Hydroxycobalamin Reveals the Involvement of Hydrogen Sulfide in the Hypoxic Responses of Rat Carotid Body Chemoreceptor Cells

 , , and
, , and
Abstract
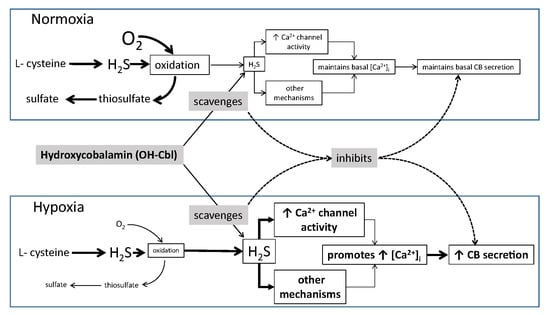
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Anesthesia Surgical Procedures
2.2. Isolation and Chemoreceptor Cell Culture
2.3. H-Catecholamine (3H-CA) Release Experiments Using Intact CBs
2.4. Intracellular Ca2+ Measurements
2.5. Statistics
3. Results
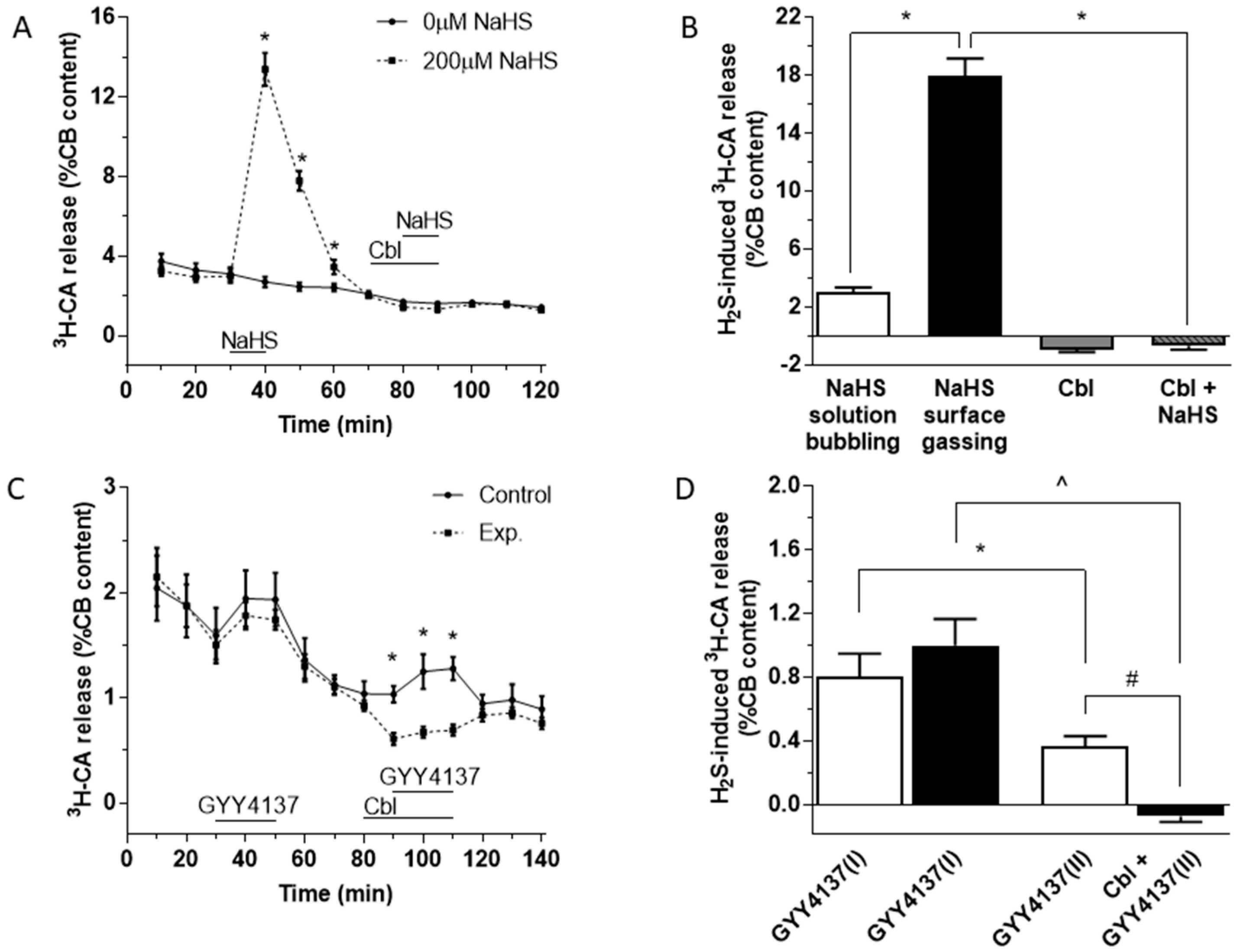
3.1. Effects of Cbl on the Release of 3H-CA Induced by Hydrogen Sulfide Donors
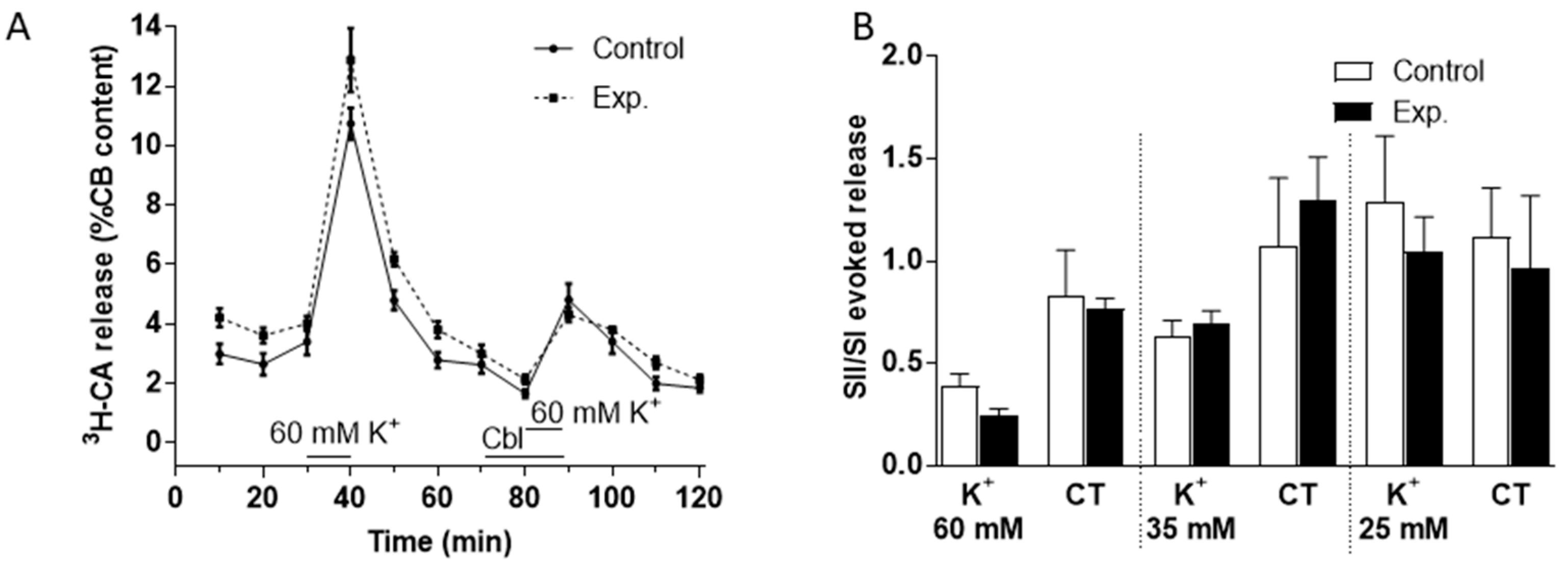
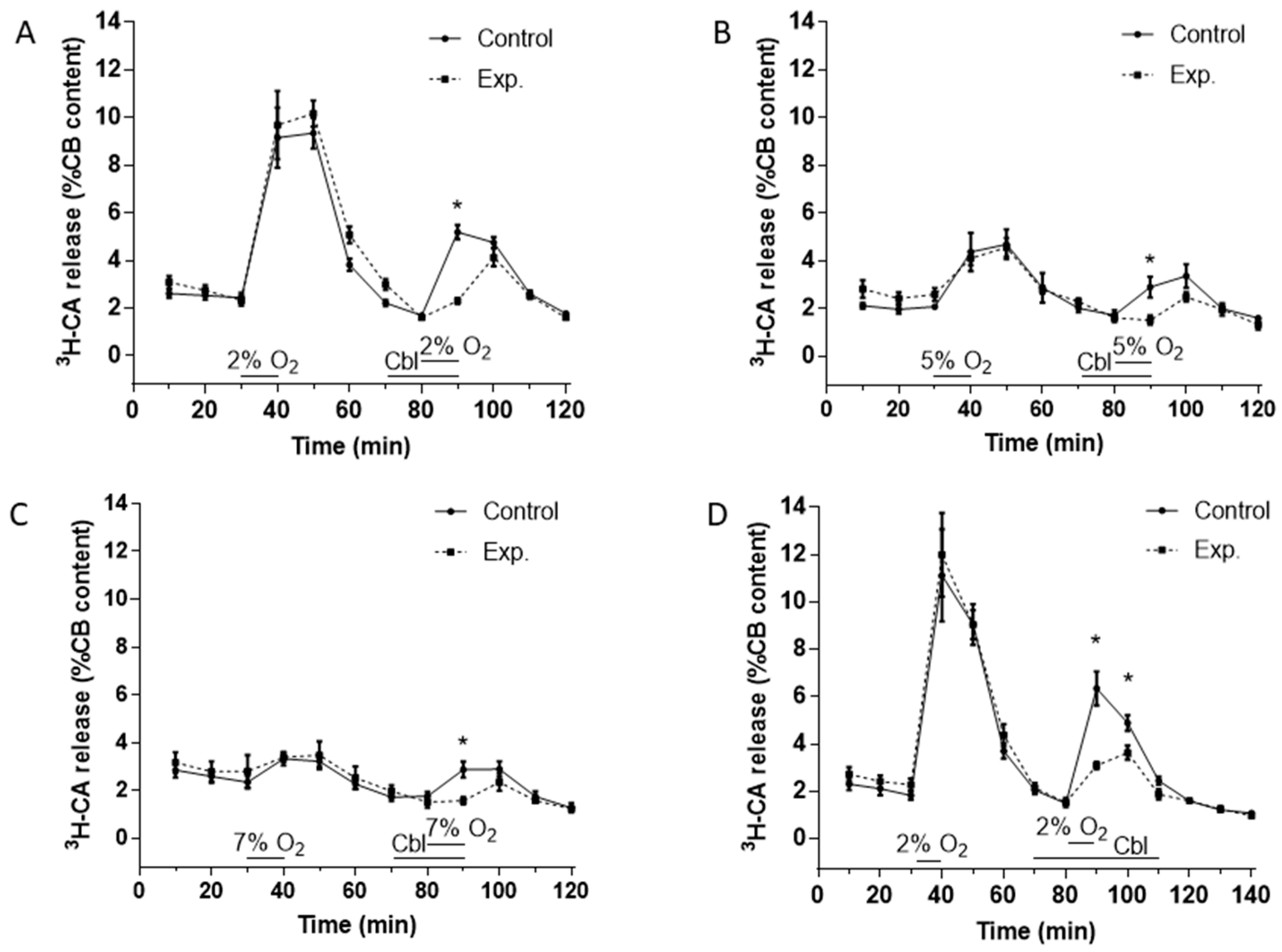
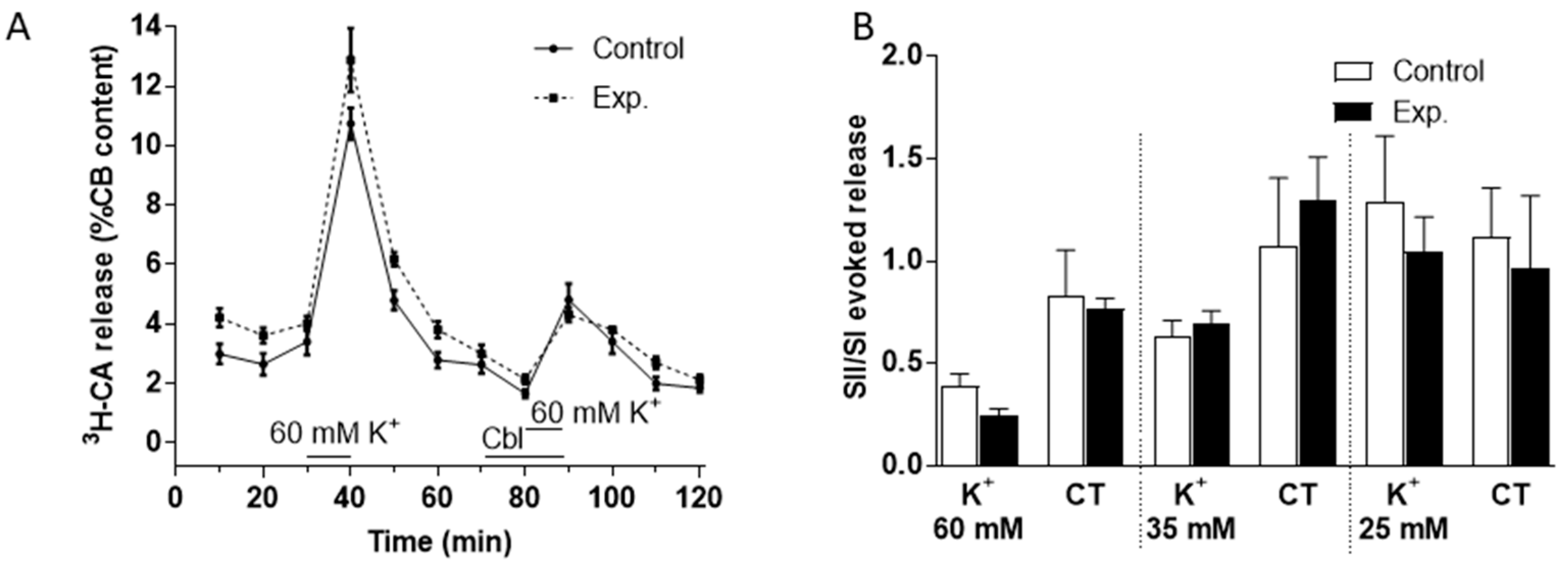
3.2. Effects of Cbl on the Release of 3H-CA Induced by Hypoxic and High External K+ Stimulation of the Carotid Body
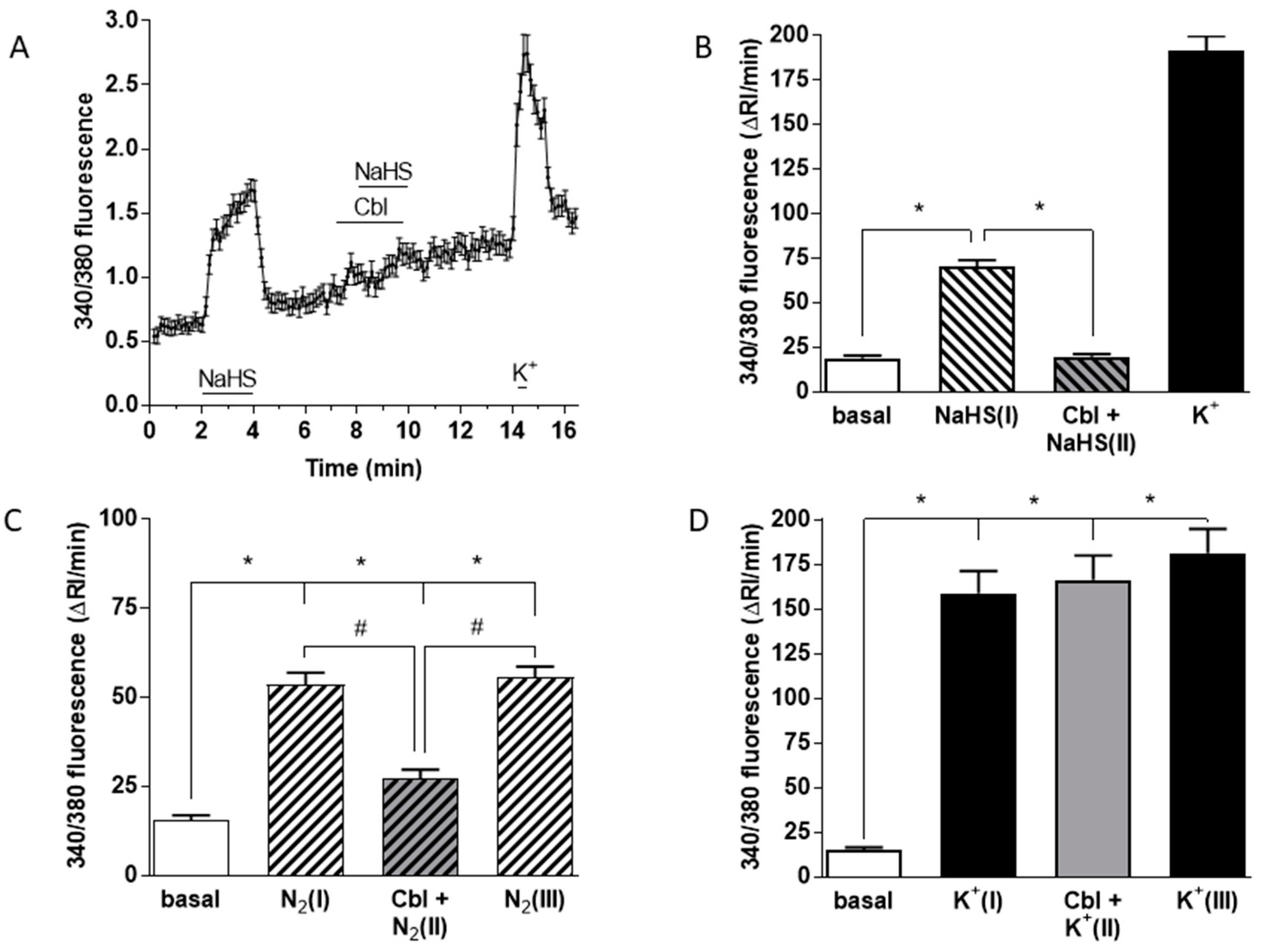
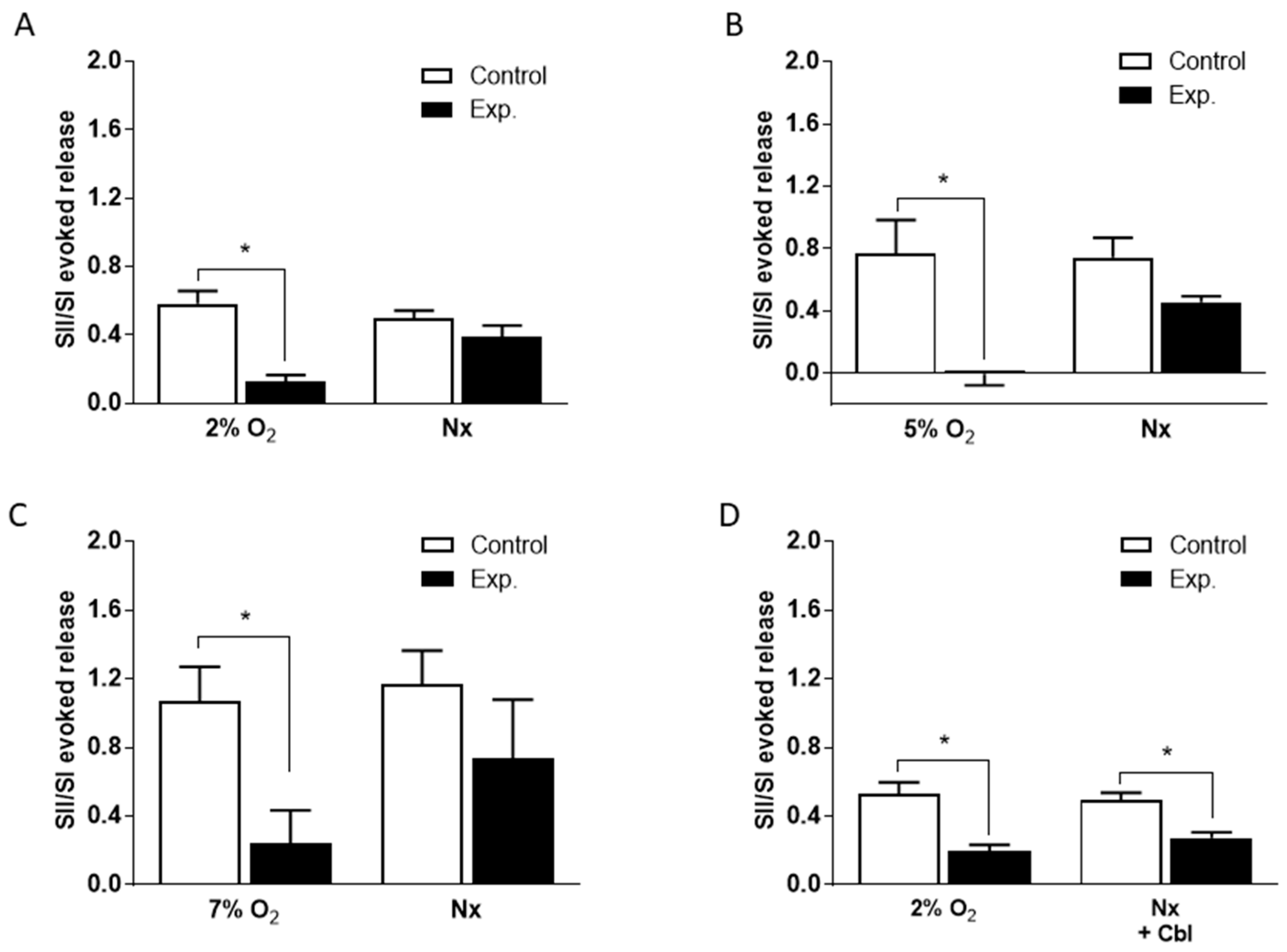
3.3. Effects of Cbl on the Intracellular Calcium Transients Induced by NaHS, Hypoxia, and High External K+
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fidone, S.; Gonzalez, C. Initiation and control of chemoreceptor activity in the carotid body. In Handbook of Physiology, Section 3, The Respiratory System, vol II, Control of Breathing; Fishman, A.P., Cherniack, N.S., Widdicombe, J.G., Geiger, S.R., Eds.; American Physiological Society: Bethesda, MD, USA, 1986; Volume 2, pp. 247–312. [Google Scholar]
- Gonzalez, C.; Almaraz, L.; Obeso, A.; Rigual, R. Carotid body chemoreceptors: From natural stimuli to sensory discharges. Physiol. Rev. 1994, 74, 829–898. [Google Scholar] [CrossRef]
