Oral Bisphenol A Worsens Liver Immune-Metabolic and Mitochondrial Dysfunction Induced by High-Fat Diet in Adult Mice: Cross-Talk between Oxidative Stress and Inflammasome Pathway

 ,
, 


 , ,
, ,  ,
,  ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. In Vivo Experimental Procedures
2.2. Biochemical, Hormone, and Hepatic Determinations
2.3. Glucose and Pyruvate Tolerance Tests
2.4. Measurements of Mitochondrial Oxidative Capacity and Enzyme Activity
2.5. ROS Assay and Malondialdehyde Measurement
2.6. Hepatic Histological Analysis
2.7. Protein Preparation and Western Blot Analysis
2.8. RNA Isolation and Real-Time PCR
2.9. Statistical Analysis
3. Results
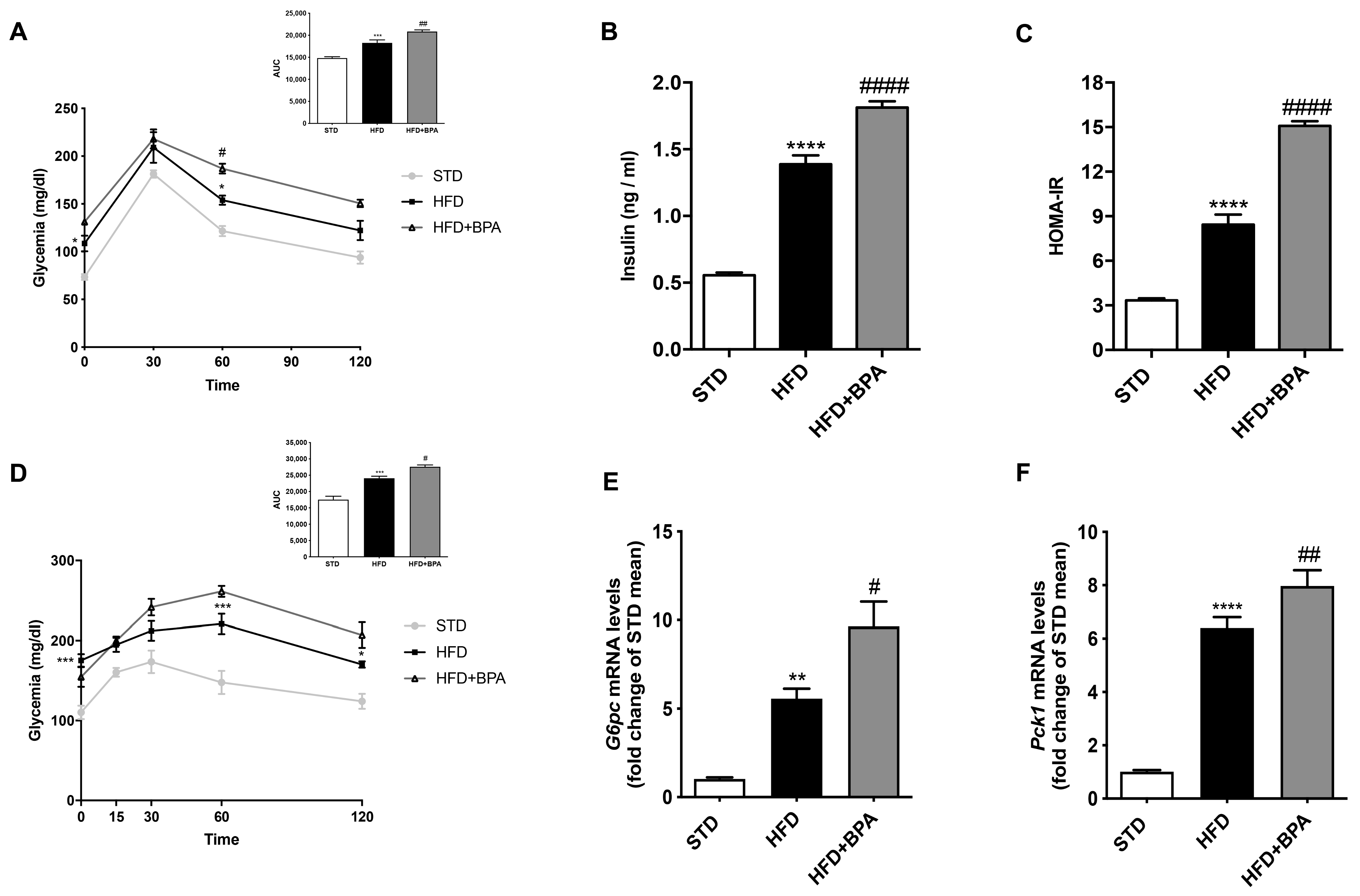
3.1. Effect of BPA Exposure on Serum Biochemical Parameters and Adipokine Profile in HFD Mice
3.2. BPA Exacerbates HFD-Induced Impairment of Glucose Homeostasis
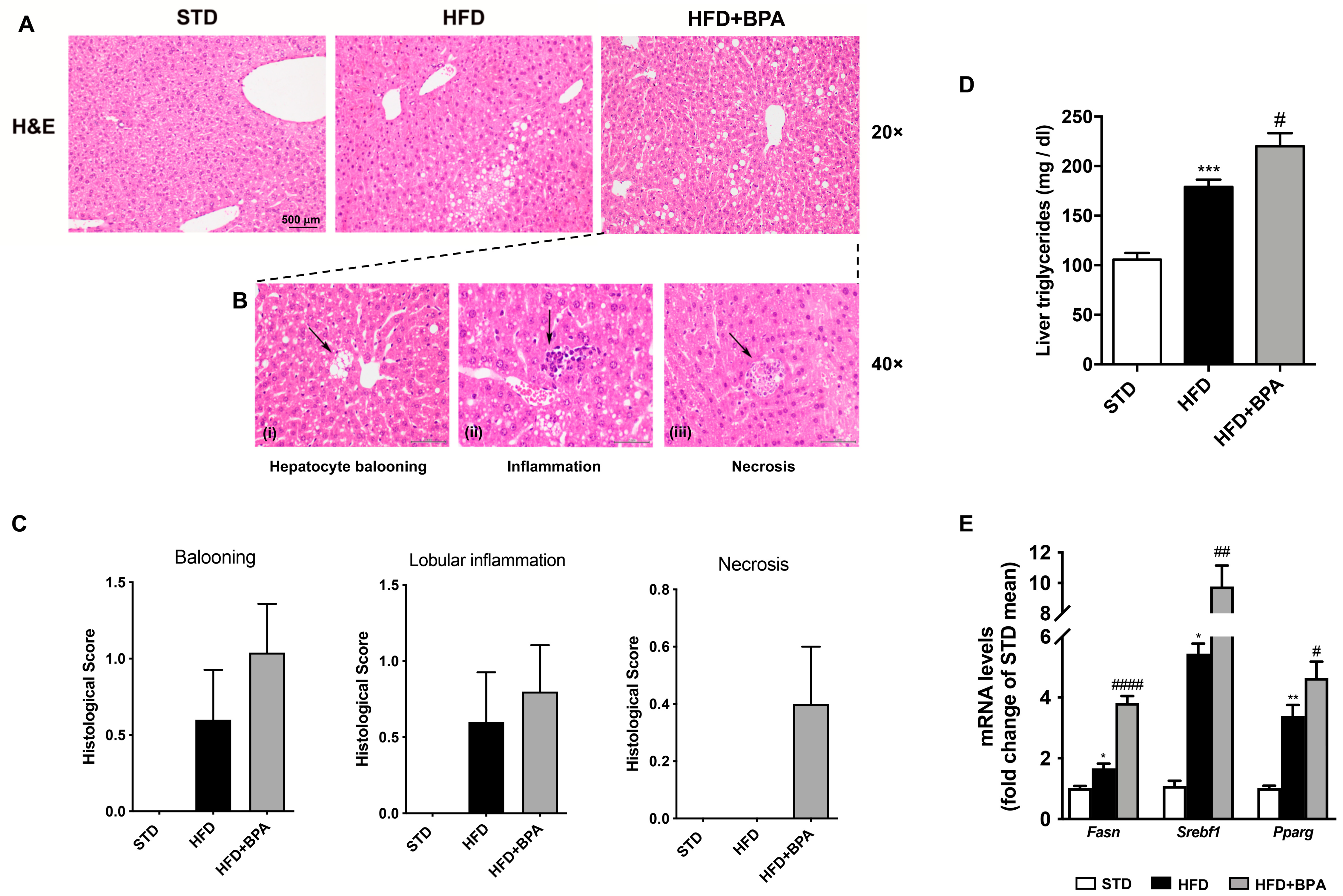
3.3. BPA Exposure Worsens Liver Inflammation in HFD-Fed Mice
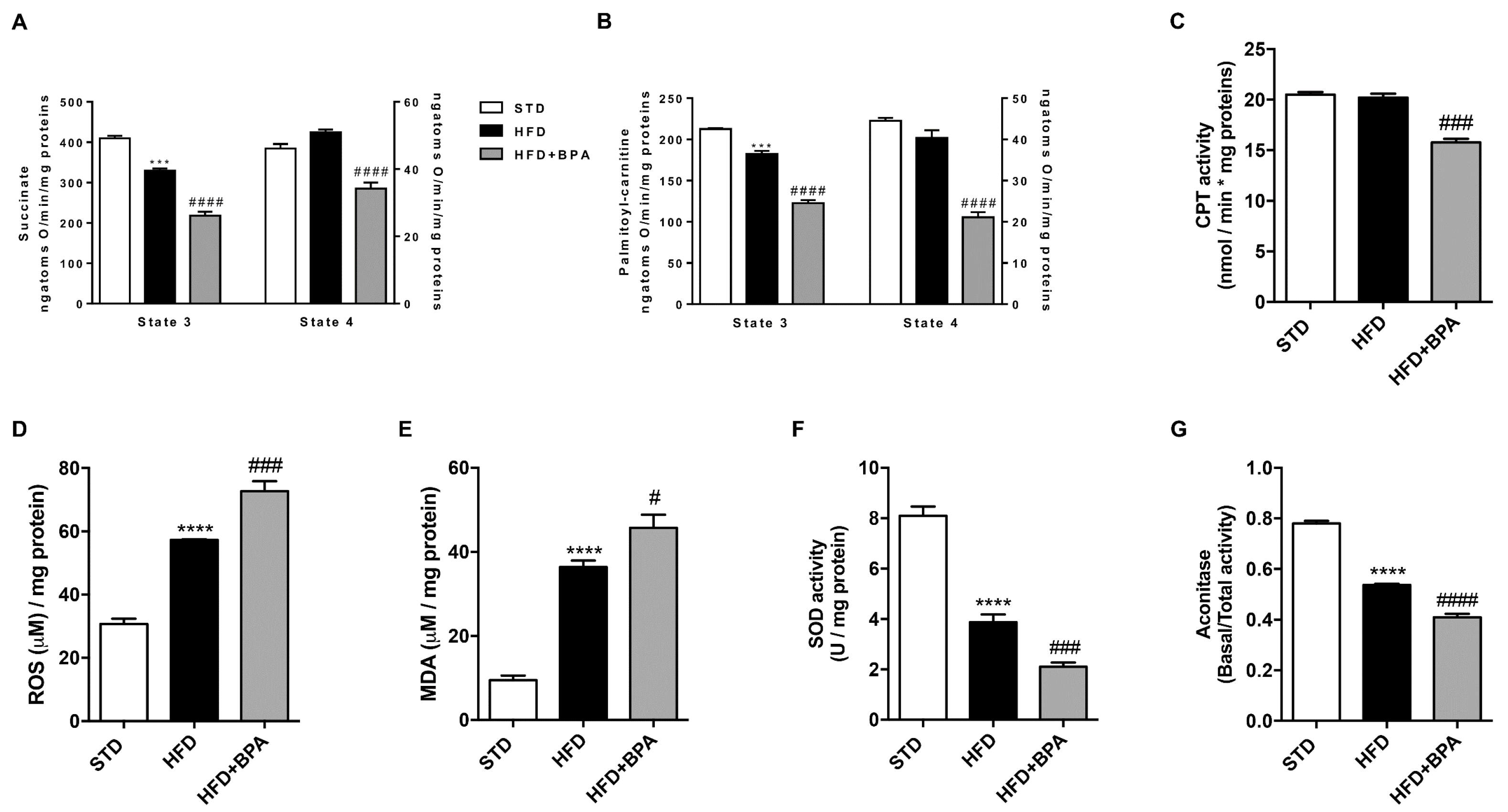
3.4. BPA Worsens Liver Mitochondrial Efficiency and Oxidative Stress Induced by HFD
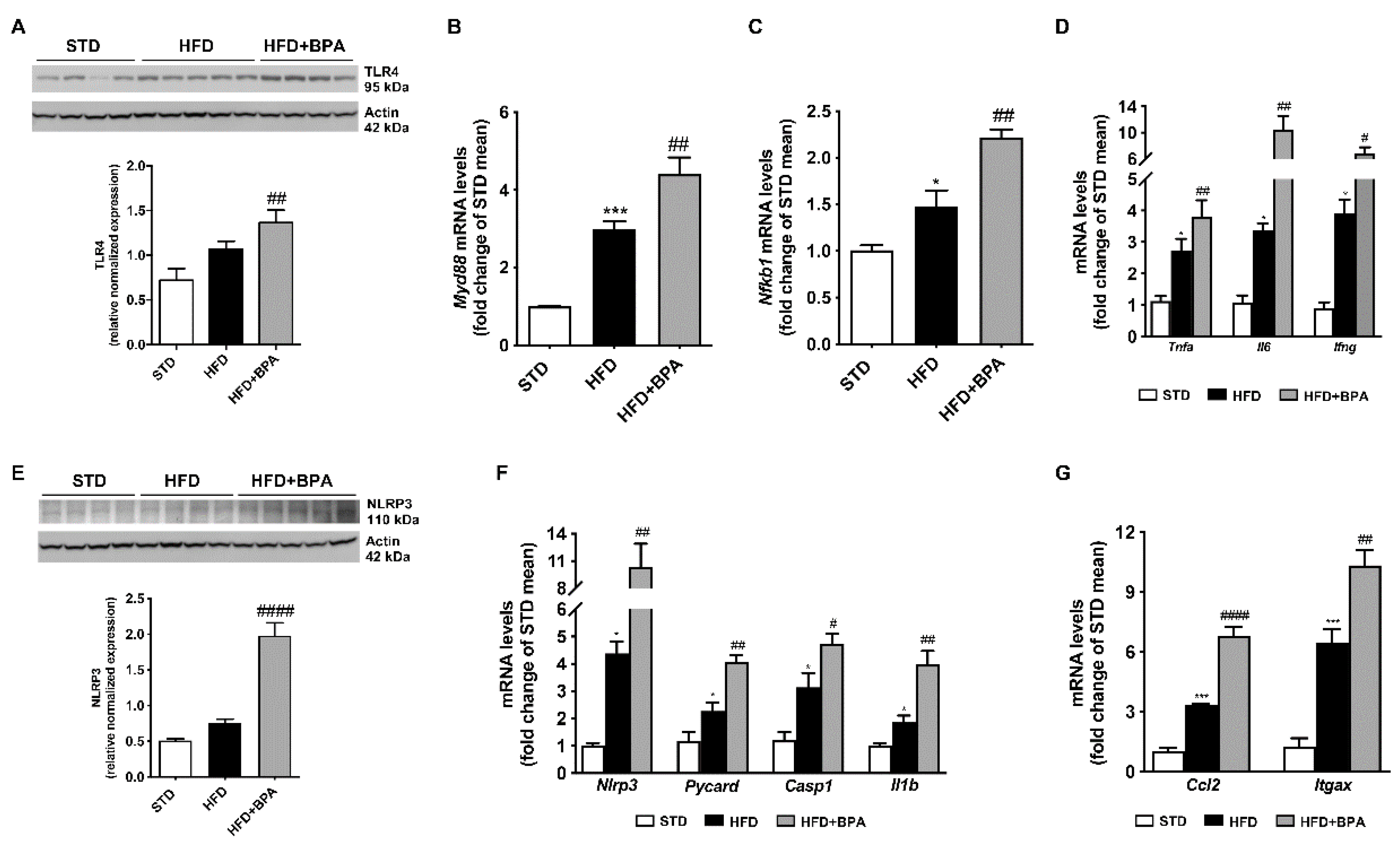
3.5. BPA Amplifies Immune/Inflammatory Response and Induces NLRP3 Inflammasome Activation in the Liver of HFD Mice
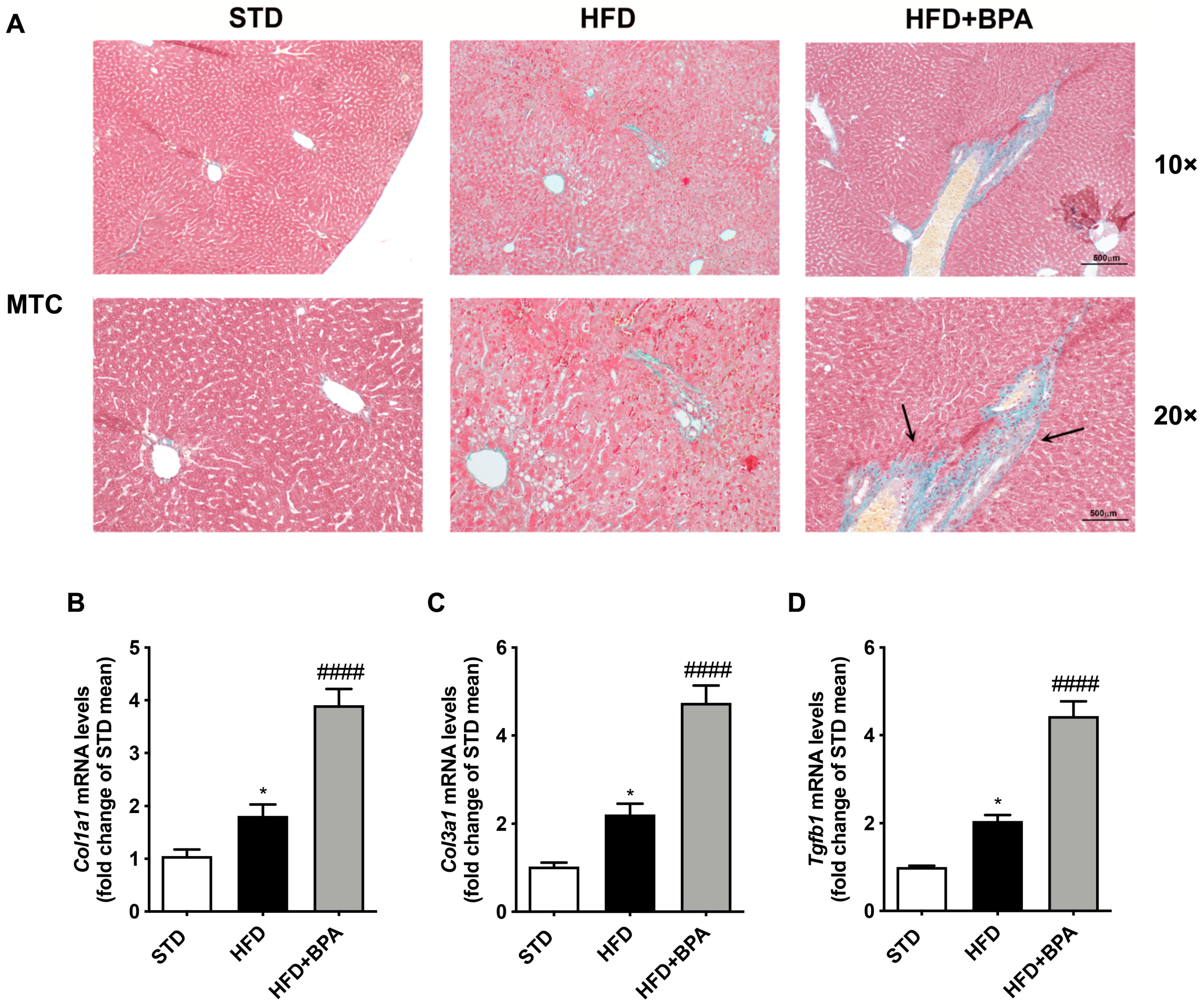
3.6. BPA Exposure Promotes Liver Fibrosis Progression in HFD-Fed Mice
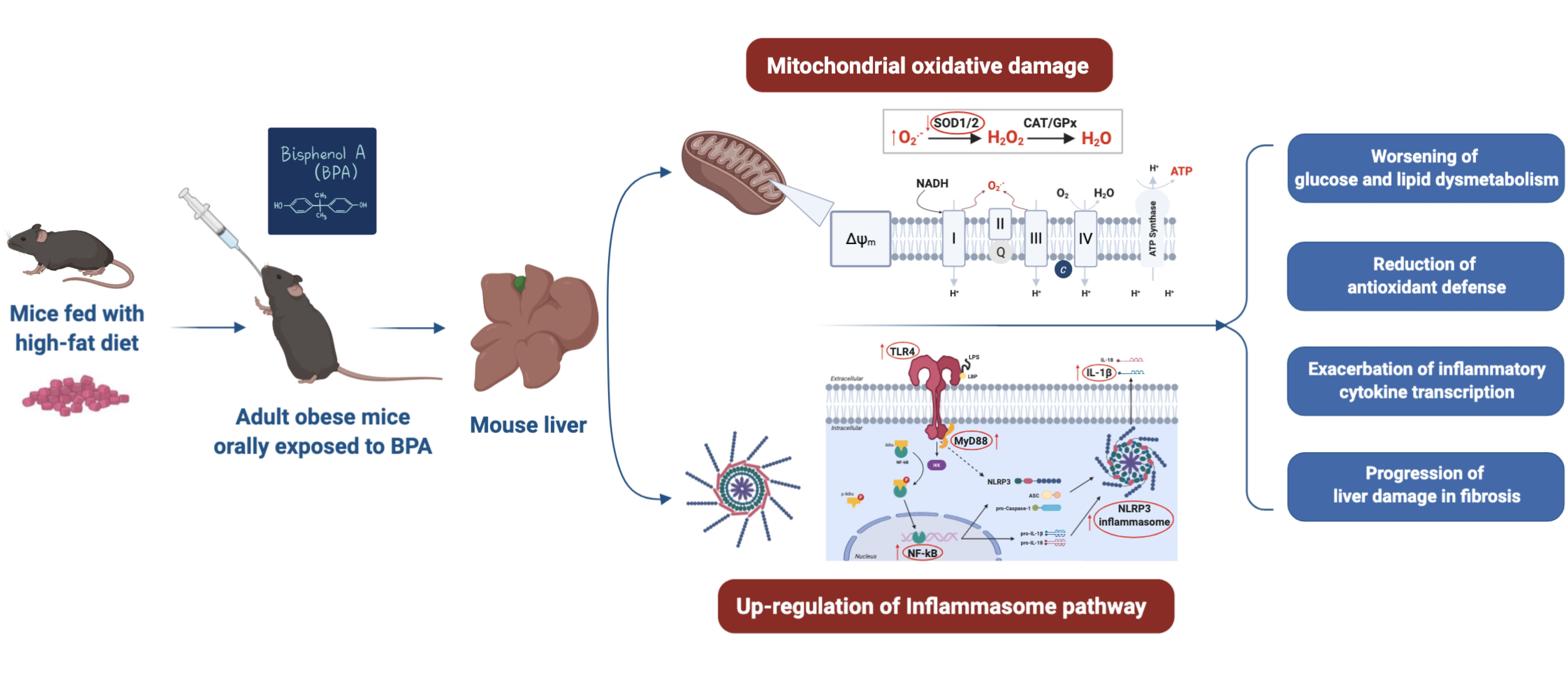
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alonso-Magdalena, P.; Rivera, F.J.; Guerrero-Bosagna, C. Bisphenol-A and metabolic diseases: Epigenetic, developmental and transgenerational basis. Environ. Epigenet. 2016, 2. [Google Scholar] [CrossRef]
- Heras-Gonzalez, L.; Latorre, J.A.; Martinez-Bebia, M.; Espino, D.; Olea-Serrano, F.; Mariscal-Arcas, M. The relationship of obesity with lifestyle and dietary exposure to endocrine-disrupting chemicals. Food Chem. Toxicol. 2020, 136, 110983. [Google Scholar] [CrossRef]
