Skeletal Muscle Mitochondrial Dysfunction and Oxidative Stress in Peripheral Arterial Disease: A Unifying Mechanism and Therapeutic Target



Abstract
:
1. Introduction
2. Pathogenesis, Diagnosis, and Treatment of PAD
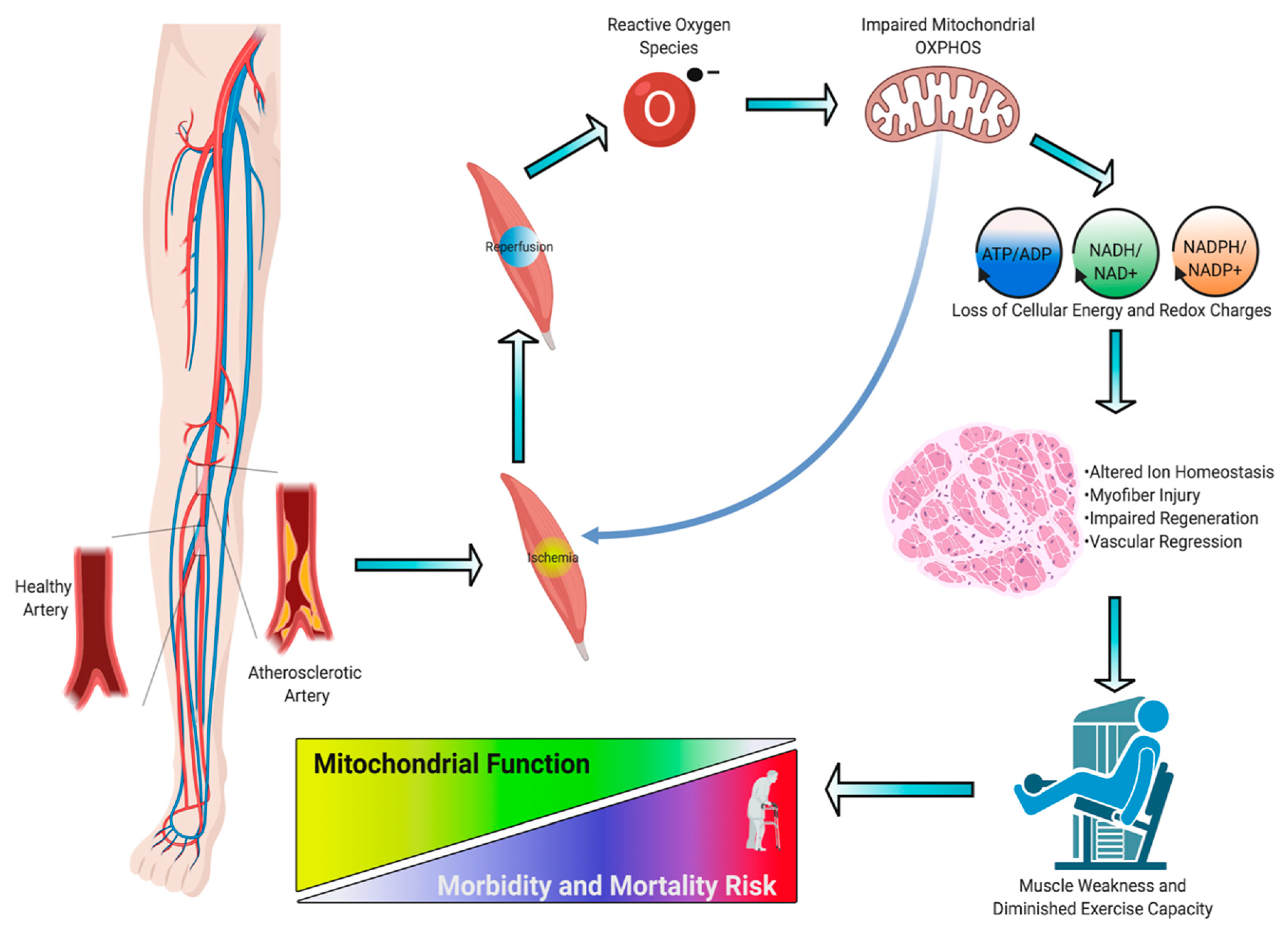
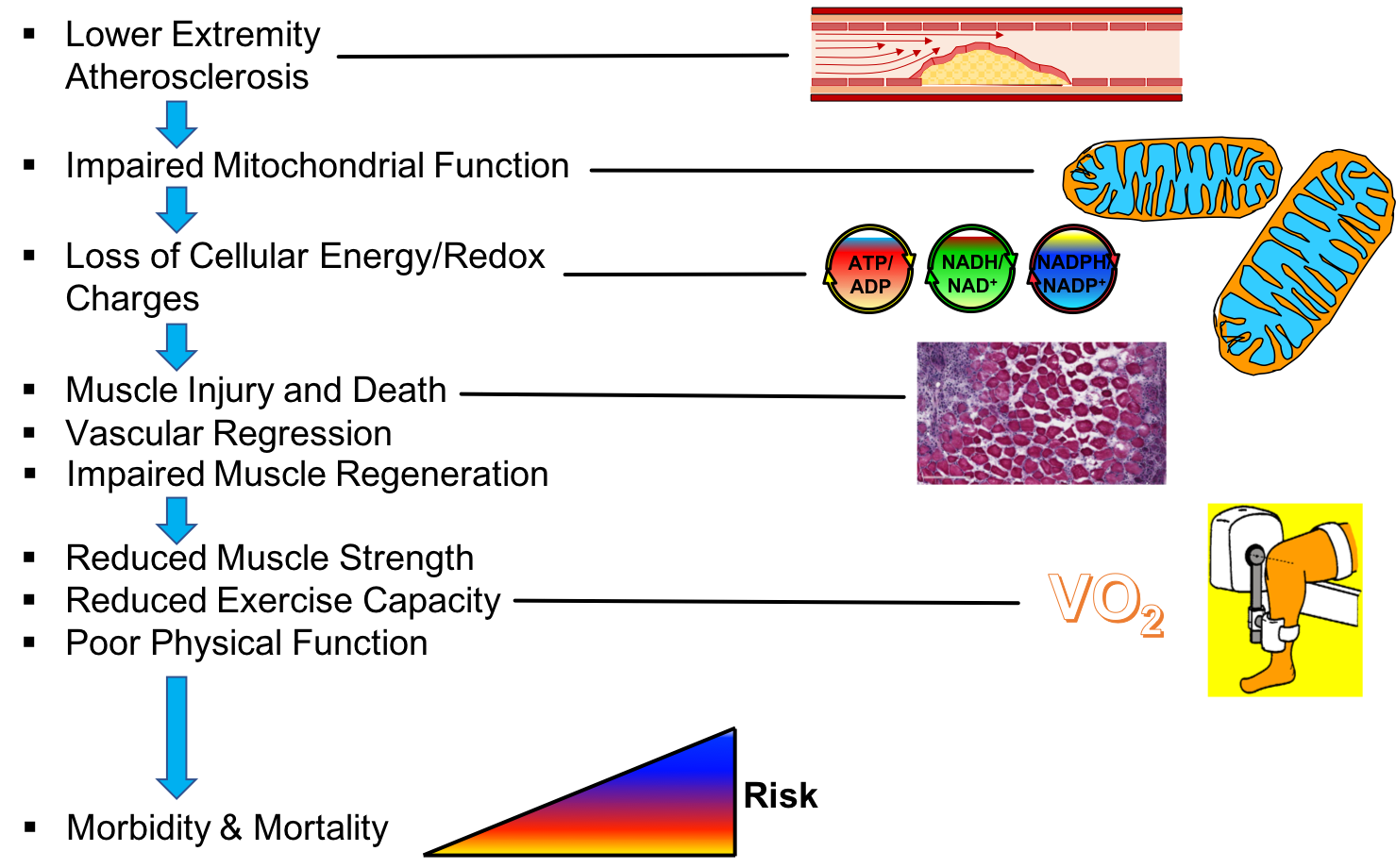
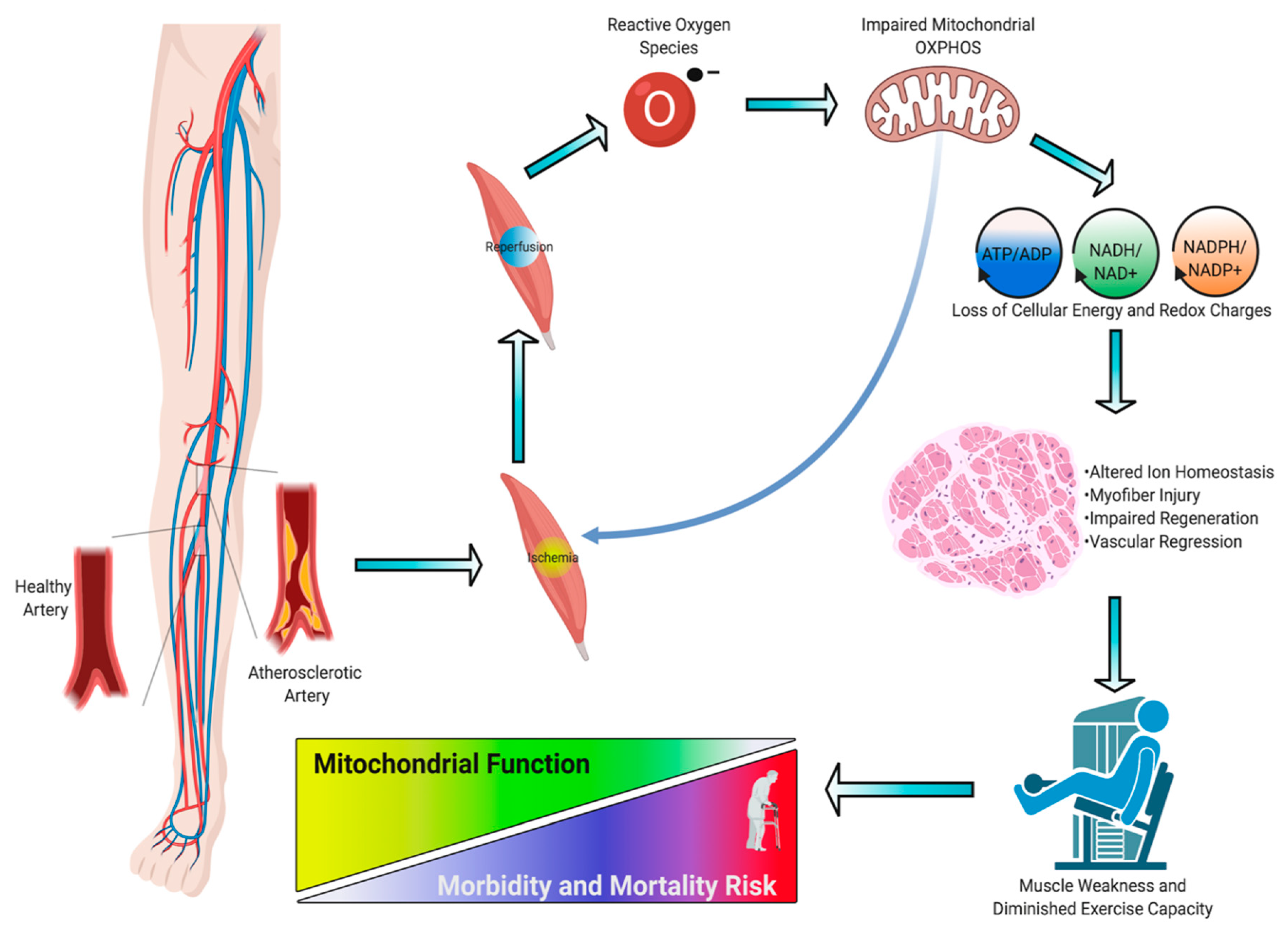
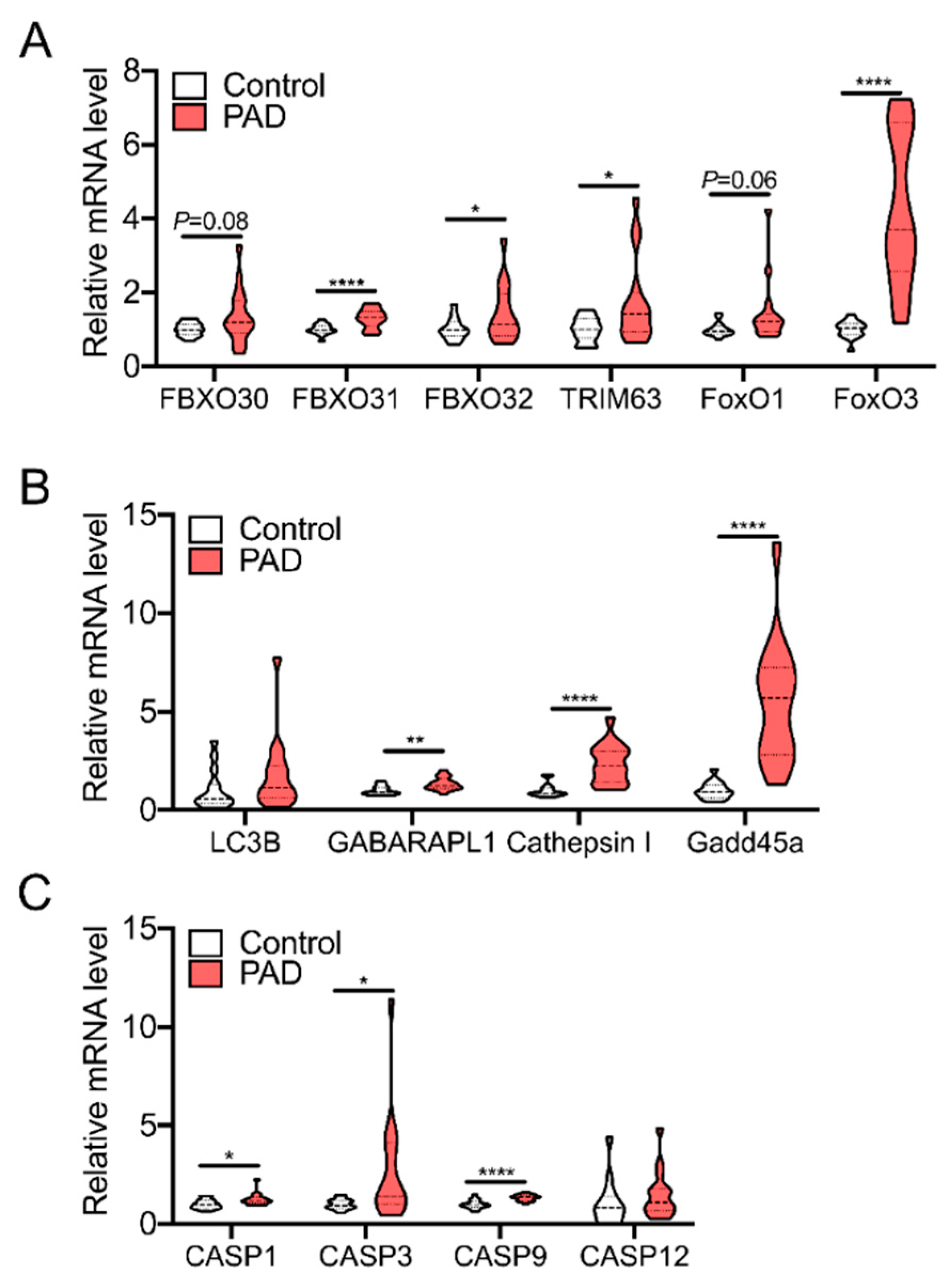
3. Skeletal Muscle Pathology in PAD
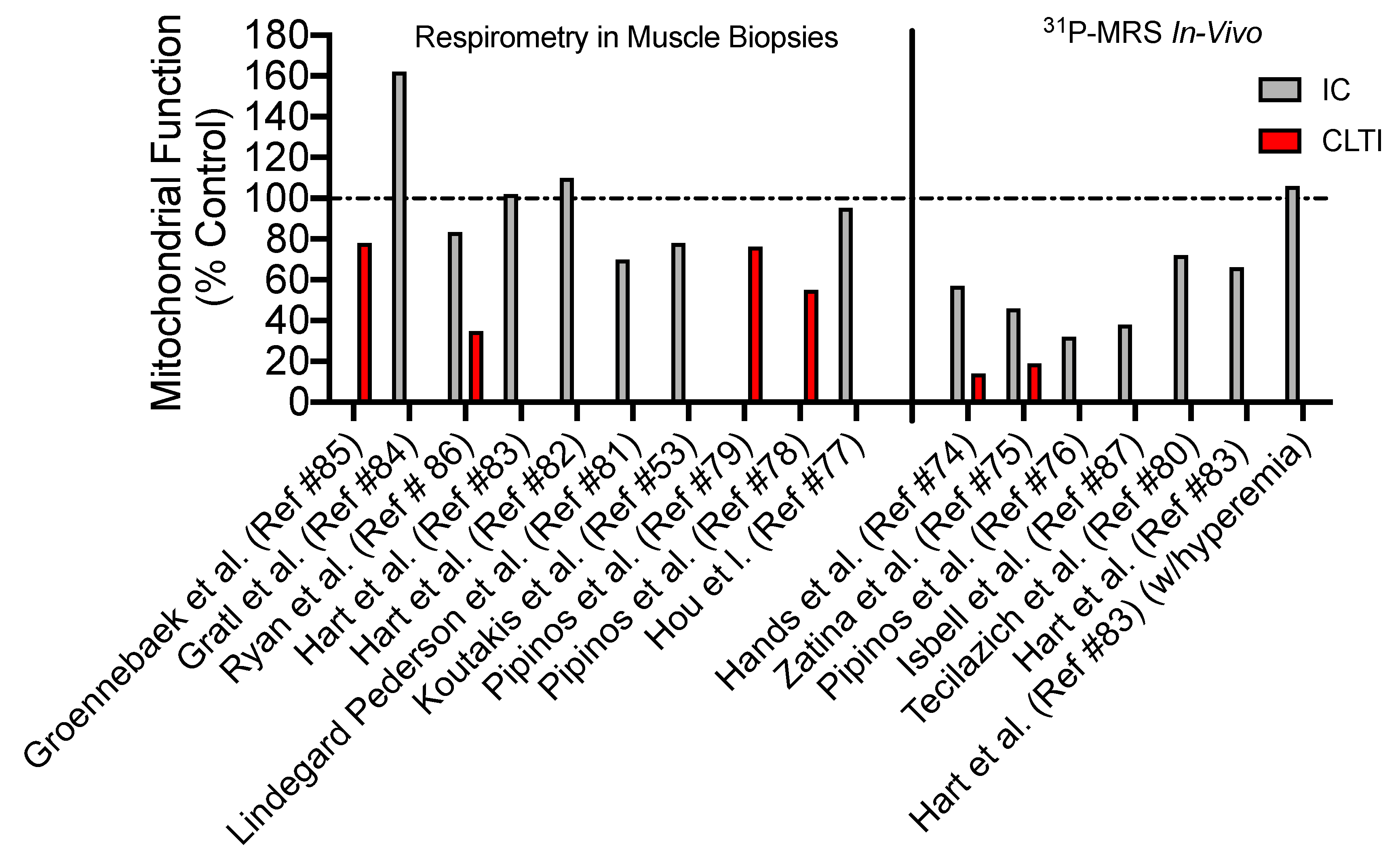
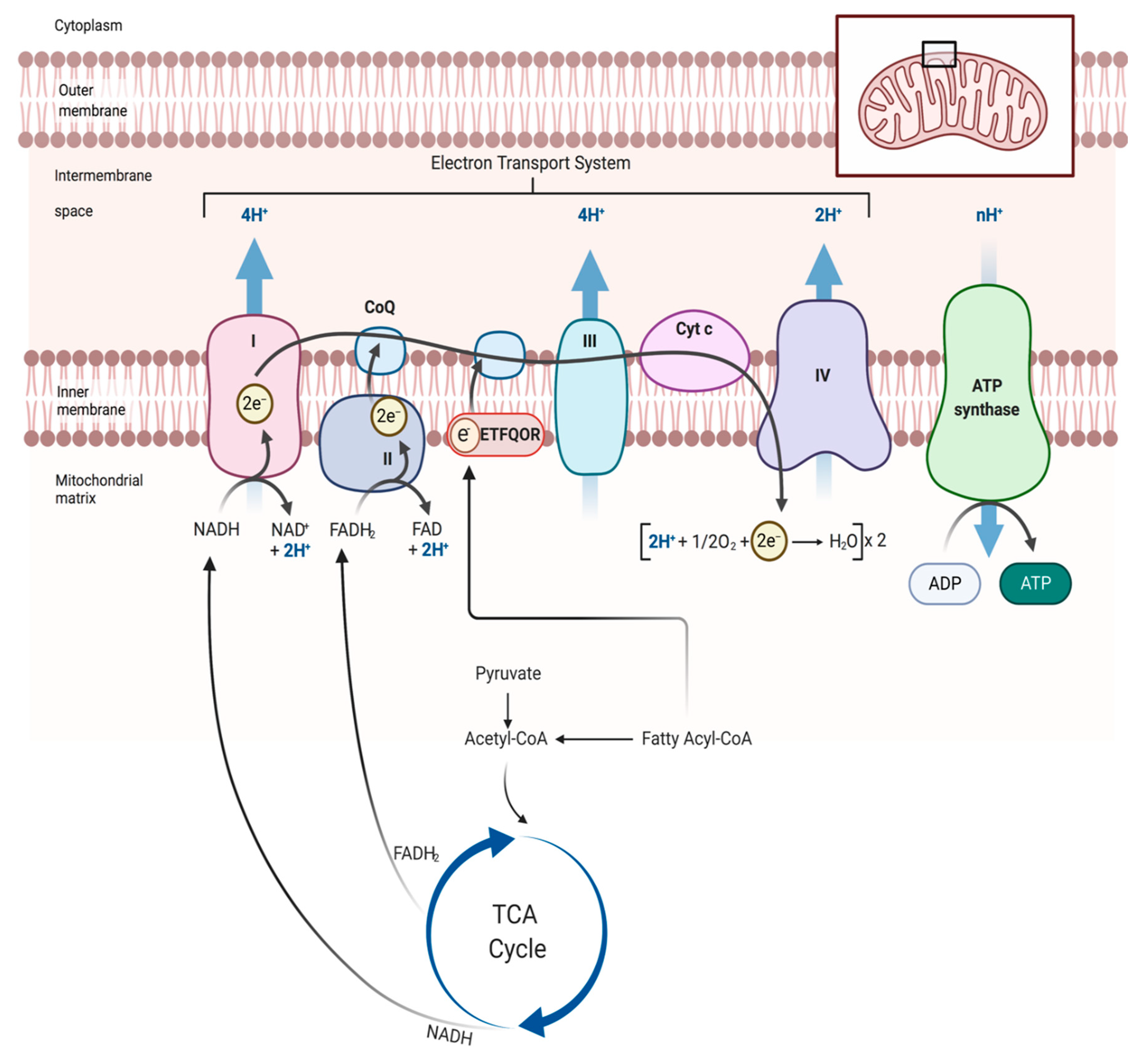
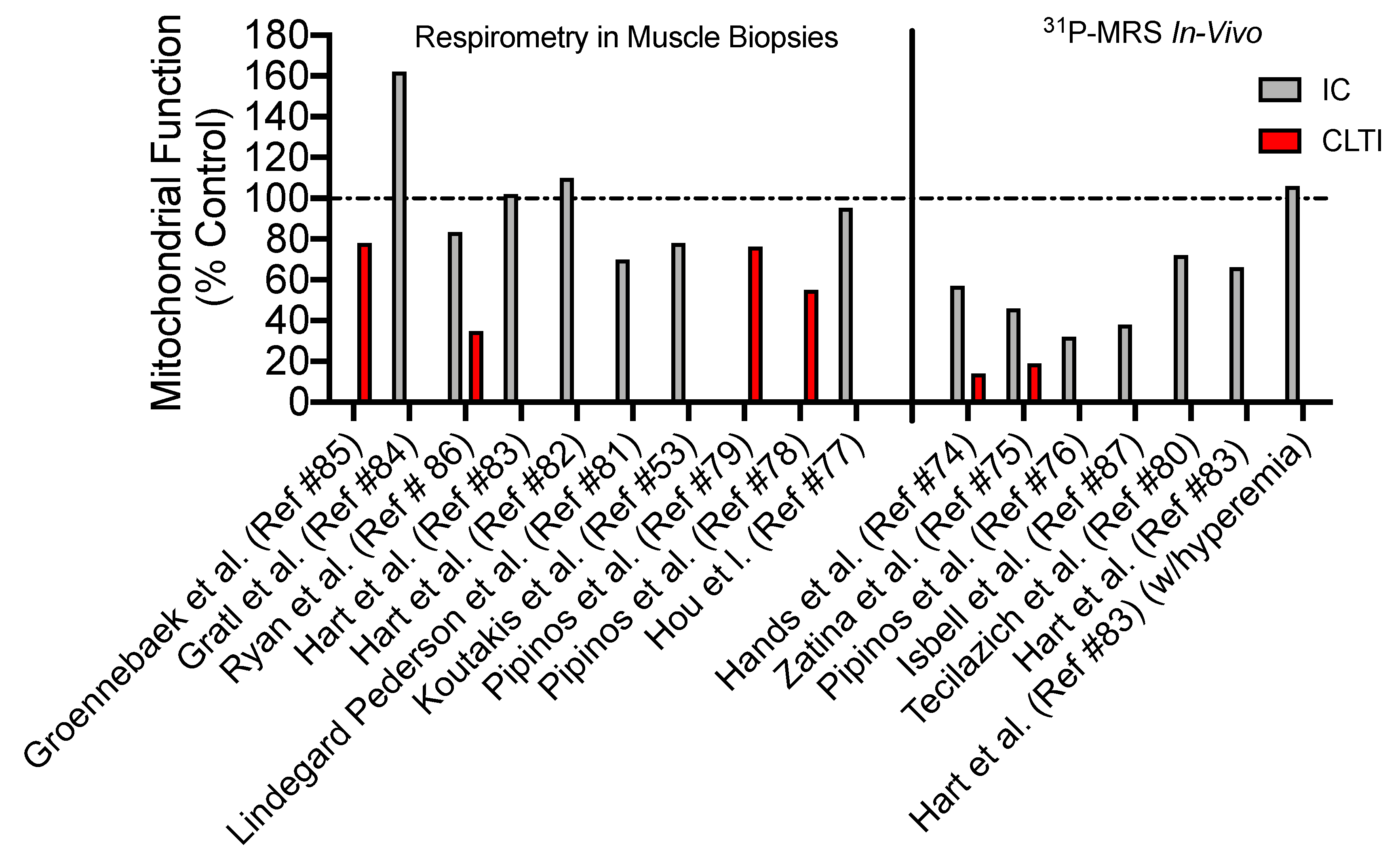
4. Skeletal Muscle Mitochondrial Function: Evidence from PAD patients
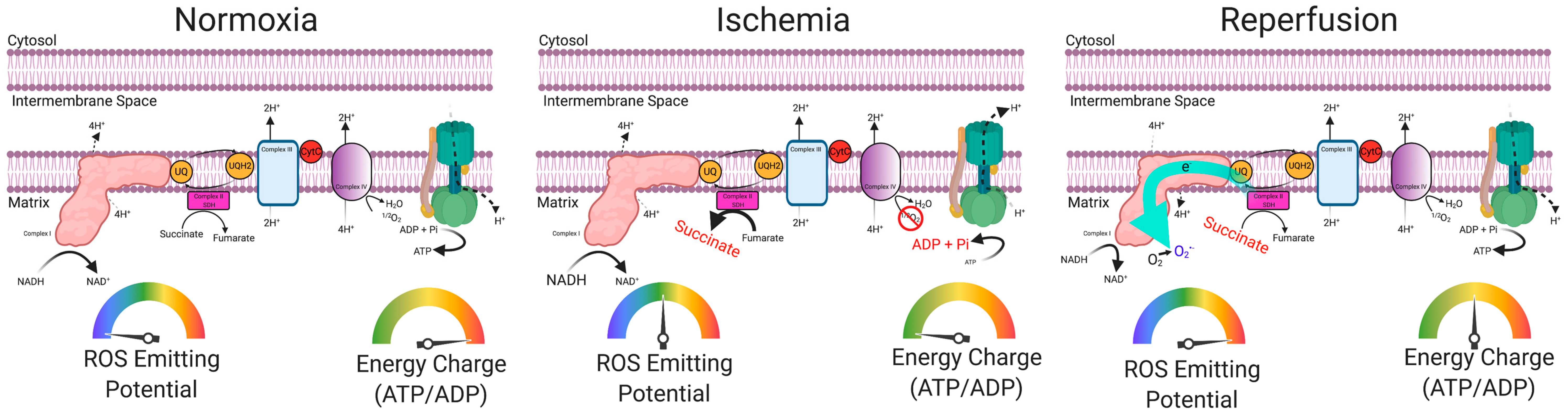
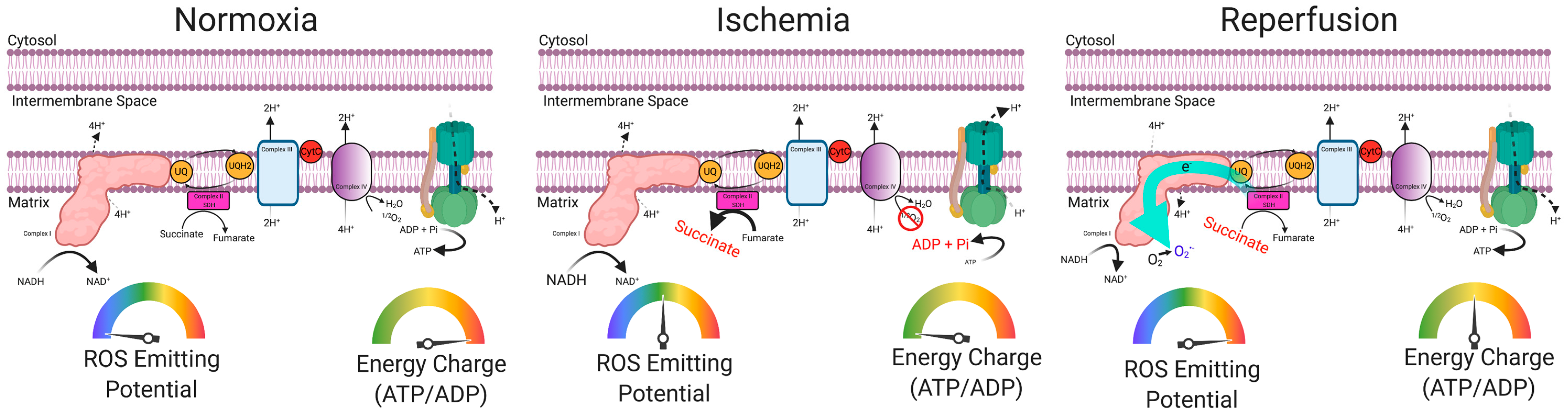
5. Skeletal Muscle Oxidative Stress in PAD
6. Preclinical Evidence Supporting a Causal Role of Mitochondria in the Pathogenesis of Limb Ischemia
7. Risk Factors and Comorbidities Converge on Muscle Mitochondria
8. Therapeutic Targeting of Mitochondria: The Future Is Bright for PAD
9. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Fowkes, F.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.A.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Tsai, T.T.; Rehring, T.F.; Rogers, R.K.; Shetterly, S.M.; Wagner, N.M.; Gupta, R.; Jazaeri, O.; Hedayati, N.; Jones, W.S.; Patel, M.R.; et al. The Contemporary Safety and Effectiveness of Lower Extremity Bypass Surgery and Peripheral Endovascular Interventions in the Treatment of Symptomatic Peripheral Arterial Disease. Circilation 2015, 132, 1999–2011. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.M.; Kalbaugh, C.A.; Blackhurst, D.W.; Cass, A.L.; Trent, E.A.; Langan, E.M.; Youkey, J.R. Determinants of functional outcome after revascularization for critical limb ischemia: An analysis of 1000 consecutive vascular interventions. J. Vasc. Surg. 2006, 44, 747–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, S.; Mohler, E.R.; Lederman, R.J.; Mendelsohn, F.O.; Saucedo, J.F.; Goldman, C.K.; Blebea, J.; Macko, J.; Kessler, P.D.; Rasmussen, H.S.; et al. Regional Angiogenesis With Vascular Endothelial Growth Factor in Peripheral Arterial Disease. Circulation 2003, 108, 1933–1938. [Google Scholar] [CrossRef]