- Olson, K.R.; Healy, M.J.; Qin, Z.; Skovgaard, N.; Vulesevic, B.; Duff, D.W.; Whitfield, N.L.; Yang, G.; Wang, R.; Perry, S.F. Hydrogen sulfide as an oxygen sensor in trout gill chemoreceptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R669–R680. [Google Scholar] [CrossRef]
- Peng, Y.J.; Nanduri, J.; Raghuraman, G.; Souvannakitti, D.; Gadalla, M.M.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. H2S mediates O2 sensing in the carotid body. Proc. Natl. Acad. Sci. USA 2010, 107, 10719–10724. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sun, B.; Wang, X.; Jin, Z.; Zhou, Y.; Dong, L.; Jiang, L.H.; Rong, W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid. Redox Signal. 2010, 12, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, V.V.; Nanduri, J.; Raghuraman, G.; Fox, A.P.; Gadalla, M.M.; Kumar, G.K.; Snyder, S.H.; Prabhakar, N.R. Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am. J. Physiol. Cell Physiol. 2012, 303, C916–C923. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Li, Q.; Sun, B.; Zhang, G.; Rong, W. Hydrogen sulfide activates the carotid body chemoreceptors in cat, rabbit and rat ex vivo preparations. Respir. Physiol. Neurobiol. 2015, 208, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, V.V.; Peng, Y.J.; Yuan, G.; Fox, A.P.; Kumar, G.K.; Nanduri, J.; Prabhakar, N.R. CaV3.2 T-type Ca2+ channels in H2S-mediated hypoxic response of the carotid body. Am. J. Physiol. Cell Physiol. 2015, 308, C146–C154. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Vasavda, C.; Peng, Y.J.; Makarenko, V.V.; Raghuraman, G.; Nanduri, J.; Gadalla, M.M.; Semenza, G.L.; Kumar, G.K.; Snyder, S.H.; et al. Protein kinase G-regulated production of H2S governs oxygen sensing. Sci. Signal. 2015, 8, ra37. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Hydrogen sulfide is an oxygen sensor in the carotid body. Respir. Physiol. Neurobiol. 2011, 179, 103. [Google Scholar] [CrossRef]
