1. Introduction
Marine macroalgae, or seaweed, is a large group of macroscopic organisms that are an important component in aquatic ecosystems. The wide diversity of marine organisms is being recognized as a rich source of functional materials and, in 2015, the global seaweed aquaculture production reached 30 million tons [
1]. Although marine algae have gained increasing attention over the last years due to the fact of their bioactive natural substances with potential health benefits, they are still identified as an underexploited resource [
2,
3,
4,
5,
6].
Natural antioxidants with multifunctional potential are of high interest, and numerous studies have focused on natural antioxidants, including polyphenols and flavonoids, from terrestrial plants [
7,
8,
9]. However, the application potential of polyphenolic analyses of marine sources suffers from several factors, most importantly, the lack of exactness with respect to quantitative and qualitative data at a molecular level. Marine plant material with analytic matrices at very low concentrations and a high and variable dissolved salt concentration makes polyphenol analyses challenging [
4,
10]. The diversity of phenolic compounds also varies from simple to highly polymerized substances which makes qualitative and quantitative procedures, involving sample preparation and extraction, difficult to standardize. Thus, this makes for a further challenge in the analyses and in furthering the research in this field.
Colorimetric assays, such as Folin-Ciocalteu, have been extensively used to quantify phlorotannins and polyphenolic content in seaweeds. However, since the assay is difficult to standardize and not selective, it has been recommended to use the assay for approximate measurements of an extract’s antioxidant potential only [
11,
12,
13,
14,
15]. Since the colorimetric assays neither separate nor give a correct quantitative measurement of the individual compounds, high-performance liquid chromatography (HPLC) has been the method of choice for separation and quantification of polyphenols in plants. The HPLC with multiple diode array UV-Visible detection (DAD) quantifies according to Lambert-Beer’s law (
A =
εcl). A compound’s ability to absorb UV-Visible light (
A) is related to the compound’s molar absorptivity value (ε) and molar concentration (
c). The diversity of molar absorptivity values of polyphenols is almost as large as the number of polyphenols existing; even within the same polyphenol class, there will be differences [
16]. In the lack of commercially available standards, one standard is often chosen when total amounts of polyphenols or phlorotannins are quantified. Gallic acid (GA) seems to be the most used standard for total polyphenolic quantification and phloroglucinol (PG) for the phlorotannin quantification in brow algae [
17,
18,
19,
20]. In addition to the limitations with commercially available standards, HPLC will also suffer from a lack of separation of complex extract matrices and loss of compound amounts due to the irreversible retention on the HPLC column during elution.
In recent years, quantitative
1H NMR (qNMR) have gained increasing attention as a method for quantitative determination of metabolites in complex biological matrices [
21,
22,
23]. According to the review by Pauli et al. (2012) [
22] and references therein, qNMR methods have proven successful when standard chromatographic methods have been ineffective [
22]. In general, qNMR can be considered a primary ratio method of measurement in which the analytes can be correlated directly to a calibration standard, and since the reference compound differs from the analytes, generating a calibration curve becomes unnecessary. However, the quantification needs to be validated with reference compounds. Some work on quantification of phlorotannins in brown algae (
Ascophyllum nodosum,
Fucus vesiculosus, and
Cystoseira tamariscifolia) with qNMR has been done using internal standards [
14,
23].
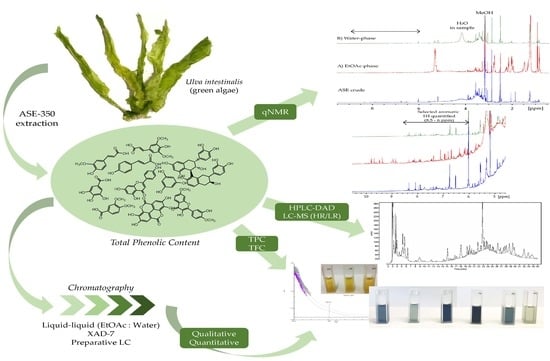
In this case study, we examined the polyphenolic content of the green algae Ulva intestinalis (syn. Enteromorpha intestinalis) collected on the west coast of Norway. An optimized extraction of the polyphenolic content was performed. The extract and semi-purified fractions were further analysed utilizing qNMR with an external reference for quantification of the total phenolic content. For comparison, HPLC-DAD and TPC assay analyses were also performed. To further explore the diverse group of polyphenols in Ulva intestinalis, qualitative analyses were performed with HPLC-DAD, HPLC-LR, and HR-MS. We entered this case study with the overarching goal of examining which analytical methods could lead to a more reliable value of polyphenolic content in seaweed and, thus, obtain a better view of the grand potential of seaweed phenolics.
