Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
1.1. Mitochondria Represent the Most Intriguing Organelle of Eucaryotic Cell
1.2. mtDNA Damage is Linked to Heterogeneous Clinical Phenotypes
1.3. Next Generation Sequencing Techniques could Reveal a Relevant Role of mtDNA Impairments in Retinal Degenerations
1.4. Evaluating mtDNA Damage by Mitochondrial Transcript Analysis in Photo-Induced Oxidative Stressed RPE Cells could Shed Light on the Role of Mitochondria in Retinal Dystrophies
2. Materials and Methods
2.1. Cell Culture Samples
2.2. MTT Assay
2.3. Whole RNA Extraction and RNA-Seq Profiling
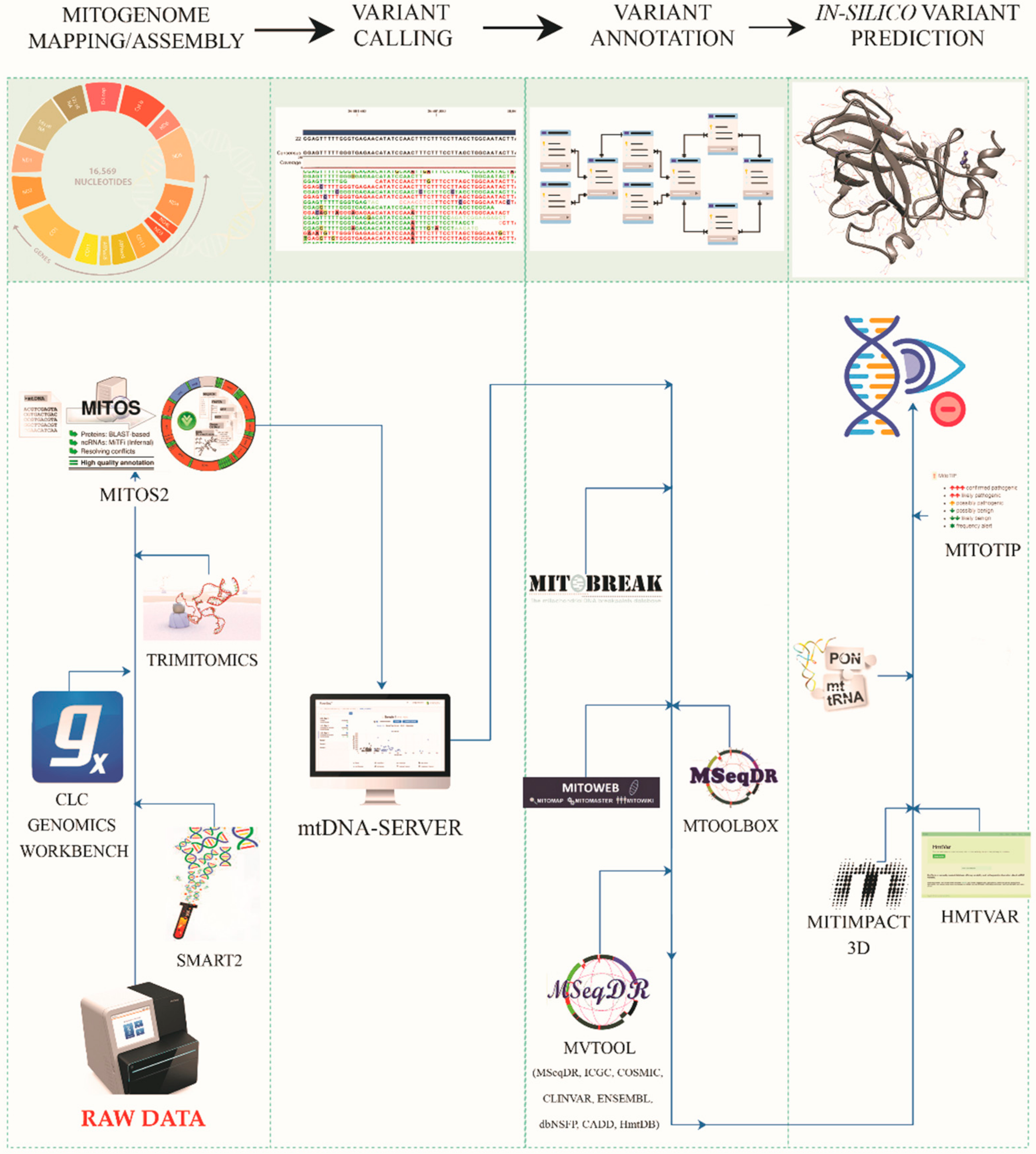
2.4. Mitogenome Assembly/Mapping
2.5. Variant Detection by mtDNA-Server
2.6. Variant Annotation and Prioritization
2.7. In Silico Predictions and Variant Consequences
2.8. Sub-Pathway Analysis
3. Results
3.1. A2E Treatment Determined a Substantial Negative Effect on RPE Cells Survival
3.2. Alignment and Assembly of mtDNAs
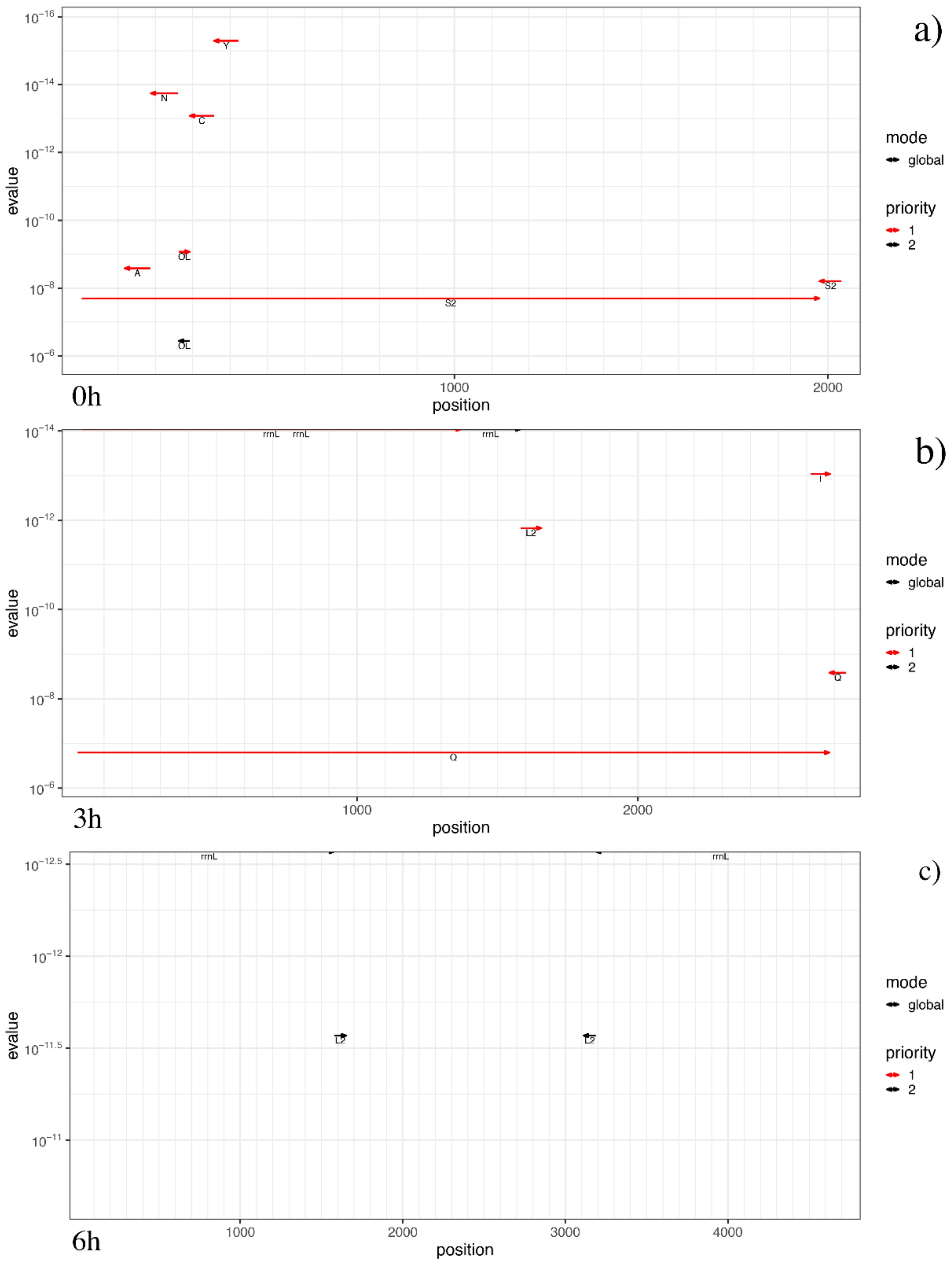
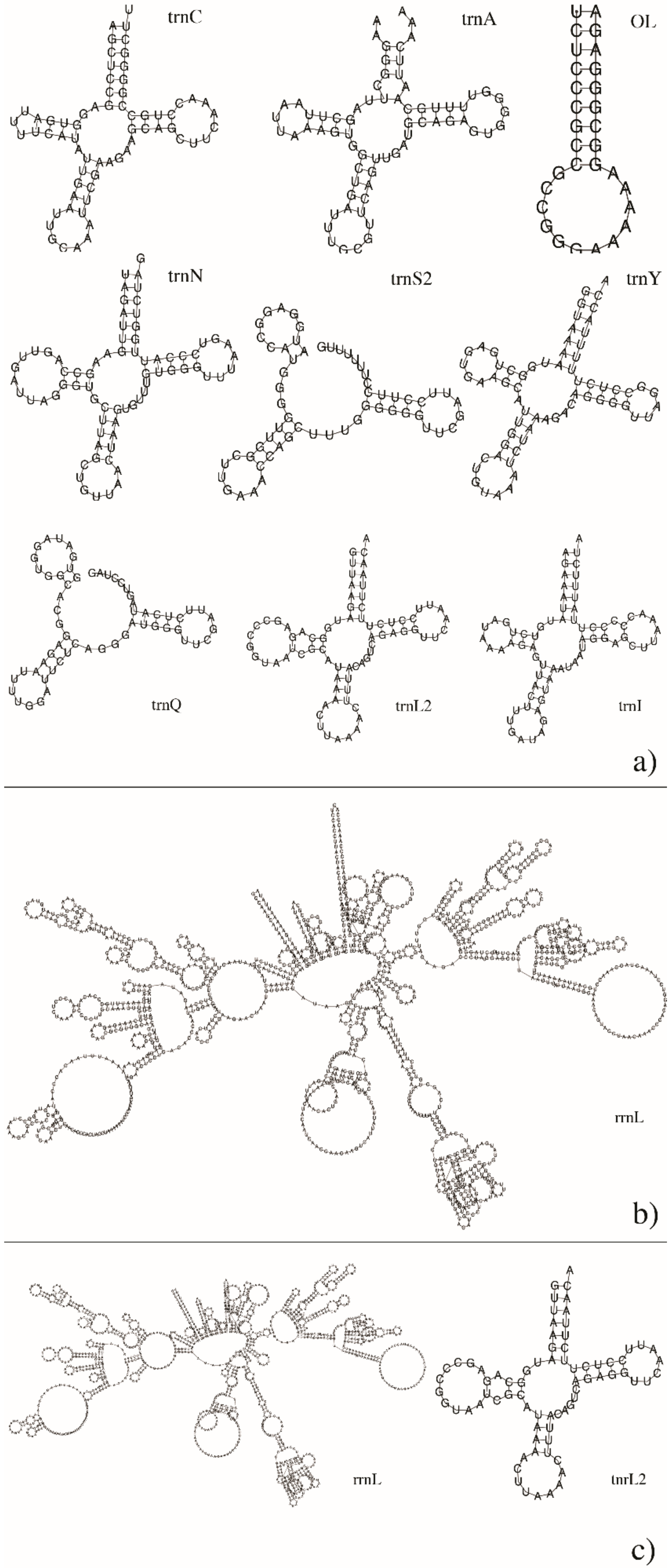
3.3. Mitogenome Annotations
3.4. MtDNA Variant Calling and Annotations
3.5. In Silico Functional Consequences and Pathogenicity Predictions
3.6. Sub-Pathways Analysis of mtDNA Mutated Genes Suggested a Positive Regulation of ATP Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Deposition
References
- Agrawal, A.; Mabalirajan, U. Rejuvenating cellular respiration for optimizing respiratory function: Targeting mitochondria. Am. J. Physiol. Lung. Cell Mol. Physiol. 2016, 310, L103–L113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennerlein, S.; Wang, C.; Rehling, P. Plasticity of Mitochondrial Translation. Trends Cell Biol. 2017, 27, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Farge, G.; Falkenberg, M. Organization of DNA in Mammalian Mitochondria. Int. J. Mol. Sci. 2019, 20, 2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.C.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Navathe, S.B.; Baldi, P.; Wallace, D.C. MITOMAP: A human mitochondrial genome database—2004 update. Nucleic Acids Res. 2005, 33, D611–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 100858. [Google Scholar] [CrossRef]