- Fitzgerald, R.S.; Shirahata, M.; Chang, I.; Kostuk, E.; Kiihl, S. The impact of hydrogen sulfide (H2S) on neurotransmitter release from the cat carotid body. Respir. Physiol. Neurobiol. 2011, 176, 80–89. [Google Scholar] [CrossRef]
- Haouzi, P.; Bell, H.; Philmon, M. Hydrogen sulfide oxidation and the arterial chemoreflex: Effect of methemoglobin. Respir. Physiol. Neurobiol. 2011, 177, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Van de Louw, A.; Haouzi, P. Inhibitory effects of hyperoxia and methemoglobinemia on H2S induced ventilatory stimulation in the rat. Respir. Physiol. Neurobiol. 2012, 181, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012, 463, 743–754. [Google Scholar] [CrossRef]
- Kim, D.; Kim, I.; Wang, J.; White, C.; Carroll, J.L. Hydrogen sulfide and hypoxia-induced changes in TASK (K2P3/9) activity and intracellular Ca2+ concentration in rat carotid body glomus cells. Respir. Physiol. Neurobiol. 2015, 215, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hogan, J.O.; Wang, R.; White, C.; Kim, D. Role of cystathionine-γ-lyase in hypoxia-induced changes in TASK activity, intracellular [Ca2+] and ventilation in mice. Respir. Physiol. Neurobiol. 2017, 246, 98–106. [Google Scholar] [CrossRef]
- Van de Louw, A.; Haouzi, P. Ferric Iron and Cobalt (III) compounds to safely decrease hydrogen sulfide in the body? Antioxid. Redox Signal. 2013, 19, 510–516. [Google Scholar] [CrossRef]