- Van Royen, N.; Schirmer, S.H.; Atasever, B.; Behrens, C.Y.; Ubbink, D.; Buschmann, E.E.; Voskuil, M.; Bot, P.; Hoefer, I.; Schlingemann, R.O.; et al. START Trial. Circulation 2005, 112, 1040–1046. [Google Scholar] [CrossRef]
- Creager, M.A.; Olin, J.W.; Belch, J.J.F.; Moneta, G.L.; Henry, T.D.; Rajagopalan, S.; Annex, B.H.; Hiatt, W.R. Effect of Hypoxia-Inducible Factor-1α Gene Therapy on Walking Performance in Patients With Intermittent Claudication. Circulation 2011, 124, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Hammer, A.; Steiner, S. Gene therapy for therapeutic angiogenesis in peripheral arterial disease-systematic review and meta-analysis of randomized, controlled trials. Vasa 2013, 42, 331–339. [Google Scholar] [CrossRef]
- McDermott, M.M. Functional Impairment in Peripheral Artery Disease and How to Improve It in 2013. Curr. Cardiol. Rep. 2013, 15, 1–8. [Google Scholar] [CrossRef]
- Gardner, A.W.; Montgomery, P.S.; Parker, D.E. Physical activity is a predictor of all-cause mortality in patients with intermittent claudication. J. Vasc. Surg. 2008, 47, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Liu, K.; Tian, L.; Criqui, M.H.; Guralnik, J.M.; Ferrucci, L.; Liao, Y.; McDermott, M.M. Leg strength predicts mortality in men but not in women with peripheral arterial disease. J. Vasc. Surg. 2010, 52, 624–631. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.M.; Liu, K.; Ferrucci, L.; Tian, L.; Guralnik, J.M.; Liao, Y.; Criqui, M.H. Decline in Functional Performance Predicts Later Increased Mobility Loss and Mortality in Peripheral Arterial Disease. J. Am. Coll. Cardiol. 2011, 57, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Liu, K.; Ferrucci, L.; Criqui, M.H.; Tian, L.; Guralnik, J.M.; Tao, H.; McDermott, M.M. The Walking Impairment Questionnaire stair-climbing score predicts mortality in men and women with peripheral arterial disease. J. Vasc. Surg. 2012, 55, 1662–1673.