Enzyme Selection and Hydrolysis under Optimal Conditions Improved Phenolic Acid Solubility, and Antioxidant and Anti-Inflammatory Activities of Wheat Bran

 ,
,  ,
, 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Enzymatic Activity Assays
2.3. Enzymatic Hydrolysis of WB
2.4. Identification and Quantification of Monosaccharides
2.5. Extraction and Quantification of Total Soluble Phenolic Compounds (TSPC)
2.6. Identification and Quantification of FA and Derivatives
2.7. Determination of Antioxidant Activity
2.8. Determination of Anti-Inflammatory Activity
2.9. Modeling and Optimization of Hydrolytic Treatments
2.10. Statistical Analysis
3. Results
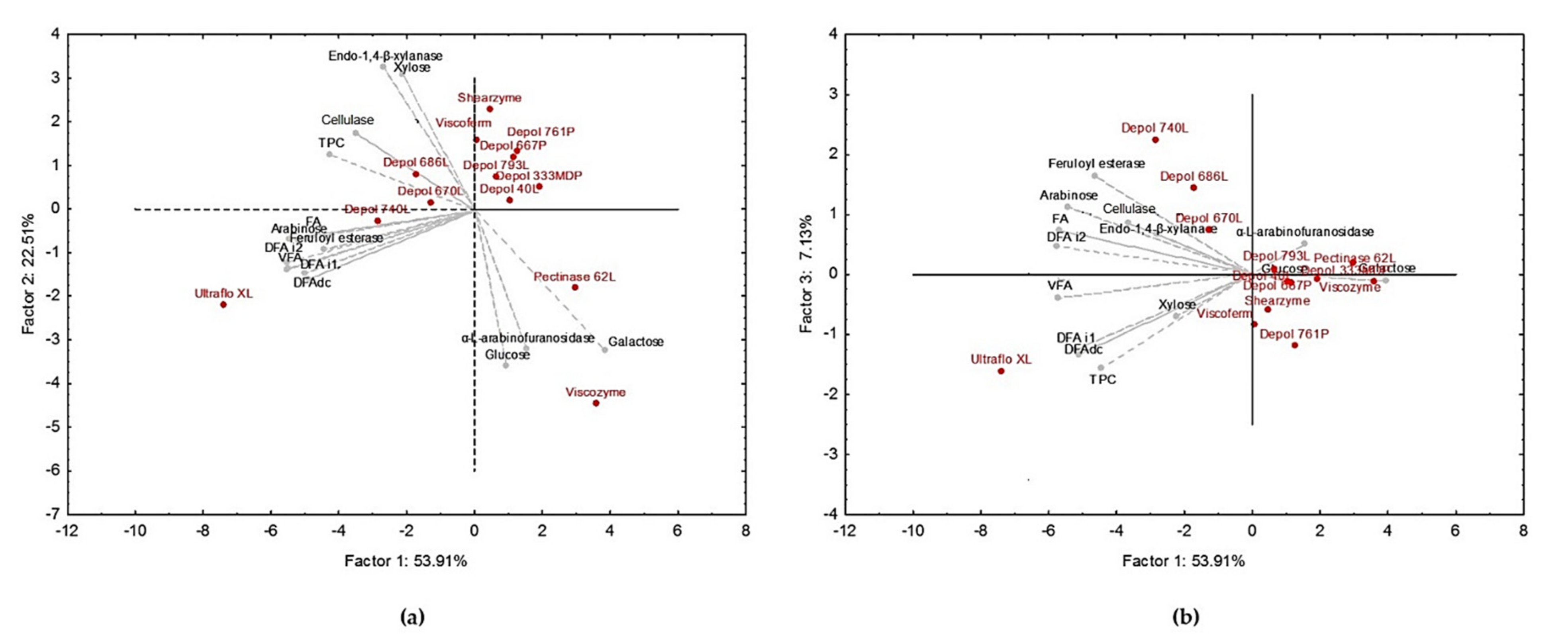
3.1. Ultraflo XL Showed Higher Efficiency for the Release of Bound Phenolic Acids from WB
3.2. Autoclave and Subsequent Ultraflo XL Treatment of WB Exerted Synergistic Effects on Solubilization of Phenolic Compounds
3.3. Hydrolysis Conditions Explained the Variation in the Solubilization Yield, Free Phenolic Compounds, and Antioxidant and Anti-Inflammatory Activities of WB Hydrolysates
3.4. Statistical Optimization of Temperature, pH, and Time during WB Enzymatic Hydrolysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barron, C.; Surget, A.; Rouau, X. Relative amounts of tissues in mature wheat (Triticum aestivum L.) grain and their carbohydrate and phenolic acid composition. J. Cereal Sci. 2007, 45, 88–96. [Google Scholar] [CrossRef]
- Rosa, N.N.; Dufour, C.; Lullien-Pellerin, V.; Micard, V. Exposure or release of ferulic acid from wheat aleurone: Impact on its antioxidant capacity. Food Chem. 2013, 141, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Yadav, M.P.; Singh, B.; Bhinder, S.; Simon, S.; Singh, N. Isolation and characterization of arabinoxylans from wheat bran and study of their contribution to wheat flour dough rheology. Carbohydr. Polym. 2019, 221, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Onipe, O.O.; Jideani, A.I.O.; Beswa, D. Composition and functionality of wheat bran and its application in some cereal food products. Int. J. Food Sci. Technol. 2015, 50, 2509–2518. [Google Scholar] [CrossRef]
- Yin, Z.; Wu, W.; Sun, C.; Lei, Z.; Chen, H.; Liu, H.; Chen, W.; Ma, J.; Min, T.; Zhang, M.; et al. Comparison of releasing bound phenolic acids from wheat bran by fermentation of three Aspergillus species. Int. J. Food Sci. Technol. 2018, 53, 1120–1130. [Google Scholar] [CrossRef]
- Maes, C.; Delcour, J.A. Structural Characterisation of Water-extractable and Water-unextractable Arabinoxylans in Wheat Bran. J. Cereal Sci. 2002, 35, 315–326. [Google Scholar] [CrossRef]