- Yang, T.C.; Yarmishyn, A.A.; Yang, Y.P.; Lu, P.C.; Chou, S.J.; Wang, M.L.; Lin, T.C.; Hwang, D.K.; Chou, Y.B.; Chen, S.J.; et al. Mitochondrial transport mediates survival of retinal ganglion cells in affected LHON patients. Hum. Mol. Genet. 2020, 29, 1454–1464. [Google Scholar] [CrossRef]
- Huang, C.H.; Su, S.L.; Hsieh, M.C.; Cheng, W.L.; Chang, C.C.; Wu, H.L.; Kuo, C.L.; Lin, T.T.; Liu, C.S. Depleted leukocyte mitochondrial DNA copy number in metabolic syndrome. J. Atheroscler. Thromb. 2011, 18, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Caston, R.A.; Demple, B. Risky repair: DNA-protein crosslinks formed by mitochondrial base excision DNA repair enzymes acting on free radical lesions. Free Radic. Biol. Med. 2017, 107, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Chen, L.; Ge, Q.; Lu, N.; Li, J.; Pan, X.; Qiao, Y.; Tu, J.; Lu, Z. Evaluating heteroplasmic variations of the mitochondrial genome from whole genome sequencing data. Gene 2019, 699, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.J.; Brilhante, V.; Suomalainen, A. Next-generation sequencing for mitochondrial disorders. Br. J. Pharmacol. 2014, 171, 1837–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaimo, A.; Linares, G.G.; Bujjamer, J.M.; Gorojod, R.M.; Alcon, S.P.; Martinez, J.H.; Baldessari, A.; Grecco, H.E.; Kotler, M.L. Toxicity of blue led light and A2E is associated to mitochondrial dynamics impairment in ARPE-19 cells: Implications for age-related macular degeneration. Arch. Toxicol. 2019, 93, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Yang, J.; Hong, Z.; Wu, Y.; Xie, Y.; Wang, G. Mechanisms of blue light-induced eye hazard and protective measures: A review. Biomed. Pharmacother. 2020, 130, 110577. [Google Scholar] [CrossRef]