2. Materials and Methods
2.1. Plant Materials
Samples of Ulva intestinalis (syn. Enteromorpha intestinalis) were collected in June from the western coast of Norway; Rogn, Ormhilleren (60°29’38.8” N 4°55’11.9” E). The voucher specimen of Ulva intestinalis was deposited in the Herbarium BG (Voucher no. BG-A-75) at the University Museum of Bergen, Bergen.
2.2. Chemicals
All chemicals used were of analytical grade. Methanol (≥99.9%), acetonitrile (≥99.8%), trifluoroacetic acid (TFA) and Folin-Ciocalteu reagent were all acquired form Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA). Formic (98–100%) and acetic (99.8%) acids were both acquired from Riedel-de Haën (Honeywell Inc., Charlotte, NC, USA). Luteolin, apigenin, myrcetin, diosmetin, quercetin, caffeic acid, coumaric acid, ferulic acid, sinapic acid, and gallic acid reference standards were all purchased from Sigma–Aldrich (Sigma-Aldrich, St. Louis, MO, USA). The analytical standard of tricin was purchased from PhytoLab (PhytoLab BmbH & Co. KG, Vestenbergsgreuth, Germany), (+)-catechin was purchased from USP (USP, Rockville, MD, USA), and DPPH free radical was purchased from Merck (Merck, Kenilworth, NJ, USA). Deionized water was deionized at the University of Bergen (Bergen, Norway).
2.3. Extraction and Purification
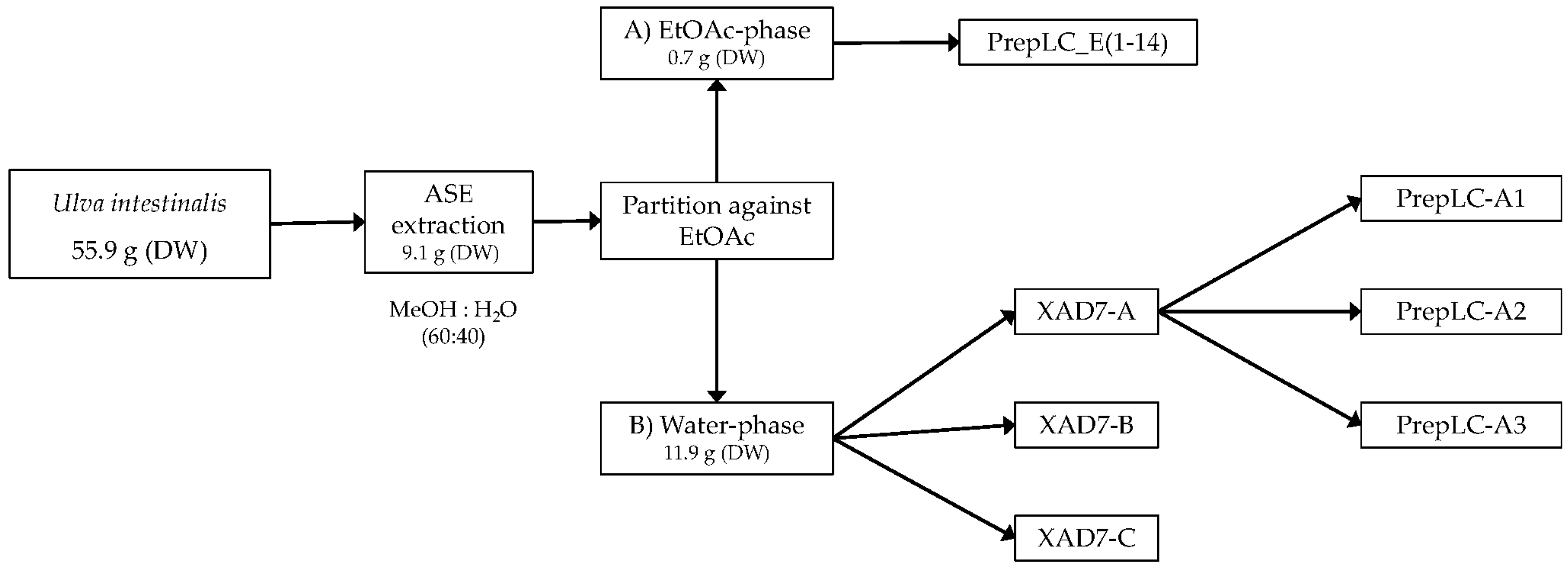
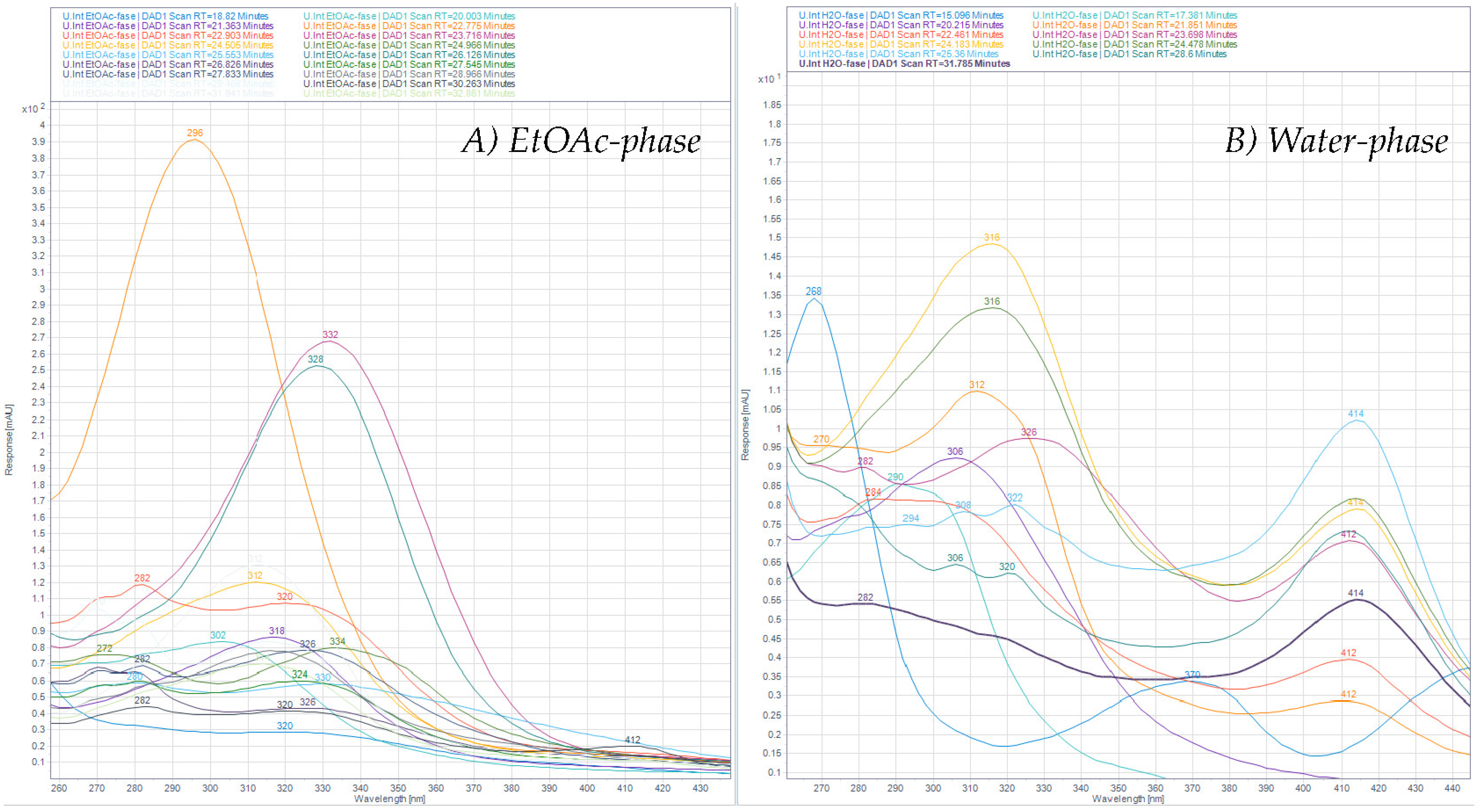