- Andersson, A.A.M.; Dimberg, L.; Aman, P.; Landberg, R. Recent findings on certain bioactive components in whole grain wheat and rye. J. Cereal Sci. 2014, 59, 294–311. [Google Scholar] [CrossRef]
- Vitaglione, P.; Mennella, I.; Ferracane, R.; Rivellese, A.A.; Giacco, R.; Ercolini, D.; Gibbons, S.M.; La Storia, A.; Gilbert, J.A.; Jonnalagadda, S.; et al. Whole-grain wheat consumption reduces inflammation in a randomized controlled trial on overweight and obese subjects with unhealthy dietary and lifestyle behaviors: Role of polyphenols bound to cereal dietary fiber. Am. J. Clin. Nutr. 2015, 101, 251–261. [Google Scholar] [CrossRef]
- Palani Swamy, S.k.; Govindaswamy, V. Therapeutical properties of ferulic acid and bioavailability enhancement through feruloyl esterase. J. Funct. Foods 2015, 17, 657–666. [Google Scholar] [CrossRef]
- Vangsøe, C.T.; Sørensen, J.F.; Bach Knudsen, K.E. Aleurone cells are the primary contributor to arabinoxylan oligosaccharide production from wheat bran after treatment with cell wall-degrading enzymes. Int. J. Food Sci. Technol. 2019, 54, 2847–2853. [Google Scholar] [CrossRef]
- Wang, Z.; Li, S.; Ge, S.; Lin, S. Review of Distribution, Extraction Methods, and Health Benefits of Bound Phenolics in Food Plants. J. Agric. Food Chem. 2020, 68, 3330–3343. [Google Scholar] [CrossRef] [PubMed]
- Kroon, P.; Garcia-Conesa, M.; Fillingham, I.; Hazlewood, G.; Williamson, G. Release of ferulic acid dehydrodimers from plant cell walls by feruloyl esterases. J. Sci. Food Agric. 1999, 79, 428–434. [Google Scholar] [CrossRef]
- Mateo Anson, N.; van den Berg, R.; Havenaar, R.; Bast, A.; Haenen, G.R.M.M. Bioavailability of ferulic acid is determined by its bioaccessibility. J. Cereal Sci. 2009, 49, 296–300. [Google Scholar] [CrossRef]
- Ribas-Agustí, A.; Martín-Belloso, O.; Soliva-Fortuny, R.; Elez-Martínez, P. Food processing strategies to enhance phenolic compounds bioaccessibility and bioavailability in plant-based foods. Crit. Rev. Food Sci. Nutr. 2018, 58, 2531–2548. [Google Scholar] [CrossRef] [PubMed]
- Ferri, M.; Happel, A.; Zanaroli, G.; Bertolini, M.; Chiesa, S.; Commisso, M.; Guzzo, F.; Tassoni, A. Advances in combined enzymatic extraction of ferulic acid from wheat bran. New Biotechnol. 2020, 56, 38–45. [Google Scholar] [CrossRef]
- Dupoiron, S.; Lameloise, M.-L.; Pommet, M.; Bennaceur, O.; Lewandowski, R.; Allais, F.; Teixeira, A.R.S.; Rémond, C.; Rakotoarivonina, H. A novel and integrative process: From enzymatic fractionation of wheat bran with a hemicellulasic cocktail to the recovery of ferulic acid by weak anion exchange resin. Ind. Crop. Prod. 2017, 105, 148–155. [Google Scholar] [CrossRef]