- Grebenova, D.; Kuzelova, K.; Smetana, K.; Pluskalova, M.; Cajthamlova, H.; Marinov, I.; Fuchs, O.; Soucek, J.; Jarolim, P.; Hrkal, Z. Mitochondrial and endoplasmic reticulum stress-induced apoptotic pathways are activated by 5-aminolevulinic acid-based photodynamic therapy in HL60 leukemia cells. J. Photochem. Photobiol. B 2003, 69, 71–85. [Google Scholar] [CrossRef]
- Lin, C.H.; Wu, M.R.; Huang, W.J.; Chow, D.S.; Hsiao, G.; Cheng, Y.W. Low-Luminance Blue Light-Enhanced Phototoxicity in A2E-Laden RPE Cell Cultures and Rats. Int. J. Mol. Sci. 2019, 20, 1799. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Scimone, C.; Alibrandi, S.; Nicocia, G.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Discovery of GLO1 New Related Genes and Pathways by RNA-Seq on A2E-Stressed Retinal Epithelial Cells Could Improve Knowledge on Retinitis Pigmentosa. Antioxidants (Basel) 2020, 9, 416. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants (Basel) 2020, 9, 318. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; D’Angelo, R.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; Scimone, C. Effects of A2E-Induced Oxidative Stress on Retinal Epithelial Cells: New Insights on Differential Gene Response and Retinal Dystrophies. Antioxidants (Basel) 2020, 9, 307. [Google Scholar] [CrossRef] [Green Version]
- FastQC - Babraham Bioinformatics. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 November 2020).
- Okonechnikov, K.; Conesa, A.; Garcia-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- CLC Genomics Workbench 20.0.4. Available online: https://digitalinsights.qiagen.com (accessed on 19 November 2020).
- Alqahtani, F.; Mandoiu, I.I. Statistical Mitogenome Assembly with RepeaTs. J. Comput. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Plese, B.; Rossi, M.E.; Kenny, N.J.; Taboada, S.; Koutsouveli, V.; Riesgo, A. Trimitomics: An efficient pipeline for mitochondrial assembly from transcriptomic reads in nonmodel species. Mol. Ecol. Resour. 2019, 19, 1230–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissensteiner, H.; Forer, L.; Fuchsberger, C.; Schopf, B.; Kloss-Brandstatter, A.; Specht, G.; Kronenberg, F.; Schonherr, S. mtDNA-Server: Next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 2016, 44, W64–W69. [Google Scholar] [CrossRef] [PubMed]