The collected plant material was washed thoroughly in fresh water and air dried. Dried plant material was stored at −20 °C when not used. Dried material was extracted using ASE (Accelerated Solvent Extraction) (Dionex™ ASE™ 350, Thermo Fisher Scientific, Waltham, MA, USA). A dried sample of Ulva intestinalis (55.9 g) was mixed with Dionex ASE prep DE sand and added to 66 mL stainless-steel cells with two glass fiber filters placed at the bottom end of the cell, before being extracted using a Dionex ASE 350 Accelerated Solvent Extractor. The extraction procedure consisted of two different methods, one being a pre-soak method, and the other being the primary extraction method. Pre-soaking consisted of extraction at 23 °C under 1500 psi. The static extraction period was 1 min with a flush volume of 50% of cell volume, purged with N2 for 70 s, and 100% deionized water was used as the solvent in the pre-soak method. The primary extraction method consisted of preheating for 5 min, and samples were then extracted at 70 °C under 1500 psi. Static extraction time was 5 min with a flush volume of 60% of the cell volume, purged with N2 for 100 sec. The solvent used for the primary extraction was a mixture of deionized water and methanol (40:60, v/v). Primary extraction was repeated two times. The volume of the combined extract was reduced using a rotavapor, and the concentrated aqueous extract was partitioned against ethyl