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.M.; Liu, K.; Tian, L.; Guralnik, J.M.; Criqui, M.H.; Liao, Y.; Ferrucci, L. Calf Muscle Characteristics, Strength Measures, and Mortality in Peripheral Arterial Disease. J. Am. Coll. Cardiol. 2012, 59, 1159–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Liu, K.; Ferrucci, L.; Criqui, M.H.; Tian, L.; Guralnik, J.M.; Tao, H.; McDermott, M.M. Declining Walking Impairment Questionnaire Scores Are Associated With Subsequent Increased Mortality in Peripheral Artery Disease. J. Am. Coll. Cardiol. 2013, 61, 1820–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeper, N.J.; Myers, J.; Zhou, M.; Nead, K.T.; Syed, A.; Kojima, Y.; Caceres, R.D.; Cooke, J.P. Exercise capacity is the strongest predictor of mortality in patients with peripheral arterial disease. J. Vasc. Surg. 2013, 57, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissanen, T.T.; Vajanto, I.; Hiltunen, M.O.; Rutanen, J.; Kettunen, M.; Niemi, M.; Leppanen, P.; Turunen, M.P.; Markkanen, J.E.; Arve, K.; et al. Expression of Vascular Endothelial Growth Factor and Vascular Endothelial Growth Factor Receptor-2 (KDR/Flk-1) in Ischemic Skeletal Muscle and Its Regeneration. Am. J. Pathol. 2002, 160, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Pipinos, I.I.; Judge, S.M.; Selsby, J.T.; Zhu, Z.; Swanson, S.A.; Nella, A.A.; Dodd, S.L. Basic Science Review: The Myopathy of Peripheral Arterial Occlusive Disease: Part 2. Oxidative Stress, Neuropathy, and Shift in Muscle Fiber Type. Vasc. Endovasc. Surg. 2008, 42, 101–112. [Google Scholar] [CrossRef]
- Criqui, M.H.; Aboyans, V. Epidemiology of Peripheral Artery Disease. Circ. Res. 2015, 116, 1509–1526. [Google Scholar] [CrossRef] [Green Version]
- Fowkes, F.G.R.; Aboyans, V.; McDermott, M.M.; Sampson, U.K.A.; Criqui, M.H. Peripheral artery disease: Epidemiology and global perspectives. Nat. Rev. Cardiol. 2017, 14, 156–170. [Google Scholar] [CrossRef]
- Krishna, S.M.; Moxon, J.V.; Golledge, J. A Review of the Pathophysiology and Potential Biomarkers for Peripheral Artery Disease. Int. J. Mol. Sci. 2015, 16, 11294–11322. [Google Scholar] [CrossRef] [Green Version]