- MSeqDR: The Mitochondrial Disease Sequence Data Resource Consortium. Available online: https://mseqdr.org (accessed on 19 November 2020).
- Shen, L.; Attimonelli, M.; Bai, R.; Lott, M.T.; Wallace, D.C.; Falk, M.J.; Gai, X. MSeqDR mvTool: A mitochondrial DNA Web and API resource for comprehensive variant annotation, universal nomenclature collation, and reference genome conversion. Hum. Mutat. 2018, 39, 806–810. [Google Scholar] [CrossRef]
- Shen, L.; Diroma, M.A.; Gonzalez, M.; Navarro-Gomez, D.; Leipzig, J.; Lott, M.T.; van Oven, M.; Wallace, D.C.; Muraresku, C.C.; Zolkipli-Cunningham, Z.; et al. MSeqDR: A Centralized Knowledge Repository and Bioinformatics Web Resource to Facilitate Genomic Investigations in Mitochondrial Disease. Hum. Mutat. 2016, 37, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Clima, R.; Preste, R.; Calabrese, C.; Diroma, M.A.; Santorsola, M.; Scioscia, G.; Simone, D.; Shen, L.; Gasparre, G.; Attimonelli, M. HmtDB 2016: Data update, a better performing query system and human mitochondrial DNA haplogroup predictor. Nucleic Acids Res. 2017, 45, D698–D706. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Pesini, E.; Lott, M.T.; Procaccio, V.; Poole, J.C.; Brandon, M.C.; Mishmar, D.; Yi, C.; Kreuziger, J.; Baldi, P.; Wallace, D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007, 35, D823–D828. [Google Scholar] [CrossRef] [Green Version]
- Belsare, S.; Levy-Sakin, M.; Mostovoy, Y.; Durinck, S.; Chaudhuri, S.; Xiao, M.; Peterson, A.S.; Kwok, P.Y.; Seshagiri, S.; Wall, J.D. Evaluating the quality of the 1000 genomes project data. BMC Genom. 2019, 20, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef] [PubMed]
- MITOS WebServer. Available online: http://mitos2.bioinf.uni-leipzig.de/index.py (accessed on 19 November 2020).