- Nowak, K.; Jablonska, E.; Ratajczak-Wrona, W. Immunomodulatory effects of synthetic endocrine disrupting chemicals on the development and functions of human immune cells. Environ. Int. 2019, 125, 350–364. [Google Scholar] [CrossRef]
- Kreitinger, J.M.; Beamer, C.A.; Shepherd, D.M. Environmental Immunology: Lessons Learned from Exposure to a Select Panel of Immunotoxicants. J. Immunol. 2016, 196, 3217–3225. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Lubrano, C.; Genovesi, G.; Specchia, P.; Costantini, D.; Mariani, S.; Petrangeli, E.; Lenzi, A.; Gnessi, L. Obesity and metabolic comorbidities: Environmental diseases? Oxidative Med. Cell Longev. 2013, 2013, 640673. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Beigh, S.; Chaudhari, B.P.; Sharma, S.; Aliul Hasan Abdi, S.; Ahmad, S.; Ahmad, F.; Parvez, S.; Raisuddin, S. Mitochondrial dysfunction induced by Bisphenol A is a factor of its hepatotoxicity in rats. Environ. Toxicol. 2016, 31, 1922–1934. [Google Scholar] [CrossRef]
- Ooe, H.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. Induction of reactive oxygen species by bisphenol A and abrogation of bisphenol A-induced cell injury by DJ-1. Toxicol. Sci. 2005, 88, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Tayama, S. Metabolism and cytotoxicity of bisphenol A and other bisphenols in isolated rat hepatocytes. Arch. Toxicol. 2000, 74, 99–105. [Google Scholar] [CrossRef]
- Huc, L.; Lemarie, A.; Gueraud, F.; Helies-Toussaint, C. Low concentrations of bisphenol A induce lipid accumulation mediated by the production of reactive oxygen species in the mitochondria of HepG2 cells. Toxicol. In Vitro 2012, 26, 709–717. [Google Scholar] [CrossRef]
- Asahi, J.; Kamo, H.; Baba, R.; Doi, Y.; Yamashita, A.; Murakami, D.; Hanada, A.; Hirano, T. Bisphenol A induces endoplasmic reticulum stress-associated apoptosis in mouse non-parenchymal hepatocytes. Life Sci. 2010, 87, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Cortese, K.; Voci, A.; Vergani, L.; Fabbri, R.; Barmo, C.; Gallo, G.; Canesi, L. Direct effects of Bisphenol A on lipid homeostasis in rat hepatoma cells. Chemosphere 2013, 91, 1123–1129. [Google Scholar] [CrossRef]