- Tsiara, S.; Elisaf, M.; Jagroop, I.A.; Mikhailidis, D.P. Platelets as Predictors of Vascular Risk: Is There a Practical Index of Platelet Activity? Clin. Appl. Thromb. 2003, 9, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Brevetti, G.; Giugliano, G.; Brevetti, L.; Hiatt, W.R. Inflammation in Peripheral Artery Disease. Circulation 2010, 122, 1862–1875. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, N.M.; Creager, M.A. Pathophysiology of Intermittent Claudication in Peripheral Artery Disease. Circ. J. 2017, 81, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A.; García-Cardeña, G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 2013, 22, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Garimella, P.S.; Hart, P.D.; O’Hare, A.; Deloach, S.; Herzog, C.A.; Hirsch, A.T. Peripheral Artery Disease and CKD: A Focus on Peripheral Artery Disease as a Critical Component of CKD Care. Am. J. Kidney Dis. 2012, 60, 641–654. [Google Scholar] [CrossRef]
- Jude, E.B.; Oyibo, S.O.; Chalmers, N.; Boulton, A.J. Peripheral Arterial Disease in Diabetic and Nondiabetic Patients: A comparison of severity and outcome. Diabetes Care 2001, 24, 1433–1437. [Google Scholar] [CrossRef] [Green Version]
- Conte, M.S.; Bradbury, A.W.; Kolh, P.; White, J.V.; Dick, F.; Fitridge, R.; Mills, J.L.; Ricco, J.-B.; Suresh, K.R.; Murad, M.H.; et al. Global vascular guidelines on the management of chronic limb-threatening ischemia. J. Vasc. Surg. 2019, 69, 3S–125S.e40. [Google Scholar] [CrossRef] [Green Version]
- Abu Dabrh, A.M.; Steffen, M.W.; Undavalli, C.; Asi, N.; Wang, Z.; Elamin, M.B.; Conte, M.S.; Murad, M.H. The natural history of untreated severe or critical limb ischemia. J. Vasc. Surg. 2015, 62, 1642–1651.e3. [Google Scholar] [CrossRef] [Green Version]
