubMed]
- Yang, Y.; Yao, J.H.; Du, Q.Y.; Zhou, Y.C.; Yao, T.J.; Wu, Q.; Liu, J.; Ou, Y.R. Connexin 32 downregulation is critical for chemoresistance in oxaliplatin-resistant HCC cells associated with EMT. Cancer Manag. Res. 2019, 11, 5133–5146. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, E.; Sato, H.; Shirai, S.; Nagashima, Y.; Fukumoto, K.; Hagiwara, H.; Negishi, E.; Ueno, K.; Omori, Y.; Yamasaki, H.; et al. Connexin32 as a tumor suppressor gene in a metastatic renal cell carcinoma cell line. Oncogene 2005, 24, 3684–3690. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Li, X.; Pan, C.; Lin, W.; Shao, R.; Liu, Y.; Zhang, J.; Luo, Y.; Qian, K.; Shi, M.; et al. ATXN2L upregulated by epidermal growth factor promotes gastric cancer cell invasiveness and oxaliplatin resistance. Cell Death Dis. 2019, 10, 173. [Google Scholar] [CrossRef] [PubMed]
- Kaehler, C.; Isensee, J.; Nonhoff, U.; Terrey, M.; Hucho, T.; Lehrach, H.; Krobitsch, S. Ataxin-2-like is a regulator of stress granules and processing bodies. PLoS ONE 2012, 7, e50134. [Google Scholar] [CrossRef]
- Yang, D.; Wang, H.; Zhang, J.; Li, C.; Lu, Z.; Liu, J.; Lin, C.; Li, G.; Qian, H. In vitro characterization of stem cell-like properties of drug-resistant colon cancer subline. Oncol. Res. 2013, 21, 51–57. [Google Scholar] [CrossRef]
- Kim, A.Y.; Kwak, J.H.; Je, N.K.; Lee, Y.H.; Jung, Y.S. Epithelial-mesenchymal Transition is Associated with Acquired Resistance to 5-Fluorocuracil in HT-29 Colon Cancer Cells. Toxicol. Res. 2015, 31, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Harada, K.; Ferdous, T.; Ueyama, Y. Establishment of 5-fluorouracil-resistant oral squamous cell carcinoma cell lines with epithelial to mesenchymal transition changes. Int. J. Oncol. 2014, 44, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ke, J.; He, Z.; Chen, Z.; Huang, Q.; Ai, W.; Wang, G.; Wei, Y.; Zou, X.; Zhang, S.; et al. HES1 Promotes Colorectal Cancer Cell Resistance To 5-Fu by Inducing Of EMT and ABC Transporter Proteins. J. Cancer 2017, 8, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ma, Q.; Shi, Y.; Li, X.; Wang, M.; Wang, J.; Ge, J.; Chen, Z.; Wang, Z.; Jiang, H. A novel 5-fluorouracil-resistant human esophageal squamous cell carcinoma cell line Eca-109/5-FU with significant drug resistance-related characteristics. Oncol. Rep. 2017, 37, 2942–2954. [Google Scholar] [CrossRef]
- Vishnoi, K.; Mahata, S.; Tyagi, A.; Pandey, A.; Verma, G.; Jadli, M.; Singh, T.; Singh, S.M.; Bharti, A.C. Human papillomavirus oncoproteins differentially modulate epithelial-mesenchymal transition in 5-FU-resistant cervical cancer cells. Tumour Biol. 2016, 37, 13137–13154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Feng, M.; Zheng, G.; Chen, Y.; Wang, X.; Pen, B.; Yin, J.; Yu, Y.; He, Z. Chemoresistance to 5-fluorouracil induces epithelial-mesenchymal transition via up-regulation of Snail in MCF7 human breast cancer cells. Biochem. Biophys. Res. Commun. 2012, 417, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, M.; Kabashima, A.; Higuchi, H.; Igarashi, T.; Sakai, G.; Iizuka, H.; Nakamura, S.; Adachi, M.; Hamamoto, Y.; Funakoshi, S.; et al. Chemoresistance is associated with cancer stem cell-like properties and epithelial-to-mesenchymal transition in pancreatic cancer cells. Anti-Cancer Res. 2012, 32, 3847–3853. [Google Scholar]
- Miyoshi, S.; Tsugawa, H.; Matsuzaki, J.; Hirata, K.; Mori, H.; Saya, H.; Kanai, T.; Suzuki, H. Inhibiting xCT Improves 5-Fluorouracil Resistance of Gastric Cancer Induced by CD44 Variant 9 Expression. Anti-Cancer Res. 2018, 38, 6163–6170. [Google Scholar] [CrossRef] [PubMed]
- Terzioglu, G.; Turksoy, O.; Bayrak, O.F. Identification of An mtDNA Setpoint Associated with Highest Levels of CD44 Positivity and Chemoresistance in HGC-27 and MKN-45 Gastric Cancer Cell Lines. Cell J. 2018, 20, 312–317. [Google Scholar] [PubMed]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.-K.; Jang, M.; Song, M.-J.; Kim, D.; Kim, Y.; Jang, H.H. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants 2019, 8, 471. https://doi.org/10.3390/antiox8100471
Kim E-K, Jang M, Song M-J, Kim D, Kim Y, Jang HH. Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants. 2019; 8(10):471. https://doi.org/10.3390/antiox8100471
Chicago/Turabian StyleKim, Eun-Kyung, MinGyeong Jang, Min-Jeong Song, Dongwoo Kim, Yosup Kim, and Ho Hee Jang. 2019. "Redox-Mediated Mechanism of Chemoresistance in Cancer Cells" Antioxidants 8, no. 10: 471. https://doi.org/10.3390/antiox8100471
APA StyleKim, E.-K., Jang, M., Song, M.-J., Kim, D., Kim, Y., & Jang, H. H. (2019). Redox-Mediated Mechanism of Chemoresistance in Cancer Cells. Antioxidants, 8(10), 471. https://doi.org/10.3390/antiox8100471