- Truong, D.H.; Mihajlovic, A.; Gunness, P.; Hindmarsh, W.; O’Brien, P.J. Prevention of hydrogen sulfide (H2S)-induced mouse lethality and cytotoxicity by hydroxocobalamin (vitamin B(12a)). Toxicology 2007, 242, 16–22. [Google Scholar] [CrossRef]
- Borron, S.W.; Baud, F.J.; Megarbane, B.; Bismuth, C. Hydroxocobalamin for severe acute cyanide poisoning by ingestion or inhalation. Am. J. Emerg. Med. 2007, 25, 551–558. [Google Scholar] [CrossRef]
- Quadros, E.V.; Sequeira, J.M. Cellular uptake of cobalamin: Transcobalamin and the TCblR/CD320 receptor. Biochimie 2013, 95, 1008. [Google Scholar] [CrossRef]
- Depeint, F.; Bruce, W.R.; Shangari, N.; Mehta, R.; O’Brien, P.J. Mitochondrial function and toxicity: Role of B vitamins on the one-carbon transfer pathways. Chem. Biol. Interact. 2006, 163, 113–132. [Google Scholar] [CrossRef]
- Berliner, N.; Rosenberg, L.E. Uptake and metabolism of free cyanocobalamin by cultured human fibroblasts from controls and a patient with transcobalamin II deficiency. Metabolism 1981, 30, 230. [Google Scholar] [CrossRef]
- Hall, C.A.; Hitzig, W.H.; Green, P.D.; Begley, J.A. Transport of therapeutic cyanocobalamin in the congenital deficiency of transcobalamin II (TC II). Blood 1979, 53, 251. [Google Scholar] [PubMed]
- Sukocheva, O.A.; Abramov, A.Y.; Levitskaya, J.O.; Gagelgans, A.I.; Carpenter, D.O. Modulation of intracellular Ca(2+) concentration by vitamin B12 in rat thymocytes. Blood Cells Mol. Dis. 2001, 27, 812. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.L.; Wang, C.C.; Huang, C.Y.; Wang, S.J. Cyanocobalamin, vitamin B12, depresses glutamate release through inhibition of voltage-dependent Ca2+ influx in rat cerebrocortical nerve terminals (synaptosomes). Eur. J. Pharmacol. 2009, 602, 230. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Hydrogen sulfide, reactive sulfur species and coping with reactive oxygen species. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef]