- Dinelli, G.; Segura-Carretero, A.; Di Silvestro, R.; Marotti, I.; Arraez-Roman, D.; Benedettelli, S.; Ghiselli, L.; Fernadez-Gutierrez, A. Profiles of phenolic compounds in modern and old common wheat varieties determined by liquid chromatography coupled with time-of-flight mass spectrometry. J. Chromatogr. A 2011, 1218, 7670–7681. [Google Scholar] [CrossRef]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. [14] Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1999; Volume 299, pp. 152–178. [Google Scholar]
- Garcia-Mora, P.; Peñas, E.; Frias, J.; Martínez-Villaluenga, C. Savinase, the most suitable enzyme for releasing peptides from lentil (Lens culinaris var. Castellana) protein concentrates with multifunctional properties. J. Agric. Food Chem. 2014, 62, 4166–4174. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free-radical method to evaluate antioxidant activity. Food Sci. Technol. -Lebensm. -Wiss. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Candioti, L.V.; De Zan, M.M.; Camara, M.S.; Goicoechea, H.C. Experimental design and multiple response optimization. Using the desirability function in analytical methods development. Talanta 2014, 124, 123–138. [Google Scholar] [CrossRef] [PubMed]
- FAO (Food and Agriculture Organization of the United Nations). FAOSTAT. Wheat Bran Production. Available online: www.faostat.org (accessed on 3 August 2020).
- Fritsch, C.; Staebler, A.; Happel, A.; Marquez, M.A.C.; Aguilo-Aguayo, I.; Abadias, M.; Gallur, M.; Cigognini, I.M.; Montanari, A.; Lopez, M.J.; et al. Processing, Valorization and Application of Bio-Waste Derived Compounds from Potato, Tomato, Olive and Cereals: A Review. Sustainability 2017, 9, 1492. [Google Scholar] [CrossRef]
- Baeza, G.; Sarriá, B.; Bravo, L.; Mateos, R. Polyphenol content, in vitro bioaccessibility and antioxidant capacity of widely consumed beverages. J. Sci. Food Agric. 2018, 98, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Malgas, S.; Mafa, M.S.; Mkabayi, L.; Pletschke, B.I. A mini review of xylanolytic enzymes with regards to their synergistic interactions during hetero-xylan degradation. World J. Microbiol. Biotechnol. 2019, 35, 187. [Google Scholar] [CrossRef]
- Spaggiari, M.; Calani, L.; Folloni, S.; Ranieri, R.; Dall’Asta, C.; Galaverna, G. The impact of processing on the phenolic acids, free betaine and choline in Triticum spp. L. whole grains and milling by-products. Food Chem. 2020, 311. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, Y.; Dong, H.; Hou, H.; Zhang, X. Distribution of Phenolic Acids and Antioxidant Activities of Different Bran Fractions from Three Pigmented Wheat Varieties. J. Chem. 2018, 2018, 6459243. [Google Scholar] [CrossRef]
- Kim, S.-M.; Lim, S.-T. Enhanced antioxidant activity of rice bran extract by carbohydrase treatment. J. Cereal Sci. 2016, 68, 116–121. [Google Scholar] [CrossRef]