- Sigvant, B.; Kragsterman, B.; Falkenberg, M.; Hasvold, P.; Johansson, S.; Thuresson, M.; Nordanstig, J. Contemporary cardiovascular risk and secondary preventive drug treatment patterns in peripheral artery disease patients undergoing revascularization. J. Vasc. Surg. 2016, 64, 1009–1017.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridh, E.B.; Andersson, M.; Thuresson, M.; Sigvant, B.; Kragsterman, B.; Johansson, S.; Hasvold, P.; Falkenberg, M.; Nordanstig, J. Amputation Rates, Mortality, and Pre-operative Comorbidities in Patients Revascularised for Intermittent Claudication or Critical Limb Ischaemia: A Population Based Study. Eur. J. Vasc. Endovasc. Surg. 2017, 54, 480–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rantner, B.; Kollerits, B.; Pohlhammer, J.; Stadler, M.; Lamina, C.; Peric, S.; Klein-Weigel, P.; Mühlthaler, H.; Fraedrich, G.; Kronenberg, F. The fate of patients with intermittent claudication in the 21st century revisited–results from the CAVASIC Study. Sci. Rep. 2017, 7, 45833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norgren, L.; Hiatt, W.R.; Dormandy, J.A.; Nehler, M.R.; Harris, K.A.; Fowkes, F.G.R. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J. Vasc. Surg. 2007, 45, S5–S67. [Google Scholar] [CrossRef] [Green Version]
- Bevan, G.H.; Solaru, K.T.W. Evidence-Based Medical Management of Peripheral Artery Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 541–553. [Google Scholar] [CrossRef]
- Conte, M.S.; Pomposelli, F.B.; Clair, D.G.; Geraghty, P.J.; McKinsey, J.F.; Mills, J.L.; Moneta, G.L.; Murad, M.H.; Powell, R.J.; Reed, A.B.; et al. Society for Vascular Surgery practice guidelines for atherosclerotic occlusive disease of the lower extremities: Management of asymptomatic disease and claudication. J. Vasc. Surg. 2015, 61, 2S–41S.e1. [Google Scholar] [CrossRef] [Green Version]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.R.; Hamburg, N.M.; Kinlay, S.; et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2017, 135, e686–e725. [Google Scholar] [CrossRef]
- Mills, J.L.; Conte, M.S.; Armstrong, D.G.; Pomposelli, F.B.; Schanzer, A.; Sidawy, A.N.; Andros, G. The Society for Vascular Surgery Lower Extremity Threatened Limb Classification System: Risk stratification based on Wound, Ischemia, and foot Infection (WIfI). J. Vasc. Surg. 2014, 59, 220–234.e2. [Google Scholar] [CrossRef] [Green Version]