- Edwards, G.; Feletou, M.; Weston, A.H. Hydrogen sulfide as an endothelium-derived hyperpolarizing factor in rodent mesenteric arteries. Circ. Res. 2012, 110, e13–e14. [Google Scholar] [CrossRef]
- Gallego-Martin, T.; Agapito, T.; Ramirez, M.; Olea, E.; Yubero, S.; Rocher, A.; Gomez-Nino, A.; Gonzalez, C.; Obeao, A. Experimental observations on the biological significance of sulphide in the carotid body chemoreception. In Proceedings of the XIXth International Society for Arterial Chemoreception (ISAC), Leeds, UK, 29 June–3 July 2014. Unpublished conference paper. [Google Scholar]
- Vicario, I.; Rigual, R.; Obeso, A.; Gonzalez, C. Characterization of the synthesis and release of catecholamine in the rat carotid body in vitro. Am. J. Physiol. Cell Physiol. 2000, 278, C490–C499. [Google Scholar] [CrossRef]
- Conde, S.V.; Obeso, A.; Vicario, I.; Rigual, R.; Rocher, A.; Gonzalez, C. Caffeine inhibition of rat carotid body chemoreceptors is mediated by A2A and A2B adenosine receptors. J. Neurochem. 2006, 98, 616–628. [Google Scholar] [CrossRef]
- Gomez-Nino, A.; Obeso, A.; Baranda, J.A.; Santo-Domingo, J.; Lopez-Lopez, J.R.; Gonzalez, C. MaxiK potassium channels in the function of chemoreceptor cells of the rat carotid body. Am. J. Physiol. Cell Physiol. 2009, 297, C715–C722. [Google Scholar] [CrossRef]
- Gallego-Martin, T.; Fernandez-Martinez, S.; Rigual, R.; Obeso, A.; Gonzalez, C. Effects of low glucose on carotid body chemoreceptor cell activity studied in cultures of intact organs and in dissociated cells. Am. J. Physiol. Cell Physiol. 2012, 302, C1128–C1140. [Google Scholar] [CrossRef]