- Xue, Y.; Cui, X.; Zhang, Z.; Zhou, T.; Gao, R.; Li, Y.; Ding, X. Effect of β-endoxylanase and α-arabinofuranosidase enzymatic hydrolysis on nutritional and technological properties of wheat brans. Food Chem. 2020, 302, 125332. [Google Scholar] [CrossRef]
- Faulds, C.B.; Mandalari, G.; LoCurto, R.; Bisignano, G.; Waldron, K.W. Arabinoxylan and mono- and dimeric ferulic acid release from brewer’s grain and wheat bran by feruloyl esterases and glycosyl hydrolases from Humicola insolens. Appl. Microbiol. Biotechnol. 2004, 64, 644–650. [Google Scholar] [CrossRef]
- Călinoiu, L.F.; Vodnar, D.C. Thermal Processing for the Release of Phenolic Compounds from Wheat and Oat Bran. Biomolecules 2020, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Huang, L.; Zhang, Y. Effect of steam explosion treatment on barley bran phenolic compounds and antioxidant activity. J. Agric. Food Chem. 2012, 60, 7177–7184. [Google Scholar] [CrossRef] [PubMed]
- Rico, D.; Villaverde, A.; Martinez-Villaluenga, C.; Gutierrez, A.L.; Caballero, P.A.; Ronda, F.; Peñas, E.; Frias, J.; Martin Diana, A.B. Application of Autoclave Treatment for Development of a Natural Wheat Bran Antioxidant Ingredient. Foods 2020, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.K.; Li, L.L.; Long, L.K.; Ding, S.J. Comprehensive evaluation of combining hydrothermal pretreatment (autohydrolysis) with enzymatic hydrolysis for efficient release of monosaccharides and ferulic acid from corn bran. Ind. Crop. Prod. 2018, 113, 348–357. [Google Scholar] [CrossRef]
- Lau, T.; Harbourne, N.; Oruña-Concha, M.J. Optimization of enzyme-assisted extraction of ferulic acid from sweet corn cob by response surface methodology. J. Sci. Food Agric. 2020, 100, 1479–1485. [Google Scholar] [CrossRef]
- Zeuner, B.; Ståhlberg, T.; van Buu, O.N.; Kunov-Kruse, A.J.; Riisager, A.; Meyer, A.S. Dependency of the hydrogen bonding capacity of the solvent anion on the thermal stability of feruloyl esterases in ionic liquid systems. Green Chem. 2011, 13, 1550–1557. [Google Scholar] [CrossRef]
- Aluko, R.E.; Girgih, A.T.; He, R.; Malomo, S.; Li, H.; Offengenden, M.; Wu, J. Structural and functional characterization of yellow field pea seed (Pisum sativum L.) protein-derived antihypertensive peptides. Food Res. Int. 2015, 77, 10–16. [Google Scholar] [CrossRef]
- Zhou, K.; Su, L.; Yu, L. Phytochemicals and Antioxidant Properties in Wheat Bran. J. Agric. Food Chem. 2004, 52, 6108–6114. [Google Scholar] [CrossRef]