- Leonardi, A.; Cofini, M.; Rigante, D.; Lucchetti, L.; Cipolla, C.; Penta, L.; Esposito, S. The Effect of Bisphenol A on Puberty: A Critical Review of the Medical Literature. Int. J. Environ. Res. Public Health 2017, 14, 1044. [Google Scholar] [CrossRef] [Green Version]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS ONE 2013, 8, e55387. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Vieira, E.; Soriano, S.; Menes, L.; Burks, D.; Quesada, I.; Nadal, A. Bisphenol A exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environ. Health Perspect. 2010, 118, 1243–1250. [Google Scholar] [CrossRef] [Green Version]
- Angle, B.M.; Do, R.P.; Ponzi, D.; Stahlhut, R.W.; Drury, B.E.; Nagel, S.C.; Welshons, W.V.; Besch-Williford, C.L.; Palanza, P.; Parmigiani, S.; et al. Metabolic disruption in male mice due to fetal exposure to low but not high doses of bisphenol A (BPA): Evidence for effects on body weight, food intake, adipocytes, leptin, adiponectin, insulin and glucose regulation. Reprod. Toxicol. 2013, 42, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Sun, X.; Chen, Y.; Li, Y.; Song, L.; Zhou, Z.; Xu, B.; Lin, Y.; Xu, S. Perinatal exposure to bisphenol A exacerbates nonalcoholic steatohepatitis-like phenotype in male rat offspring fed on a high-fat diet. J. Endocrinol. 2014, 222, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Moon, M.K.; Jeong, I.K.; Jung Oh, T.; Ahn, H.Y.; Kim, H.H.; Park, Y.J.; Jang, H.C.; Park, K.S. Long-term oral exposure to bisphenol A induces glucose intolerance and insulin resistance. J. Endocrinol. 2015, 226, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Arevalo, M.; Alonso-Magdalena, P.; Servitja, J.M.; Boronat-Belda, T.; Merino, B.; Villar-Pazos, S.; Medina-Gomez, G.; Novials, A.; Quesada, I.; Nadal, A. Maternal Exposure to Bisphenol-A During Pregnancy Increases Pancreatic beta-Cell Growth During Early Life in Male Mice Offspring. Endocrinology 2016, 157, 4158–4171. [Google Scholar] [CrossRef]