acetate (EtOAc) four times. The contents of both the EtOAc phase and the water phase were examined using HPLC-DAD, HPLC-LRMS, HPLC-HRMS, and colorimetric assays including Total Phenolic Content Assay (TPC) and Total Flavonoid Content Assay (TFC). Before analysis, all phases were carefully reduced to dryness using rotavapor, and, finally, the samples were dried under N2 gas.
The aqueous extract was applied to an Amberlite XAD-7 column and washed with distilled water. Methanol was applied for elution. The pre-eluted washing water was analyzed for polyphenols with HPLC. Collected methanolic fractions (XAD7-A, XAD7-B, XAD7-C) were reduced using a rotavapor and analyzed on analytical HPLC. The XAD-7 fraction A contained the highest number of polyphenols and was chosen to be submitted to preparative HPLC to obtain three purified fractions; prepLC-A1, -A2, and -A3 (
Figure 1).
2.4. General Instrumentation
2.4.1. Preparative HPLC
The preparative HPLC system consisted of a Gilson 321 pump (Gilson Inc., Middleton, WI, USA), an Ultimate 3000 variable wavelength detector (Dionex, Thermo Fisher Scientific, Sunnyvale, CA, USA), and a 25 × 2.12 cm (10 µm) UniverSil C18 column (Fortis Technologies Ltd., Neston, UK). Two solvents were used: (A) super distilled water (0.1% acetic acid) and (B) acetonitrile (0.1% acetic acid) with initial conditions of 90% A and 10% B followed by an isocratic elution