- Stoner, M.C.; Calligaro, K.D.; Chaer, R.A.; Dietzek, A.M.; Farber, A.; Guzman, R.J.; Hamdan, A.D.; Landry, G.J.; Yamaguchi, D.J. Reporting standards of the Society for Vascular Surgery for endovascular treatment of chronic lower extremity peripheral artery disease. J. Vasc. Surg. 2016, 64, e1–e21. [Google Scholar] [CrossRef] [Green Version]
- Mills, J.L. BEST-CLI trial on the homestretch. J. Vasc. Surg. 2019, 69, 313–314. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, T.; Oba, Y.; Koshida, R.; Suzuki, Y.; Murata, A.; Ito, T. Prediction of the Technical Success of Endovascular Therapy in Patients with Critical Limb Threatening Ischaemia Using the Global Limb Anatomical Staging System. Eur. J. Vasc. Endovasc. Surg. 2020, 60, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.W.; Adam, D.J.; Bell, J.; Forbes, J.F.; Fowkes, F.G.R.; Gillespie, I.; Ruckley, C.V.; Raab, G.M. Bypass versus Angioplasty in Severe Ischaemia of the Leg (BASIL) trial: An intention-to-treat analysis of amputation-free and overall survival in patients randomized to a bypass surgery-first or a balloon angioplasty-first revascularization strategy. J. Vasc. Surg. 2010, 51, 5S–17S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, W.P.; Loretz, L.; Hanesian, C.; Flahive, J.; Bostrom, J.; Lunig, N.; Schanzer, A.; Messina, L. Society for Vascular Surgery Wound, Ischemia, foot Infection (WIfI) score correlates with the intensity of multimodal limb treatment and patient-centered outcomes in patients with threatened limbs managed in a limb preservation center. J. Vasc. Surg. 2017, 66, 488–498.e2. [Google Scholar] [CrossRef] [PubMed]
- Almasri, J.; Adusumalli, J.; Asi, N.; Lakis, S.; Alsawas, M.; Prokop, L.J.; Bradbury, A.; Kolh, P.; Conte, M.S.; Murad, M.H. A systematic review and meta-analysis of revascularization outcomes of infrainguinal chronic limb-threatening ischemia. J. Vasc. Surg. 2018, 68, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, Y.; Matsumoto, T.; Aoyagi, Y.; Tanaka, S.; Okadome, J.; Morisaki, K.; Shirabe, K.; Maehara, Y. Sarcopenia