- Calabrese, C.; Simone, D.; Diroma, M.A.; Santorsola, M.; Gutta, C.; Gasparre, G.; Picardi, E.; Pesole, G.; Attimonelli, M. MToolBox: A highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 2014, 30, 3115–3117. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; Carneiro, J.; Amorim, A.; Pereira, F. MitoBreak: The mitochondrial DNA breakpoints database. Nucleic Acids Res. 2014, 42, D1261–D1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellana, S.; Ronai, J.; Mazza, T. MitImpact: An exhaustive collection of pre-computed pathogenicity predictions of human mitochondrial non-synonymous variants. Hum. Mutat. 2015, 36, E2413–E2422. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Yang, J.; Chung, B.H.; Lau, Y.L.; Yang, W. EFIN: Predicting the functional impact of nonsynonymous single nucleotide polymorphisms in human genome. BMC Genom. 2014, 15, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, Y.; Yachdav, G.; Rost, B. SNAP predicts effect of mutations on protein function. Bioinformatics 2008, 24, 2397–2398. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Frousios, K.; Iliopoulos, C.S.; Schlitt, T.; Simpson, M.A. Predicting the functional consequences of non-synonymous DNA sequence variants—Evaluation of bioinformatics tools and development of a consensus strategy. Genomics 2013, 102, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.C.; Zhang, Z. SNPdryad: Predicting deleterious non-synonymous human SNPs using only orthologous protein sequences. Bioinformatics 2014, 30, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, D.; Tanyalcin, I.; Ferte, J.; Gazzo, A.; Orlando, G.; Lenaerts, T.; Rooman, M.; Vranken, W. DEOGEN2: Prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. 2017, 45, W201–W206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Navarro, A.; Gaudioso-Simon, A.; Alvarez-Jarreta, J.; Montoya, J.; Mayordomo, E.; Ruiz-Pesini, E. Machine learning classifier for identification of damaging missense mutations exclusive to human mitochondrial DNA-encoded polypeptides. BMC Bioinform. 2017, 18, 158. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.C.; Joyce, C.; Ritchie, G.R.; John, S.L.; Cunningham, F.; Asimit, J.; Zeggini, E. A combined functional annotation score for non-synonymous variants. Hum. Hered. 2012, 73, 47–51. [Google Scholar] [CrossRef]
- Capriotti, E.; Altman, R.B.; Bromberg, Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genom. 2013, 14 (Suppl. 3), S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Bai, J.; Zhang, J.; Yang, L.; Duan, J.; Li, Y.; Gao, M. CONDEL: Detecting Copy Number Variation and Genotyping Deletion Zygosity from Single Tumor Samples using Sequence Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Castellana, S.; Fusilli, C.; Mazzoccoli, G.; Biagini, T.; Capocefalo, D.; Carella, M.; Vescovi, A.L.; Mazza, T. High-confidence assessment of functional impact of human mitochondrial non-synonymous genome variations by APOGEE. PLoS Comput. Biol. 2017, 13, e1005628. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, R.; Krumholz, K.; Huang, Y.F.; Siepel, A. PhastWeb: A web interface for evolutionary conservation scoring of multiple sequence alignments using phastCons and phyloP. Bioinformatics 2019, 35, 2320–2322. [Google Scholar] [CrossRef]
- Accetturo, M.; Santamaria, M.; Lascaro, D.; Rubino, F.; Achilli, A.; Torroni, A.; Tommaseo-Ponzetta, M.; Attimonelli, M. Human mtDNA site-specific variability values can act as haplogroup markers. Hum. Mutat. 2006, 27, 965–974. [Google Scholar] [CrossRef]
- Simonetti, F.L.; Teppa, E.; Chernomoretz, A.; Nielsen, M.; Marino Buslje, C. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013, 41, W8–W14. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Perez, A.; Deu-Pons, J.; Lopez-Bigas, N. Improving the prediction of the functional impact of cancer mutations by baseline tolerance transformation. Genome Med. 2012, 4, 89. [Google Scholar] [CrossRef] [Green Version]
- Carter, H.; Karchin, R. Predicting the functional consequences of somatic missense mutations found in tumors. Methods Mol. Biol. 2014, 1101, 135–159. [Google Scholar] [CrossRef] [PubMed]
- EVmutation Effects Predicted from Sequence Co-Variation. Available online: https://marks.hms.harvard.edu/evmutation/index.html (accessed on 19 November 2020).