for the first 5 minutes, and the subsequent linear gradient conditions, 5–18 min: to 16% B, 18–22 min: to 18% B, 26–31 min: to 28% B, 31–32 min: to 40% B, 32–40 min: isocratic at 40% B, 40–43 min: to 10% B. The flow rate was 15 mL/min, and the aliquots of 750 µL were injected.
2.4.2. Analytical HPLC-DAD
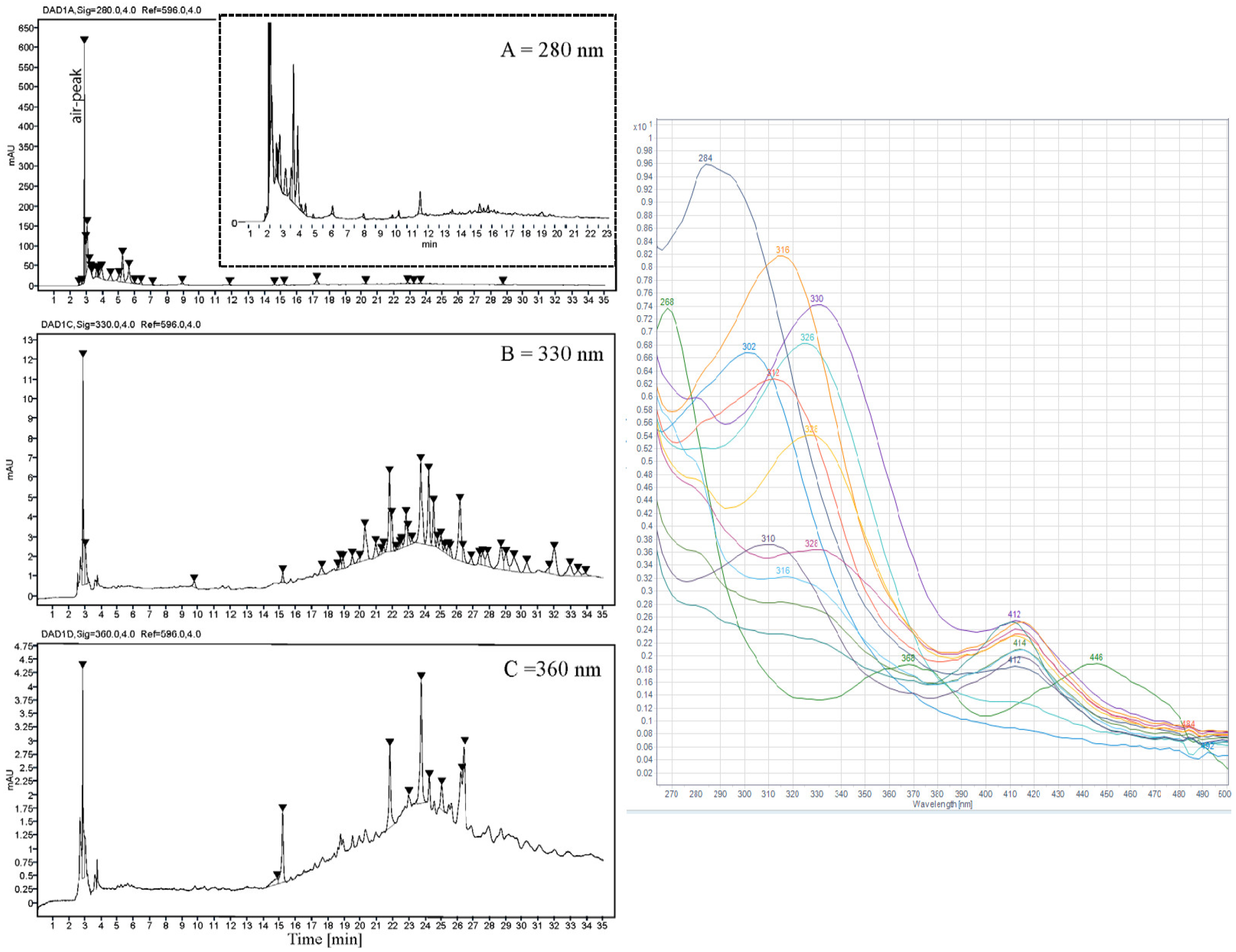
All HPLC-DAD analyses were performed on an Agilent 1260 Infinity HPLC system (Agilent Technologies, Santa Clara, CA, USA) equipped with a 1260 diode array detector (DAD) and a 200× C analysis was performed using two solvents, (A) super distilled water (0.5% TFA) and (B) acetonitrile (0.5% TFA), in a gradient (0–10 min: 95% A + 5% B, 10–20 min: 85% A + 15% B, 20–34 min: 60% A + 40% B. 34–35 min: 95% A + 5% B). The flow rate was 1.0 mL/min, and aliquots of 20 µL were injected with an Agilent 1260 vial sampler. UV-Vis absorption spectra were recorded during the HPLC analysis over the wavelength range of 200–600 nm in steps of 2 nm.
The established HPLC method was validated for linearity, sensitivity, precision, and accuracy.
Table 1 presents data for calibration curves, test ranges, limit of detection (LOD), and limit of quantification (LOQ) for gallic acid. The LOD and LOQ were calculated based on the standard deviation of
y-intercepts of the regression line (S
y) and the slope (S), using the equations LOD = 3.3 × S
y/S and LOQ = 10 × S
y/S.