- Hao, M.; Ding, L.; Xuan, L.; Wang, T.; Li, M.; Zhao, Z.; Lu, J.; Xu, Y.; Chen, Y.; Wang, W.; et al. Urinary bisphenol A concentration and the risk of central obesity in Chinese adults: A prospective study. J. Diabetes 2018, 10, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, R.W.; Myers, J.P.; Taylor, J.A.; Nadal, A.; Dyer, J.A.; Vom Saal, F.S. Experimental BPA Exposure and Glucose-Stimulated Insulin Response in Adult Men and Women. J. Endocr. Soc. 2018, 2, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Vom Saal, F.S.; Hughes, C. An extensive new literature concerning low-dose effects of bisphenol A shows the need for a new risk assessment. Environ. Health Perspect. 2005, 113, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, C.; Lama, A.; Pirozzi, C.; Cavaliere, G.; Trinchese, G.; Di Guida, F.; Nitrato Izzo, A.; Cimmino, F.; Paciello, O.; De Biase, D.; et al. Palmitoylethanolamide counteracts hepatic metabolic inflexibility modulating mitochondrial function and efficiency in diet-induced obese mice. FASEB J. 2020, 34, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef]
- Cavaliere, G.; Trinchese, G.; Bergamo, P.; De Filippo, C.; Mattace Raso, G.; Gifuni, G.; Putti, R.; Moni, B.H.; Canani, R.B.; Meli, R.; et al. Polyunsaturated Fatty Acids Attenuate Diet Induced Obesity and Insulin Resistance, Modulating Mitochondrial Respiratory Uncoupling in Rat Skeletal Muscle. PLoS ONE 2016, 11, e0149033. [Google Scholar] [CrossRef] [Green Version]
- Pirozzi, C.; Lama, A.; Simeoli, R.; Paciello, O.; Pagano, T.B.; Mollica, M.P.; Di Guida, F.; Russo, R.; Magliocca, S.; Canani, R.B.; et al. Hydroxytyrosol prevents metabolic impairment reducing hepatic inflammation and restoring duodenal integrity in a rat model of NAFLD. J. Nutr. Biochem. 2016, 30, 108–115. [Google Scholar] [CrossRef]