- Iserte, J.; Simonetti, F.L.; Zea, D.J.; Teppa, E.; Marino-Buslje, C. I-COMS: Interprotein-COrrelated Mutations Server. Nucleic Acids Res. 2015, 43, W320–W325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preste, R.; Vitale, O.; Clima, R.; Gasparre, G.; Attimonelli, M. HmtVar: A new resource for human mitochondrial variations and pathogenicity data. Nucleic Acids Res. 2019, 47, D1202–D1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonney, S.; Leipzig, J.; Lott, M.T.; Zhang, S.; Procaccio, V.; Wallace, D.C.; Sondheimer, N. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput. Biol. 2017, 13, e1005867. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Vihinen, M. PON-mt-tRNA: A multifactorial probability-based method for classification of mitochondrial tRNA variations. Nucleic Acids Res. 2016, 44, 2020–2027. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kreutzer, D.A.; Essigmann, J.M. Oxidized, deaminated cytosines are a source of C --> T transitions in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 3578–3582. [Google Scholar] [CrossRef] [Green Version]
- Otten, A.B.C.; Sallevelt, S.; Carling, P.J.; Dreesen, J.; Drusedau, M.; Spierts, S.; Paulussen, A.D.C.; de Die-Smulders, C.E.M.; Herbert, M.; Chinnery, P.F.; et al. Mutation-specific effects in germline transmission of pathogenic mtDNA variants. Hum. Reprod. 2018, 33, 1331–1341. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.C.P.; Ferrasa, A.; Muotri, A.R.; Herai, R.H. Frequency and association of mitochondrial genetic variants with neurological disorders. Mitochondrion 2019, 46, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Nicocia, G.; Denaro, L.; Robledo, R.; Sidoti, A.; D’Angelo, R. GLO1 gene polymorphisms and their association with retinitis pigmentosa: A case-control study in a Sicilian population. Mol. Biol. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Rinaldi, C.; Aragona, P.; Briuglia, S.; D’Ascola, A.; D’Angelo, R.; Sidoti, A. Stargardt Phenotype Associated With Two ELOVL4 Promoter Variants and ELOVL4 Downregulation: New Possible Perspective to Etiopathogenesis? Investig. Ophthalmol. Vis. Sci. 2018, 59, 843–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scimone, C.; Donato, L.; Esposito, T.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. A novel RLBP1 gene geographical area-related mutation present in a young patient with retinitis punctata albescens. Hum. Genom. 2017, 11, 18. [Google Scholar] [CrossRef]
- Nashine, S.; Nesburn, A.B.; Kuppermann, B.D.; Kenney, M.C. Age-related macular degeneration (AMD) mitochondria modulate epigenetic mechanisms in retinal pigment epithelial cells. Exp. Eye Res. 2019, 189, 107701. [Google Scholar] [CrossRef]
- Marie, M.; Bigot, K.; Angebault, C.; Barrau, C.; Gondouin, P.; Pagan, D.; Fouquet, S.; Villette, T.; Sahel, J.A.; Lenaers, G.; et al. Light action spectrum on oxidative stress and mitochondrial damage in A2E-loaded retinal pigment epithelium cells. Cell Death Dis. 2018, 9, 287. [Google Scholar] [CrossRef]
- Shaban, H.; Borras, C.; Vina, J.; Richter, C. Phosphatidylglycerol potently protects human retinal pigment epithelial cells against apoptosis induced by A2E, a compound suspected to cause age-related macula degeneration. Exp. Eye Res. 2002, 75, 99–108. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Mehrzadi, S.; Hemati, K.; Reiter, R.J.; Hosseinzadeh, A. Mitochondrial dysfunction in age-related macular degeneration: Melatonin as a potential treatment. Expert. Opin. Ther. Targets 2020, 24, 359–378. [Google Scholar] [CrossRef]
- Machatha, S.G.W.; Young, S. Aldehyde Trapping Compounds and Uses Thereof. 2020. Available online: https://www.freepatentsonline.com (accessed on 20 November 2020).