2.4.3. HPLC-LRMS and HPLC-HRMS
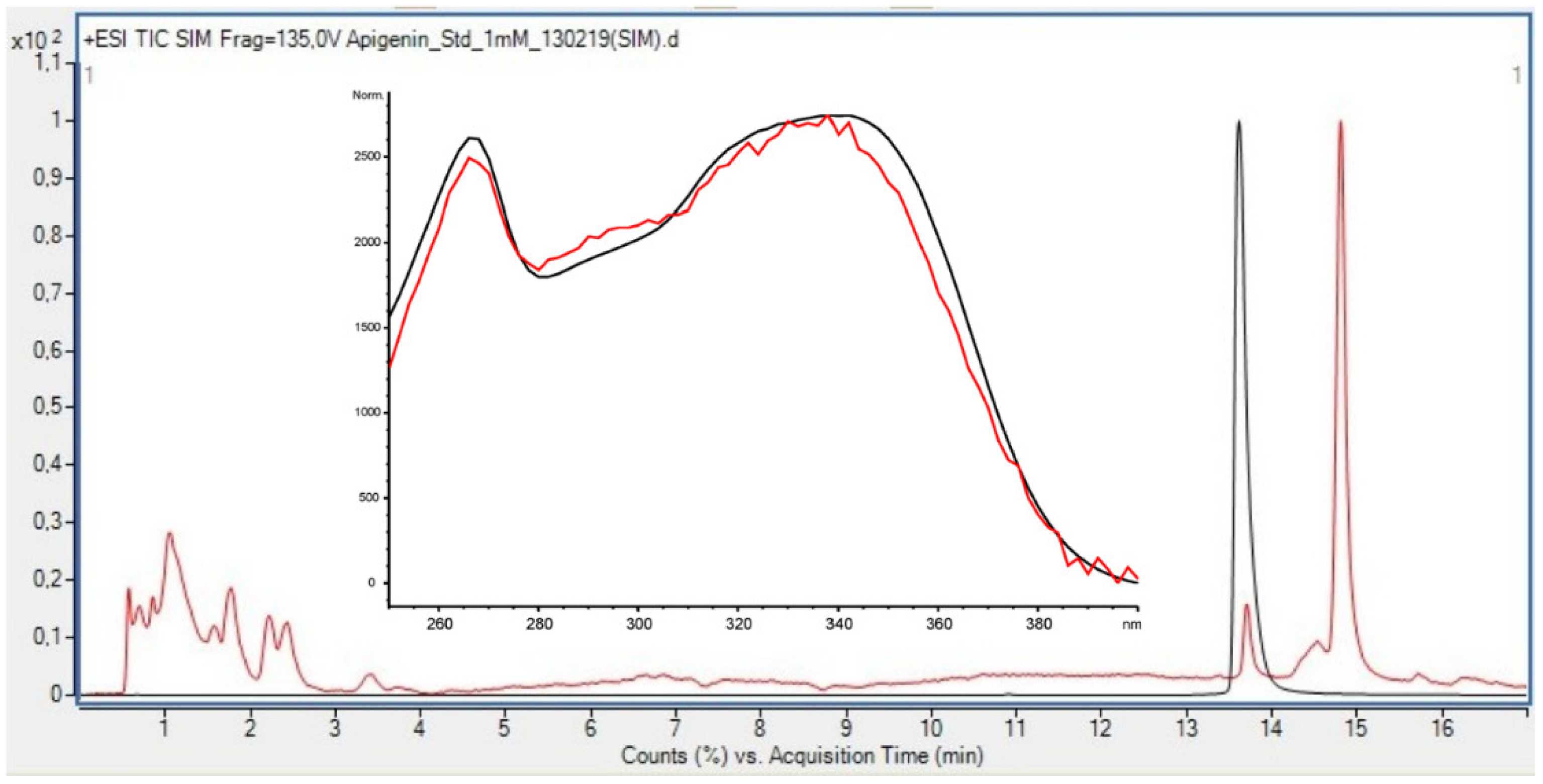
Liquid chromatography low-resolution mass spectrometry (HPLC-LRMS) (ESI+/ESI−) was performed using an Agilent Technologies 1260 Infinity Series system and an Agilent Technologies 6420A triple quadrupole mass spectrometry detector. The following conditions were applied: ionization mode: positive/negative, capillary voltage = 3000 V, gas temperature = 300 °C, gas flow rate = 3.0 L/min, acquisition range = 100–800
m/z. The elution profile for HPLC consisted of the following gradient: 0–3 min: 90%A + 10%B, 3–11 min: 86%A + 14%B, 11–15.5 min: 60%A + 40%B, 15.5–17 min: 90%A + 10%B, at a flowrate = 0.3 mL/min, where solvent A was super distilled water (0.5% formic acid), and solvent B was acetonitrile (0.5% formic acid). A 50 × 2.1 mm internal diameter, 1.8 µm Agilent Zorbax SB-C18 column was used for separation. Calibration curve of Apigenin ran on HPLC-LRMS and used for quantification is listed in
Table 2.