- Obeso, A.; Almaraz, L.; Gonzalez, C. Effects of cyanide and uncouplers on chemoreceptor activity and ATP content of the cat carotid body. Brain Res. 1989, 481, 250–257. [Google Scholar] [CrossRef]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): New insights into the biology of hydrogen sulfide. Circulation 2008, 117, 2351–2360. [Google Scholar] [CrossRef]
- Buckler, K.J.; Vaughan-Jones, R.D. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. J. Physiol. 1994, 476, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Kemp, P.J.; Telezhkin, V. Oxygen sensing by the carotid body: Is it all just rotten eggs? Antioxid. Redox Signal. 2014, 20, 794–804. [Google Scholar] [CrossRef]
- Garrod, M.G.; Green, R.; Allen, L.H.; Mungas, D.M.; Jagust, W.J.; Haan, M.N.; Miller, J.W. Fraction of total plasma vitamin B12 bound to transcobalamin correlates with cognitive function in elderly Latinos with depressive symptoms. Clin. Chem. 2008, 54, 1210. [Google Scholar] [CrossRef] [PubMed]
- Hitzig, W.H.; Dohmann, U.; Pluss, H.J.; Vischer, D. Hereditary transcobalamin II deficiency: Clinical findings in a new family. J. Pediatr. 1974, 85, 622. [Google Scholar] [CrossRef]
- Rocher, A.; Geijo-Barrientos, E.; Caceres, A.I.; Rigual, R.; Gonzalez, C.; Almaraz, L. Role of voltage-dependent calcium channels in stimulus-secretion coupling in rabbit carotid body chemoreceptor cells. J. Physiol. 2005, 562 Pt 2, 407. [Google Scholar] [CrossRef]
- Caceres, A.I.; Gonzalez-Obeso, E.; Gonzalez, C.; Rocher, A. RT-PCR and pharmacological analysis of L-and T-type calcium channels in rat carotid body. Adv. Exp. Med. Biol. 2009, 648, 105–112. [Google Scholar] [CrossRef]