- Si, D.; Shang, T.; Liu, X.; Zheng, Z.; Hu, Q.; Hu, C.; Zhang, R. Production and characterization of functional wheat bran hydrolysate rich in reducing sugars, xylooligosaccharides and phenolic acids. Biotechnol. Rep. 2020, 27, e00511. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, H.; Wu, L.; Ma, W.; Xie, Y. Antioxidant properties of feruloylated oligosaccharides of different degrees of polymerization from wheat bran. Glycoconj. J. 2018, 35, 547–559. [Google Scholar] [CrossRef]
- Amorati, R.; Pedulli, G.F.; Cabrini, L.; Zambonin, L.; Landi, L. Solvent and pH effects on the antioxidant activity of caffeic and other phenolic acids. J. Agric. Food Chem. 2006, 54, 2932–2937. [Google Scholar] [CrossRef] [PubMed]
- Neacsu, M.; McMonagle, J.; Fletcher, R.J.; Hulshof, T.; Duncan, S.H.; Scobbie, L.; Duncan, G.J.; Cantlay, L.; Horgan, G.; de Roos, B.; et al. Availability and dose response of phytophenols from a wheat bran rich cereal product in healthy human volunteers. Mol. Nutr. Food Res. 2017, 61, 1600202. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.-Y.; Chen, Y.-K.; Chen, H.-H.; Lin, S.-Y.; Fang, Y.-T. Immunomodulatory effects of feruloylated oligosaccharides from rice bran. Food Chem. 2012, 134, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bai, J.; Fan, M.; Li, T.; Li, Y.; Qian, H.; Wang, L.; Zhang, H.; Qi, X.; Rao, Z. Cereal-derived arabinoxylans: Structural features and structure–activity correlations. Trends Food Sci. Technol. 2020, 96, 157–165. [Google Scholar] [CrossRef]
- Sørensen, H.R.; Pedersen, S.; Meyer, A.S. Characterization of solubilized arabinoxylo-oligosaccharides by MALDI-TOF MS analysis to unravel and direct enzyme catalyzed hydrolysis of insoluble wheat arabinoxylan. Enzym. Microb. Technol. 2007, 41, 103–110. [Google Scholar] [CrossRef]


{kind=link}
| Enzyme | Glucose | Xylose | Arabinose | Galactose | SUM | YS |
|---|---|---|---|---|---|---|
| Control | 20.36 ± 6.21 e | 0.38 ± 0.20 j | 0.33 ± 0.04 e | 2.27 ± 0.14 def | 23.34 | 51.32 |
| Depol 333MDP | 44.62 ± 13.07 bcd | 12.61 ± 1.19 gh | 1.11 ± 0.17 de | 2.89 ± 0.63 bcd | 61.23 | 55.10 |
| Depol 40L | 58.67 ± 15.28 b | 15.35 ± 2.98 efg | 2.17 ± 0.77 d | 3.22 ± 0.55 bc | 79.41 | 64.59 |
| Depol 667P | 46.01 ± 12.75 bcd | 11.37 ± 1.67 h | 1.05 ± 0.10 de | 2.40 ± 0.17 de | 60.83 | 61.07 |
| Depol 670L | 46.59 ± 9.46 bcd | 20.25 ± 3.34 c | 6.06 ± 0.96 bc | 2.03 ± 0.62 ef | 74.93 | 59.95 |
| Depol 686L | 46.55 ± 2.92 bcd | 17.55 ± 1.23 cde | 6.94 ± 1.96 b | 1.54 ± 0.46 f | 72.54 | 64.56 |
| Depol 740L | 52.58 ± 6.93 bcd | 16.06 ± 0.65 def | 11.38 ± 1.89 a | 2.28 ± 0.00 def | 82.30 | 62.54 |
| Depol 761P | 37.78 ± 2.43 d | 18.47 ± 2.47 cd | 1.20 ± 0.17 de | 2.45 ± 0.30 cde | 59.90 | 53.59 |
| Depol 793L | 48.94 ± 7.43 bcd | 13.36 ± 1.63 fgh | 1.17 ± 0.81 de | 2.45 ± 0.55 de | 65.92 | 57.43 |