- Marnett, L.J. Oxy radicals, lipid peroxidation and DNA damage. Toxicology 2002, 181–182, 219–222. [Google Scholar] [CrossRef]
- Suter, M.; Reme, C.; Grimm, C.; Wenzel, A.; Jaattela, M.; Esser, P.; Kociok, N.; Leist, M.; Richter, C. Age-related macular degeneration. The lipofusion component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J. Biol. Chem. 2000, 275, 39625–39630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambowitz, A.M.; Belfort, M. Introns as mobile genetic elements. Annu. Rev. Biochem. 1993, 62, 587–622. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Calton, M.A.; Tang, S.; Vollrath, D. Depletion of Mitochondrial DNA in Differentiated Retinal Pigment Epithelial Cells. Sci. Rep. 2019, 9, 15355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadeja, R.N.; Thounaojam, M.C.; Bartoli, M.; Martin, P.M. Implications of NAD(+) Metabolism in the Aging Retina and Retinal Degeneration. Oxid. Med. Cell Longev. 2020, 2020, 2692794. [Google Scholar] [CrossRef] [PubMed]
- Saadat, K.A.; Murakami, Y.; Tan, X.; Nomura, Y.; Yasukawa, T.; Okada, E.; Ikeda, Y.; Yanagi, Y. Inhibition of autophagy induces retinal pigment epithelial cell damage by the lipofuscin fluorophore A2E. FEBS Open Biol. 2014, 4, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Anand, M.; Shiraz, A.K.; Magrane, J.; Gao, J.; Vollmer-Snarr, H.R.; Manfredi, G.; Finnemann, S.C. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J. Biol. Chem. 2008, 283, 24770–24780. [Google Scholar] [CrossRef] [Green Version]
- Agnez-Lima, L.F.; Melo, J.T.; Silva, A.E.; Oliveira, A.H.; Timoteo, A.R.; Lima-Bessa, K.M.; Martinez, G.R.; Medeiros, M.H.; Di Mascio, P.; Galhardo, R.S.; et al. DNA damage by singlet oxygen and cellular protective mechanisms. Mutat. Res. Rev. Mutat. Res. 2012, 751, 15–28. [Google Scholar] [CrossRef]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Menck, C.F. Singlet oxygen induced DNA damage. Mutat. Res. 1992, 275, 367–375. [Google Scholar] [CrossRef]
- Udar, N.; Atilano, S.R.; Memarzadeh, M.; Boyer, D.S.; Chwa, M.; Lu, S.; Maguen, B.; Langberg, J.; Coskun, P.; Wallace, D.C.; et al. Mitochondrial DNA haplogroups associated with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2966–2974. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.N.Y.; Kim, S.S.; Jo, Y.H. Deregulated Mitochondrial DNA in Diseases. DNA Cell Biol. 2020, 39, 1385–1400. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics Feature | 0h_RPE | 3h_RPE | 6h_RPE |
|---|---|---|---|
| Overall Passed Reads | 52,752,353 | 26,262,324 | 19,520,749 |
| Passed FWD Reads | 51,913,969 | 16,056,486 | 19,301,263 |
| Passed REV Reads | 838,384 | 205,838 | 219,486 |
| Mapping Quality OK | 440,014 | 132,054 | 162,126 |
| Mapping Quality BAD | 29,372 | 10,603 | 15,517 |
| Unmapped Reads | 0 | 0 | 0 |
| Base Read Quality OK | 52,752,353 | 16,262,324 | 19,520,749 |
| Base Read Quality BAD | 15,127,187 | 3,805,671 | 4,905,387 |
| Bad Alignment | 0 | 0 | 0 |
| Duplicates | 0 | 0 | 0 |
| Short Reads (<25 bp) | 0 | 0 | 0 |
| Mitochondrial Transcripts | 29,123 | 27,095 | 25,471 |
| 0h_RPE | 3h_RPE | 6h_RPE | |||
| Heteroplasmic Variants (Global) | 26 | 320 | 195 | ||
| Unique annotated variants (Global) | 34 | 29 | 45 | ||
| mvTool | MSeqDR Community Data Population | N° Variants | 20 | 21 | 21 |
| Mitomap Disease | 4 | 4 | 4 | ||
| Mitomap Status | 3 Reported, 1 Conflicting | 3 Reported, 1 Conflicting | 3 Reported, 1 Conflicting | ||
| HmtDB Pathogenicity | 8 Pending, 4 Benign | 8 Pending, 4 Benign | 9 Pending, 4 Benign | ||
| Disease and Phenotypes | dbSNP | 17 | 17 | 18 | |
| MSeqDR Clinical Significance | 4 Likely Pathogenic, 2 Not Provided | 4 Likely Pathogenic, 2 Not Provided | 4 Likely Pathogenic, 2 Not Provided | ||
| HmtDB Disease | 4 | 4 | 4 | ||
| COSMIC | 1 | 1 | 1 | ||
| ICGC | 2 | 2 | 2 | ||