- Conde, S.V.; Caceres, A.I.; Vicario, I.; Rocher, A.; Obeso, A.; Gonzalez, C. An overview on the homeostasis of Ca2+ in chemoreceptor cells of the rabbit and rat carotid bodies. Adv. Exp. Med. Biol. 2006, 580, 215. [Google Scholar] [PubMed]
- Olson, K.R.; DeLeon, E.R.; Liu, F. Controversies and conundrums in hydrogen sulfide biology. Nitric Oxide 2014, 41, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Sitdikova, G.F.; Fuchs, R.; Kainz, V.; Weiger, T.M.; Hermann, A. Phosphorylation of BK channels modulates the sensitivity to hydrogen sulfide (H2S). Front. Physiol. 2014, 5, 431. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Ogasawara, Y. Sulfur Atom in its Bound State Is a Unique Element Involved in Physiological Functions in Mammals. Molecules 2016, 21, 1753. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J. A novel oxygen-sensitive potassium current in rat carotid body type I cells. J. Physiol. 1997, 498 Pt 3, 649–662. [Google Scholar] [CrossRef]
- Donnelly, D.F. Are oxygen dependent K+ channels essential for carotid body chemo-transduction? Respir. Physiol. 1997, 110, 211. [Google Scholar] [CrossRef]
- Bijlenga, P.; Liu, J.H.; Espinos, E.; Haenggeli, C.A.; Fischer-Lougheed, J.; Bader, C.R.; Bernheim, L. T-type alpha 1H Ca2+ channels are involved in Ca2+ signaling during terminal differentiation (fusion) of human myoblasts. Proc. Natl. Acad. Sci. USA 2000, 97, 7627–7632. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.; Lory, P.; Mironneau, C.; Macrez, N.; Quignard, J.F. T-type CaV3.3 calcium channels produce spontaneous low-threshold action potentials and intracellular calcium oscillations. Eur. J. Neurosci. 