- Pirozzi, C.; Lama, A.; Annunziata, C.; Cavaliere, G.; De Caro, C.; Citraro, R.; Russo, E.; Tallarico, M.; Iannone, M.; Ferrante, M.C.; et al. Butyrate prevents valproate-induced liver injury: In vitro and in vivo evidence. FASEB J. 2020, 34, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Lama, A.; Annunziata, C.; Coretti, L.; Pirozzi, C.; Di Guida, F.; Nitrato Izzo, A.; Cristiano, C.; Mollica, M.P.; Chiariotti, L.; Pelagalli, A.; et al. N-(1-carbamoyl-2-phenylethyl) butyramide reduces antibiotic-induced intestinal injury, innate immune activation and modulates microbiota composition. Sci. Rep. 2019, 9, 4832. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M. Early-life exposure to EDCs: Role in childhood obesity and neurodevelopment. Nat. Rev. Endocrinol. 2017, 13, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, D.; Osborne, N.J.; Henley, W.E.; Cipelli, R.; Young, A.; Money, C.; McCormack, P.; Luben, R.; Khaw, K.T.; Wareham, N.J.; et al. Urinary bisphenol A concentration and risk of future coronary artery disease in apparently healthy men and women. Circulation 2012, 125, 1482–1490. [Google Scholar] [CrossRef] [Green Version]
- Meli, R.; Monnolo, A.; Annunziata, C.; Pirozzi, C.; Ferrante, M.C. Oxidative Stress and BPA Toxicity: An Antioxidant Approach for Male and Female Reproductive Dysfunction. Antioxidants 2020, 9, 405. [Google Scholar] [CrossRef]
- Usman, A.; Ahmad, M. From BPA to its analogues: Is it a safe journey? Chemosphere 2016, 158, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1062–1079. [Google Scholar] [CrossRef]
- Meli, R.; Mattace Raso, G.; Calignano, A. Role of innate immune response in non-alcoholic Fatty liver disease: Metabolic complications and therapeutic tools. Front. Immunol. 2014, 5, 177. [Google Scholar] [CrossRef] [Green Version]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate Regulates Liver Mitochondrial Function, Efficiency, and Dynamics in Insulin-Resistant Obese Mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhao, Z.; Ji, W. Bisphenol A induces apoptosis, oxidative stress and inflammatory response in colon and liver of mice in a mitochondria-dependent manner. Biomed. Pharmacother. 2019, 117, 109182. [Google Scholar] [CrossRef]