| Pectinase 62L | 49.21 ± 3.83 bcd | 5.67 ± 0.94 i | 1.31 ± 0.73 de | 3.27 ± 0.29 b | 59.46 | 59.32 |
| Shearzyme | 41.07 ± 6.01 cd | 28.39 ± 2.89 b | 1.93 ± 1.03 d | 1.85 ± 0.13 ef | 73.24 | 65.88 |
| Ultraflo XL | 56.94 ± 18.31 bc | 17.49 ± 1.27 de | 11.05 ± 2.18 a | 1.78 ± 0.64 ef | 87.26 | 79.15 |
| Viscoferm | 58.76 ± 6.45 b | 31.53 ± 3.30 a | 4.86 ± 0.29 c | 1.88 ± 0.25 ef | 97.03 | 68.42 |
| Viscozyme | 90.33 ± 6.60 a | 4.28 ± 0.18 i | 1.28 ± 0.60 de | 5.06 ± 0.18 a | 100.95 | 68.88 |
| Enzyme | TSPC | FA | DFA i1 | DFA i2 | DFA dc | VFA |
|---|---|---|---|---|---|---|
| Control | 2.80 ± 0.33 e | 11.64 ± 4.64 f | nd | nd | nd | 3.58 ± 1.03 c |
| Depol 333MDP | 3.30 ± 0.09 de | 53.40 ± 4.48 ef | 24.45 ± 2.16 b | 32.30 ± 0.15 d | 3.30 ± 0.82 d | 4.98 ± 5.81 c |
| Depol 40L | 4.36 ± 0.48 b | 120.10 ± 17.82 de | 19.18 ± 0.85 b | 39.75 ± 0.82 d | 8.63 ± 2.81 d | 20.64 ± 11.46 c |
| Depol 667P | 4.07 ± 0.13 bc | 43.51 ± 3.33 ef | 29.64 ± 2.75 b | 31.02 ± 0.03 d | 2.77 ± 0.75 d | 10.86 ± 1.72 c |
| Depol 670L | 4.38 ± 0.37 b | 528.93 ± 84.87 b | 60.27 ± 3.56 b | 340.92 ± 28.40 c | 73.96 ± 19.76 bc | 63.66 ± 6.46 b |
| Depol 686L | 3.61 ± 0.09 cd | 497.17 ± 67.03 b | 60.77 ± 3.51 b | 419.88 ± 0.27 bc | 85.35 ± 13.46 bc | 76.87 ± 3.73 b |
| Depol 740L | 4.14 ± 0.45 bc | 498.19 ± 82.85 b | 80.51 ± 5.15 b | 498.87 ± 1.65 b | 101.56 ± 12.66 b | 75.53 ± 29.96 b |
| Depol 761P | 4.52 ± 0.09 b | 49.97 ± 1.59 ef | 30.44 ± 0.04 b | 31.56 ± 4.43 d | 4.24 ± 1.66 d | 18.27 ± 13.98 c |
| Depol 793L | 4.27 ± 0.45 b | 240.56 ± 47.63 c | 25.41 ± 3.21 b | 42.57 ± 2.10 d | 15.81 ± 6.10 d | 11.35 ± 1.62 c |
| Pectinase 62L | 3.11 ± 0.10 de | 6.55 ± 1.79 f | 1.86 ± 0.39 b | 5.96 ± 0.06 d | 0.22 ± 0.31 d | 1.77 ± 0.49 c |
| Shearzyme | 4.33 ± 0.41 b | 111.43 ± 11.24 de | 28.00 ± 1.72 b | 38.42 ± 2.25 d | 15.72 ± 2.38 d | 8.20 ± 0.28 c |
| Ultraflo XL | 5.49 ± 0.64 a | 799.50 ± 34.19 a | 555.53 ± 16.12 a | 586.46 ± 13.06 a | 124.37 ± 87.85 a | 233.91 ± 13.96 a |
| Viscoferm | 4.65 ± 0.51 b | 179.75 ± 20.38 cd | 29.90 ± 2.73 b | 37.38 ± 2.58 d | 31.76 ± 10.27 cd | 13.34 ± 1.28 c |
| Viscozyme | 3.16 ± 0.14 de | 51.00 ± 5.35 ef | 12.89 ± 0.83 b | 31.57 ± 3.78 d | 3.99 ± 1.19 d | 8.19 ± 2.52 c |
| Treatment | TSPC | FA |
|---|---|---|
| Control | 2.80 ± 0.33 c | 11.64 ± 4.64 d |
| Autoclave | 6.85 ± 0.90 b | 478.24 ± 4.34 c |
| Ultraflo XL | 5.49 ± 0.64 b | 799.50 ± 34.19 b |
| Autoclave + Ultraflo XL | 12.61 ± 1.06 a | 3835.34 ± 119.78 a |
| T | pH | t | Ys | TSPC | FA | ORAC1 | ABTS1 | DPPH1 | FRAP2 | MCP-13 | IL-63 | TNF-α3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 55 | 3.32 | 14 | 42.9 ± 0.04 d | 564.6 ± 37.9 bc | 4.0 ± 0.7 h | 13.78 ± 2.01 de | 13.26 ± 1.08 cde | 4.21 ± 0.72 defg | 0.75 ± 0.11 fg | 28.55 ± 4.1 c | 2.74 ± 3.26 e | 50.6 ± 1.4 cd |