| VEP | Impact | 11 Low, 4 Moderate, 8 Modifier | 11 Low, 5 Moderate, 8 Modifier | 11 Low, 4 Moderate, 13 Modifier | |
| Biotype | 23 Protein Coding | 24 Protein Coding | 28 Protein Coding, 1 mt_rRNA | ||
| Consequence Terms | 16 Synonymous, 3 Missense, 4 Upstream | 14 Synonymous, 5 Missense, 4 Upstream | 11 Synonymous, 12 Missense, 4 Upstream, 1 Non-coding Transcript Exon | ||
| CADD | Raw Raknscore | 1 | 1 | 1 | |
| HmtDB Patho Table | N° Variants | 3 | 3 | 3 | |
| Pathogenicity | 1 | 1 | 1 | ||
| MitoBreak | N° Deletions | 0 | 1 | 0 | |
| Healthy Tissue | 0 | Aged Tissues | 0 | ||
| Del of replication origins | 0 | None | 0 | ||
| Location of the deleted regions | 0 | Inside the major arc | 0 | ||
| MitoMaster | N° Variants | 20 Transitions | 20 Transitions, 1 Transversion | 21 Transitions | |
| Mut Type | 15 Coding, 5 Non-coding | 17 Coding, 4 Non-coding | 16 Coding, 5 Non-coding | ||
| Patient Report | 4 | 4 | 4 | ||
| MitImpact 3D | 0h_RPE | 3h_RPE | 6h_RPE | MitImpact 3D | 0h_RPE | 3h_RPE | 6h_RPE | MitImpact 3D | 0h_RPE | 3h_RPE | 6h_RPE | |||
| N° Variants | 34 | 29 | 45 | N° Variants | 34 | 29 | 45 | N° Variants | 34 | 29 | 45 | |||
| PolyPhen 2 | Benign | 9 | 7 | 8 | FatHmm | Neutral | 9 | 8 | 10 | Mutation Assessor | Neutral Impact | 5 | 3 | 4 |
| Probably damaging | 5 | 5 | 8 | Deleterious | 5 | 4 | 6 | Low impact | 3 | 2 | 4 | |||
| SIFT | Neutral | 12 | 9 | 13 | PROVEAN | Neutral | 9 | 8 | 10 | Medium impact | 2 | 2 | 4 | |
| Deleterious | 2 | 3 | 3 | Deleterious | 5 | 4 | 9 | High impact | 3 | 3 | 3 | |||
| EFIN_SP | Neutral | 12 | 10 | 13 | CADD | Neutral | 7 | 4 | 6 | PhD-SNP | Neutral | 7 | 4 | 10 |
| Damaging | 2 | 2 | 3 | Deleterious | 7 | 8 | 10 | Disease | 7 | 8 | 6 | |||
| EFIN_HD | Neutral | 11 | 9 | 12 | PANTHER | Neutral | 10 | 7 | 10 | SNAP | Neutral | 8 | 5 | 8 |
| Damaging | 3 | 3 | 4 | Disease | 4 | 3 | 6 | Disease | 6 | 7 | 8 | |||
| Meta-SNP | Neutral | 8 | 6 | 8 | Condel | Neutral | 4 | 4 | 6 | MToolBox | Neutral | 6 | 5 | 6 |
| Disease | 6 | 6 | 8 | Deleterious | 10 | 8 | 10 | Deleterious | 8 | 7 | 10 | |||
| CAROL | Neutral | 10 | 8 | 10 | COVEC_WMV | Neutral | 10 | 8 | 10 | APOGEE | N | 11 | 10 | 11 |
| Deleterious | 4 | 4 | 6 | Deleterious | 4 | 4 | 6 | P | 3 | 2 | 5 | |||
| Mutation Taster | Polymorphism | 10 | 9 | 13 | CLINVAR | Yes | 2 | 2 | 2 | Mitoclass1 | Neutral | 10 | 7 | 9 |
| Disease Causing | 2 | 3 | 2 | No | 12 | 10 | 14 | Damaging | 4 | 5 | 7 | |||
| dbSNP | SNP | 4 | 4 | 6 | Mitomap | Yes | 0 | 0 | 2 | DDG_Intra | Yes | 8 | 6 | 8 |
| New Mutation | 10 | 8 | 10 | No | 14 | 12 | 14 | No | 6 | 6 | 8 | |||
| DDG_Inter | Yes | 2 | 1 | 1 | EV_Mutation | Yes | 8 | 5 | 10 | Pon-mt-tRNA | N° Variants | 0 | 0 | 2 |
| No | 12 | 11 | 17 | No | 6 | 7 | 6 | Neutral or Likely Neutral | / | / | 2 | |||
| HmtVAR | N° Variants | 13 | 19 | 14 | HmtVAR | N° Variants | 13 | 19 | 14 | HmtVAR | N° Variants | 13 | 19 | 14 |
| CDS | 7 | 14 | 8 | rRNA | 3 | 1 | 1 | Polymorphic | 2 | 2 | 0 | |||
| Regulatory | 3 | 4 | 4 | tRNA | 0 | 0 | 2 | Likely Polymorphic | 1 | 0 | 1 | |||
| Likely Pathogenic | 1 | 0 | 2 | Pathogenic | 0 | 1 | 0 | Unavailable | 9 | 16 | 12 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, L.; Scimone, C.; Alibrandi, S.; Pitruzzella, A.; Scalia, F.; D’Angelo, R.; Sidoti, A. Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline. Antioxidants 2020, 9, 1158. https://doi.org/10.3390/antiox9111158
Donato L, Scimone C, Alibrandi S, Pitruzzella A, Scalia F, D’Angelo R, Sidoti A. Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline. Antioxidants. 2020; 9(11):1158. https://doi.org/10.3390/antiox9111158
Chicago/Turabian StyleDonato, Luigi, Concetta Scimone, Simona Alibrandi, Alessandro Pitruzzella, Federica Scalia, Rosalia D’Angelo, and Antonina Sidoti. 2020. "Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline" Antioxidants 9, no. 11: 1158. https://doi.org/10.3390/antiox9111158