Liquid chromatography high-resolution mass spectrometry (HPLC-HRMS) (ESI+/TOF) was performed using an AccuTOF JMS-T100LC (JEOL, Peabody, USA) mass spectrometer in combination with an Agilent Technologies 1200 Series HPLC system. The following instrumental settings/conditions were used: ionization mode: positive, ion source temperature = 220 °C, needle voltage = 2500 V, desolvation gas flow = 4 L/min, nebulizing gas flow = 3 L/min, orifice1 temperature = 125 °C, orifice2 voltage = 10 V, ring lens voltage = 20 V, ion guide RF voltage = 1600 V, detector voltage = 2350 V, acquisition range = 15–1000 m/z, spectral recording interval = 0.50 sec, wait time = 0.033 nsec, and data sampling interval = 2 nsec. The elution profile for HPLC consisted of the same gradient and column as described for HPLC-LRMS, but the flowrate was increased to 0.35 mL/min.
2.4.4. NMR Spectroscopy
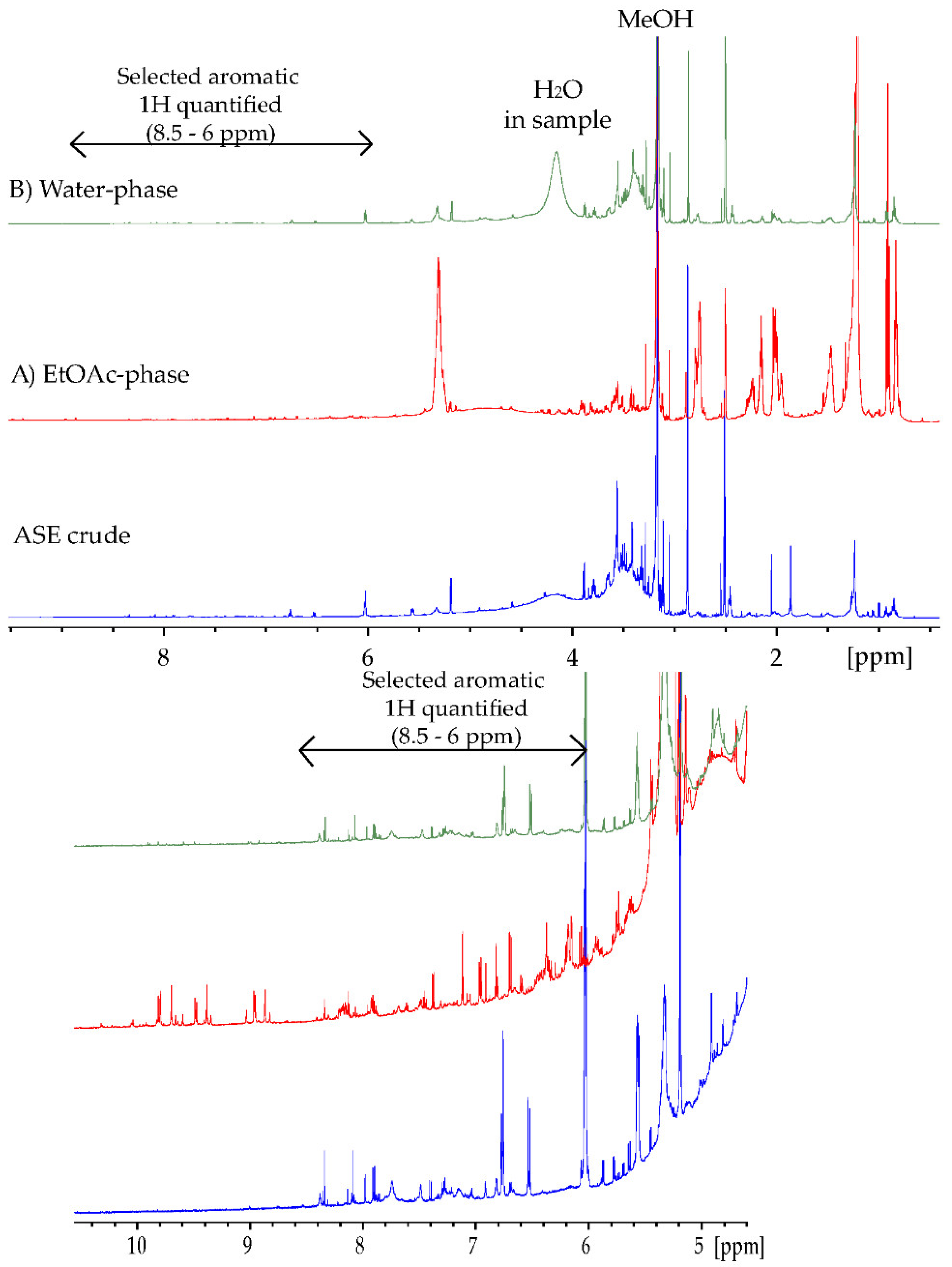
Quantification of the extracts of Ulva intestinalis was performed using 1H NMR analyses on a Bruker 600 MHz instrument (Bruker BioSpin, Zürich, Switzerland). All spectra were recorded in DMSO-d6 at 25 °C. The pulse sequence applied was zg30 with the following acquisition parameters: sweep width of 19.8 ppm, 64 k data points, 16 scans, and 2 dummy scans. The relaxation delay, d1, was set to 40 sec (equal to 5 × T1,max) to ensure complete relaxation between scans. The spectra were processed using a line broadening of 0.3 Hz. The crude extract was used for T1 measurements, utilizing the t1ir pulse sequence with a sweep width of 19.8 ppm, 16 k data points, 8 scans, 2 dummy scans, and 9 different inversion recovery delays between 1 ms and 5 s. Measured T1 values ranged from 1.0–8.1 s.