- Azevedo, L.F.; Porto Dechandt, C.R.; Cristina de Souza Rocha, C.; Hornos Carneiro, M.F.; Alberici, L.C.; Barbosa, F., Jr. Long-term exposure to bisphenol A or S promotes glucose intolerance and changes hepatic mitochondrial metabolism in male Wistar rats. Food Chem. Toxicol. 2019, 132, 110694. [Google Scholar] [CrossRef]
- Jiang, Y.; Xia, W.; Zhu, Y.; Li, X.; Wang, D.; Liu, J.; Chang, H.; Li, G.; Xu, B.; Chen, X.; et al. Mitochondrial dysfunction in early life resulted from perinatal bisphenol A exposure contributes to hepatic steatosis in rat offspring. Toxicol. Lett. 2014, 228, 85–92. [Google Scholar] [CrossRef]
- Bindhumol, V.; Chitra, K.C.; Mathur, P.P. Bisphenol A induces reactive oxygen species generation in the liver of male rats. Toxicology 2003, 188, 117–124. [Google Scholar] [CrossRef]
- Savastano, S.; Tarantino, G.; D’Esposito, V.; Passaretti, F.; Cabaro, S.; Liotti, A.; Liguoro, D.; Perruolo, G.; Ariemma, F.; Finelli, C.; et al. Bisphenol-A plasma levels are related to inflammatory markers, visceral obesity and insulin-resistance: A cross-sectional study on adult male population. J. Transl. Med. 2015, 13, 169. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Miura, K.; Seki, E.; Ohnishi, H.; Brenner, D.A. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic Fatty liver disease. Gastroenterol. Res. Pract. 2010, 2010, 362847. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic fibrosis—Overview. Toxicology 2008, 254, 120–129. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H. A critical role for the NLRP3 inflammasome in NASH. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 197. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Gutierrez-Garcia, A.K.; Choudhury, M. Mono-(2-ethylhexyl) Phthalate Aggravates Inflammatory Response via Sirtuin Regulation and Inflammasome Activation in RAW 264.7 Cells. Chem. Res. Toxicol. 