| 50 | 4 | 6 | 46.6 ± 10.2 bcd | 514.2 ± 39.4 cd | 77.1 ± 6.7 g | 16.99 ± 1.93 bcd | 14.61 ± 0.84 bcd | 6.09 ± 1.35 a | 0.85 ± 0.02 cd | 20.30 ± 7.7 d | 35.9 ± 10.4 b | 52.8 ± 9.8 bcd |
| 60 | 4 | 6 | 45.7 ± 8.8 cd | 447.6 ± 42.8 def | 6.7 ± 0.7 h | 15.16 ± 2.01 bcde | 13.28 ± 0.82 cde | 4.31 ± 1.30 def | 0.77 ± 0.01 ef | 41.02 ± 6.0 b | 17.1 ± 2.80 cd | 45.3 ± 4.2 d |
| 50 | 4 | 22 | 64.8 ± 5.6 abcd | 703.3 ± 25.8 a | 125.8 ± 1.5 f | 22.67 ± 2.07 a | 17.04 ± 0.53 a | 5.90 ± 0.28 a | 0.94 ± 0.02 a | 36.13 ± 2.0 ef | 60.1 ± 5.77 a | nd |
| 60 | 4 | 22 | 65.4 ± 11.9 abcd | 588.6 ± 35.3 b | 7.1 ± 0.5 h | 17.99 ± 1.57 bc | 15.37 ± 2.52 abc | 5.00 ± 0.16 bcd | 0.87 ± 0.01 bc | nd | 10.9 ± 0.9 de | nd |
| 55 | 5 | 0.5 | 61.1 ± 15.0 abcd | 498.4 ± 34.6 cde | 88.6 ± 3.0 g | 13.72 ± 3.41 de | 11.26 ± 1.47 ef | 4.18 ± 0.45 defg | 0.74 ± 0.05 fg | 5.29 ± 9.1 fg | 22.7 ± 9.9 c | 0.3 ± 0.5 ef |
| 47 | 5 | 14 | 64.1 ± 19.4 abcd | 562.3 ± 16.4 bc | 287.1 ± 3.9 c | 14.44 ± 3.36 cde | 14.61 ± 1.42 bcd | 5.50 ± 0.55 ab | 0.85 ± 0.01 bcd | 52.03 ± 6.6 a | 20.9 ± 9.0 cd | 63.5 ± 5.8 a |
| 55 | 5 | 14 | 69.6 ± 10.1 abc | 524.2 ± 52.3 bc | 271.6 ± 10.0 d | 15.30 ± 3.27 bcde | 14.20 ± 1.30 bcd | 5.29 ± 0.35 abc | 0.85 ± 0.04 bcd | 51.67 ± 1.3 a | 39.1 ± 4.2 b | 60.9 ± 2.7 ab |
| 63 | 5 | 14 | 59.3 ± 7.0 abcd | 528.7 ± 42.0 bc | 141.0 ± 11.0 e | 15.09 ± 1.34 bcde | 11.69 ± 1.57 ef | 4.64 ± 0.33 cde | 0.81 ± 0.04 de | 48.93 ± 2.3 a | 35.3 ± 3.9 b | 57.7 ± 2.4 abc |
| 55 | 5 | 27.5 | 71.9 ± 7.0 ab | 696.1 ± 40.7 a | 284.2 ± 7.0 cd | 18.42 ± 6.17 b | 15.94 ± 1.69 ab | 5.72 ± 0.14 ab | 0.91 ± 0.04 ab | 0.4 ± 0.0 g | nd | 0.3 ± 0.0 f |
| 50 | 6 | 6 | 66.6 ± 10.2 abcd | 377.1 ± 30.0 gh | 325.6 ± 27.4 a | 11.79 ± 1.04 e | 10.92 ± 0.72 f | 3.55 ± 0.25 fgh | 0.66 ± 0.04 h | 29.6 ± 5.9 c | 39.5 ± 10.9 b | 48.7 ± 15.5 cd |
| 60 | 6 | 6 | 68.1 ± 13.0 abcd | 350.2 ± 18.8 h | 305.1 ± 7.4 b | 11.57 ± 0.64 e | 10.94 ± 0.64 f | 3.49 ± 0.24 fgh | 0.66 ± 0.01 h | 44.9 ± 5.5 ab | 18.2 ± 4.09 cd | 55.9 ± 1.0 abc |
| 50 | 6 | 22 | 84.7 ± 9.9 a | 432.1 ± 34.5 efg | 334.4 ± 3.1 a | 14.31 ± 2.60 cde | 11.59 ± 0.76 ef | 3.22 ± 0.17 h | 0.67 ± 0.03 h | 24.9 ± 3.9 cd | 56.4 ± 6.1 a | 6.5 ± 5.2 ef |
| 60 | 6 | 22 | 83.7 ± 9.9 a | 354.0 ± 28.9 h | 311.5 ± 10.5 b | 14.01 ± 3.26 de | 10.78 ± 1.01 f | 4.12 ± 1.08 efg | 0.66 ± 0.02 h | 17.4 ± 7.9 de | 54.9 ± 4.7 a | 10.6 ± 2.3 e |
| 55 | 6.7 | 14 | 78.6 ± 15.9 a | 387.2 ± 37.9 fgh | 305.2 ± 10.7 b | 13.48 ± 2.68 ed | 12.63 ± 2.27 def | 3.41 ± 0.16 gh | 0.69 ± 0.05 gh | 2.1 ± 4.1 g | 1.5 ± 0.3 e | 10.4 ± 4.2 e |
| MANOVA | ||||||||||||