Quantification using the 1H NMR spectra was performed using the ERETIC2 function in TopSpin with DMSO2 (10 mM) as an external reference. The DMSO2 signal (~3.0 ppm) was integrated and defined as the ERETIC reference (No. H = 6, Mm = 94.13 g/mol, V(sample) = 0.75 mL, C = 10 mM).
Reference compounds for validation were gallic acid (GA), p-coumaric acid, ferulic acid, (+)-catechin, and luteolin (10 mM, DMSO-d6). An average standard deviation of < 10% was observed. The integrations were repeated three times.
Two-dimensional heteronuclear single quantum coherence (1H-13C HSQC), heteronuclear multiple bond correlation (1H-13C HMBC), and double quantum filtered correlation (1H-1H DQF COSY) spectra were also recorded on the Bruker 600 MHz instrument.
2.5. Total Phenolic Content Assay
For the determination of total phenolic content, the Folin-Ciocalteu total phenolic content assay (TPC) was used. The method used was adapted from Ainsworth and Gillespie (2007) [
24]. 200 µL of the sample or standard was added to the cuvettes (10 × 45 mm, 3 mL), followed by 400 µL 10% (
v/v) Folin–Ciocalteu reagent in super distilled water. Further, 1600 µL 700 mM Na
2CO
3 in super distilled water was added to the cuvettes. The mixture was incubated for 30 minutes, and the absorbance was measured at 765 nm using a Shimadzu UV-1800 UV spectrophotometer and a Shimadzu CPS-100 cell positioner (Shimadzu, Kyoto, Japan). Data was expressed as gallic acid equivalents (GAE). An incubation time of 2 h was also tested.
2.6. Total Flavonoid Content Assay
For the determination of the total flavonoid content, 2 mL test solution (standard or sample) was added to four cuvettes (10 × 45 mm, 3 mL) and the absorbance measured at 425 nm with solvent in the reference cuvette. An aliquot of AlCl3 solution (0.5 mL, 1%,
w/v) was added to three of the four cuvettes, and the same volume of solvent was added to the fourth (blank sample). The content of the cuvettes was stirred thoroughly, and the absorbance measured at 1 minute intervals at 425 nm for 10 minutes at 22 °C. For quantitative analysis apigenin was chosen as the reference compound (concentration range of 1–500 μg/mL). Procedure modified from Pękal and Pyrzynska (2014) [
25].
4. Conclusion
This case study provides an optimized extraction process for polyphenolic extraction of algae. The total polyphenolic content was quantified with qNMR (5.3%), HPLC-DAD (1.1%), and TPC (0.4%). Flavonoids and polyphenolic acids were tentatively identified in Ulva intestinalis samples. Apigenin was confirmed in one of the semi-purified fractions.
The same samples yielded different total phenolic contents when utilizing the different analytical methods, highlighting the difficulties related to polyphenolic quantification in extracts. All methods utilized in this study depend on assumptions and, thus, also uncertainty. This will be of special importance when analyzing complex samples at low concentrations as is the case for the polyphenolic content in marine algae. Further standardization and optimization of total phenolic quantifications of marine algae samples should be researched.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}