2019, 32, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Panchanathan, R.; Liu, H.; Leung, Y.K.; Ho, S.M.; Choubey, D. Bisphenol A (BPA) stimulates the interferon signaling and activates the inflammasome activity in myeloid cells. Mol. Cell Endocrinol. 2015, 415, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef] [Green Version]
- Hotta, K.; Kitamoto, T.; Kitamoto, A.; Ogawa, Y.; Honda, Y.; Kessoku, T.; Yoneda, M.; Imajo, K.; Tomeno, W.; Saito, S.; et al. Identification of the genomic region under epigenetic regulation during non-alcoholic fatty liver disease progression. Hepatol. Res. 2018, 48, E320–E334. [Google Scholar] [CrossRef] [Green Version]
- Menezo, Y.J.; Silvestris, E.; Dale, B.; Elder, K. Oxidative stress and alterations in DNA methylation: Two sides of the same coin in reproduction. Reprod. Biomed. Online 2016, 33, 668–683. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Xia, W.; Yang, J.; Zhu, Y.; Chang, H.; Liu, J.; Huo, W.; Xu, B.; Chen, X.; Li, Y.; et al. BPA-induced DNA hypermethylation of the master mitochondrial gene PGC-1alpha contributes to cardiomyopathy in male rats. Toxicology 2015, 329, 21–31. [Google Scholar] [CrossRef]
- Zhang, P.; Chu, T.; Dedousis, N.; Mantell, B.S.; Sipula, I.; Li, L.; Bunce, K.D.; Shaw, P.A.; Katz, L.S.; Zhu, J.; et al. DNA methylation alters transcriptional rates of differentially expressed genes and contributes to pathophysiology in mice fed a high fat diet. Mol. Metab. 2017, 6, 327–339. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serum Parameters | STD | HFD | HFD + BPA |
|---|---|---|---|
| ALT (U/L) | 65.0 ± 2.12 | 139.2 ± 4.03 **** | 183.2 ± 3.16 #### |
| ALP (U/L) | 155.0 ± 3.44 | 265.6 ± 4.58 **** | 288.2 ± 6.41 # |
| Triglycerides (mg/dL) | 114.6 ± 5.391 | 218.6 ± 6.87 **** | 258.3 ± 6.86 ## |
| MCP-1 (pg/mL) | 28.50 ± 1.28 | 61.0 ± 2.00 **** | 74.6 ± 3.55 ## |
| LPS (U/mL) | 0.62 ± 0.02 | 1.53 ± 0.02 **** | 1.84 ± 0.04 ## |
| IL-10 (ng/mL) | 0.044 ± 0.002 | 0.034 ± 0.003 * | 0.019 ± 0.001 ## |