| T | MS | 12.7 | 20,266 *** | 29,010 *** | 2.8 x 107 *** | 1.3 x 107 *** | 2.2 x 106 ** | 0.009 ** | 2897.7 *** | 3373.4 *** | 2680.1 *** | |
| pH | MS | 1375.9 *** | 164,046 *** | 385,120 *** | 2.2 x 108 *** | 7.1 x 107 *** | 1.6 x 107 *** | 0.199 *** | 671.6 ** | 869.9 | 126.9 | |
| t | MS | 700.1 ** | 58,439 *** | 15,733 *** | 1.1 x 108 *** | 1.2 x 107 *** | 9.6 x 105 * | 0.028 *** | 5165.1 *** | 472.9 | 9522.4 *** | |
| Response Variable | Mathematical Models * | R2 | R2-adj |
|---|---|---|---|
| TPSC | 0.78 | 0.76 | |
| FA | 0.93 | 0.92 | |
| ORAC | 0.80 | 0.79 | |
| DPPH | 0.81 | 0.79 | |
| FRAP | 0.79 | 0.77 | |
| MCP-1 (i%) | 0.80 | 0.79 |
| Optimum D Value | Factors at Optimum D Value | Response Variables | Predicted Values | Experimental Values | Control |
|---|---|---|---|---|---|
| 0.74 | 47 °C, pH = 4.4, 20.8 h | TSPC (mg FAE/100g) | 664.80 | 703.3 ± 25.8 | 301.52 ± 16.11 |
| FA (mg/100 g) | 222.27 | 125.8 ± 1.5 | 52.66 ± 1.58 | ||
| ORAC (g TE/100 g) | 21.5 | 22.67 ± 2.07 | 14.65 ± 2.48 | ||
| DPPH (g TE/100 g) | 6.10 | 5.90 ± 0.28 | 6.37 ± 0.40 | ||
| FRAP (mmol Fe2+/100 g) | 0.91 | 0.94 ± 0.02 | 0.32 ± 0.01 | ||
| MCP-1 (i%) | 35.09 | 36.1 ± 2.0 | 17.34 ± 2.32 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bautista-Expósito, S.; Tomé-Sánchez, I.; Martín-Diana, A.B.; Frias, J.; Peñas, E.; Rico, D.; Casas, M.J.G.; Martínez-Villaluenga, C. Enzyme Selection and Hydrolysis under Optimal Conditions Improved Phenolic Acid Solubility, and Antioxidant and Anti-Inflammatory Activities of Wheat Bran. Antioxidants 2020, 9, 984. https://doi.org/10.3390/antiox9100984
Bautista-Expósito S, Tomé-Sánchez I, Martín-Diana AB, Frias J, Peñas E, Rico D, Casas MJG, Martínez-Villaluenga C. Enzyme Selection and Hydrolysis under Optimal Conditions Improved Phenolic Acid Solubility, and Antioxidant and Anti-Inflammatory Activities of Wheat Bran. Antioxidants. 2020; 9(10):984. https://doi.org/10.3390/antiox9100984
Chicago/Turabian StyleBautista-Expósito, Sara, Irene Tomé-Sánchez, Ana Belén Martín-Diana, Juana Frias, Elena Peñas, Daniel Rico, María Jesús García Casas, and Cristina Martínez-Villaluenga. 2020. "Enzyme Selection and Hydrolysis under Optimal Conditions Improved Phenolic Acid Solubility, and Antioxidant and Anti-Inflammatory Activities of Wheat Bran" Antioxidants 9, no. 10: 984. https://doi.org/10.3390/antiox9100984
APA StyleBautista-Expósito, S., Tomé-Sánchez, I., Martín-Diana, A. B., Frias, J., Peñas, E., Rico, D., Casas, M. J. G., & Martínez-Villaluenga, C. (2020). Enzyme Selection and Hydrolysis under Optimal Conditions Improved Phenolic Acid Solubility, and Antioxidant and Anti-Inflammatory Activities of Wheat Bran. Antioxidants, 9(10), 984. https://doi.org/10.3390/antiox9100984