2006, 23, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J.; Williams, B.A.; Honore, E. An oxygen-, acid- and anaesthetic-sensitive TASK-like background potassium channel in rat arterial chemoreceptor cells. J. Physiol. 2000, 525 Pt 1, 135–142. [Google Scholar] [CrossRef]
- Telezhkin, V.; Brazier, S.P.; Cayzac, S.; Muller, C.T.; Riccardi, D.; Kemp, P.J. Hydrogen sulfide inhibits human BK(Ca) channels. Adv. Exp. Med. Biol. 2009, 648, 65–72. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego-Martin, T.; Prieto-Lloret, J.; Aaronson, P.I.; Rocher, A.; Obeso, A. Hydroxycobalamin Reveals the Involvement of Hydrogen Sulfide in the Hypoxic Responses of Rat Carotid Body Chemoreceptor Cells. Antioxidants 2019, 8, 62. https://doi.org/10.3390/antiox8030062
Gallego-Martin T, Prieto-Lloret J, Aaronson PI, Rocher A, Obeso A. Hydroxycobalamin Reveals the Involvement of Hydrogen Sulfide in the Hypoxic Responses of Rat Carotid Body Chemoreceptor Cells. Antioxidants. 2019; 8(3):62. https://doi.org/10.3390/antiox8030062
Chicago/Turabian StyleGallego-Martin, Teresa, Jesus Prieto-Lloret, Philip I. Aaronson, Asuncion Rocher, and Ana Obeso. 2019. "Hydroxycobalamin Reveals the Involvement of Hydrogen Sulfide in the Hypoxic Responses of Rat Carotid Body Chemoreceptor Cells" Antioxidants 8, no. 3: 62. https://doi.org/10.3390/antiox8030062
APA StyleGallego-Martin, T., Prieto-Lloret, J., Aaronson, P. I., Rocher, A., & Obeso, A. (2019). Hydroxycobalamin Reveals the Involvement of Hydrogen Sulfide in the Hypoxic Responses of Rat Carotid Body Chemoreceptor Cells. Antioxidants, 8(3), 62. https://doi.org/10.3390/antiox8030062