| Leptin (ng/mL) | 1.08 ± 0.04 | 12.88 ± 0.36 **** | 15.65 ± 0.42 ### |
| Adiponectin (mg/mL) | 2.87 ± 0.09 | 1.26 ± 0.06 **** | 1.07 ± 0.07 **** |
| Lep/Adipo ratio | 0.38 ± 0.02 | 9.63 ± 0.08 **** | 15.59 ± 0.81 #### |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirozzi, C.; Lama, A.; Annunziata, C.; Cavaliere, G.; Ruiz-Fernandez, C.; Monnolo, A.; Comella, F.; Gualillo, O.; Stornaiuolo, M.; Mollica, M.P.; et al. Oral Bisphenol A Worsens Liver Immune-Metabolic and Mitochondrial Dysfunction Induced by High-Fat Diet in Adult Mice: Cross-Talk between Oxidative Stress and Inflammasome Pathway. Antioxidants 2020, 9, 1201. https://doi.org/10.3390/antiox9121201
Pirozzi C, Lama A, Annunziata C, Cavaliere G, Ruiz-Fernandez C, Monnolo A, Comella F, Gualillo O, Stornaiuolo M, Mollica MP, et al. Oral Bisphenol A Worsens Liver Immune-Metabolic and Mitochondrial Dysfunction Induced by High-Fat Diet in Adult Mice: Cross-Talk between Oxidative Stress and Inflammasome Pathway. Antioxidants. 2020; 9(12):1201. https://doi.org/10.3390/antiox9121201
Chicago/Turabian StylePirozzi, Claudio, Adriano Lama, Chiara Annunziata, Gina Cavaliere, Clara Ruiz-Fernandez, Anna Monnolo, Federica Comella, Oreste Gualillo, Mariano Stornaiuolo, Maria Pina Mollica, and et al. 2020. "Oral Bisphenol A Worsens Liver Immune-Metabolic and Mitochondrial Dysfunction Induced by High-Fat Diet in Adult Mice: Cross-Talk between Oxidative Stress and Inflammasome Pathway" Antioxidants 9, no. 12: 1201. https://doi.org/10.3390/antiox9121201
APA StylePirozzi, C., Lama, A., Annunziata, C., Cavaliere, G., Ruiz-Fernandez, C., Monnolo, A., Comella, F., Gualillo, O., Stornaiuolo, M., Mollica, M. P., Raso, G. M., Ferrante, M. C., & Meli, R. (2020). Oral Bisphenol A Worsens Liver Immune-Metabolic and Mitochondrial Dysfunction Induced by High-Fat Diet in Adult Mice: Cross-Talk between Oxidative Stress and Inflammasome Pathway. Antioxidants, 9(12), 1201. https://doi.org/10.3390/antiox9121201