is a prognostic factor for overall survival in patients with critical limb ischemia. J. Vasc. Surg. 2015, 61, 945–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.M.; Peterson, C.A.; Sufit, R.; Ferrucci, L.; Guralnik, J.; Kibbe, M.R.; Polonsky, T.S.; Tian, L.; Criqui, M.H.; Zhao, L.; et al. Peripheral artery disease, calf skeletal muscle mitochondrial DNA copy number, and functional performance. Vasc. Med. 2018, 23, 340–348. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.R.; Swanson, S.A.; Haynatzki, G.; Koutakis, P.; Johanning, J.M.; Reppert, P.R.; Papoutsi, E.; Miserlis, D.; Zhu, Z.; Casale, G.P.; et al. Protein Concentration and Mitochondrial Content in the Gastrocnemius Predicts Mortality Rates in Patients With Peripheral Arterial Disease. Ann. Surg. 2015, 261, 605–610. [Google Scholar] [CrossRef]
- White, S.H.; McDermott, M.M.; Sufit, R.L.; Kosmac, K.; Bugg, A.W.; Gonzalez-Freire, M.; Ferrucci, L.; Tian, L.; Zhao, L.; Gao, Y.; et al. Walking performance is positively correlated to calf muscle fiber size in peripheral artery disease subjects, but fibers show aberrant mitophagy: An observational study. J. Transl. Med. 2016, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.M.; Hoff, F.; Ferrucci, L.; Pearce, W.H.; Guralnik, J.M.; Tian, L.; Liu, K.; Schneider, J.R.; Sharma, L.; Tan, J.; et al. Lower Extremity Ischemia, Calf Skeletal Muscle Characteristics, and Functional Impairment in Peripheral Arterial Disease. J. Am. Geriatr. Soc. 2007, 55, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, M.; Šabovič, M.; Gregoric, I.; Simunic, B.; Pisot, R. Increased Fatigability of the Gastrocnemius Medialis Muscle in Individuals with Intermittent Claudication. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Scott-Okafor, H.R.; Silver, K.K.C.; Parker, J.; Almy-Albert, T.; Gardner, A.W. Lower extremity strength deficits in peripheral arterial occlusive disease patients with intermittent claudication. Angiology 2001, 52, 7–14. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.M.; Ferrucci, L.; Guralnik, J.; Tian, L.; Liu, K.; Hoff, F.; Liao, Y.; Criqui, M.H. Pathophysiological Changes in Calf Muscle Predict Mobility Loss at 2-Year Follow-Up in Men and Women With Peripheral Arterial Disease. Circulation 2009, 120, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutakis, P.; Myers, S.A.; Cluff, K.; Ha, D.M.; Haynatzki, G.; McComb, R.D.; Uchida, K.; Miserlis, D.; Papoutsi, E.; Johanning, J.M.; et al. Abnormal myofiber morphology and limb dysfunction in claudication. J. Surg. Res. 2015, 196, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, D.R.; Skalina, T.A.; Singh, T.P.; Moxon, J.V.; Golledge, J. Association of Computed Tomographic Leg Muscle Characteristics With Lower Limb and Cardiovascular Events in Patients With Peripheral Artery Disease. J. Am. Hear. Assoc. 2018, 7, e009943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, M.M. Lower Extremity Manifestations of Peripheral Artery Disease. Circ. Res. 2015, 116, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.D.; Epstein, F.H.; Meyer, C.H.; Hagspiel, K.D.; Wang, H.; Berr, S.S.; Harthun, N.L.; Weltman, A.; Dimaria, J.M.; West, A.M.; et al. Multifactorial determinants of functional capacity in peripheral arterial disease: Uncoupling of calf muscle perfusion and metabolism. J. Am. Coll. Cardiol. 2009, 54, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Regensteiner, J.G.; Wolfel, E.E.; Brass, E.P.; Carry, M.R.; Ringel, S.P.; Hargarten, M.E.; Stamm, E.R.; Hiatt, W.R. Chronic changes in skeletal muscle histology and function in peripheral arterial disease. Circulation 1993, 87, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Garg, P.K.; Liu, K.; Ferrucci, L.; Guralnik, J.M.; Criqui, M.H.; Tian, L.; Sufit, R.; Nishida, T.; Tao, H.; Liao, Y.; et al. Lower Extremity Nerve Function, Calf Skeletal Muscle Characteristics, and Functional Performance in Peripheral Arterial Disease. J. Am. Geriatr. Soc. 2011, 59, 1855–1863. [Google Scholar] [CrossRef]
- Askew, C.D.; Green, S.C.; Walker, P.J.; Kerr, G.K.; Green, A.A.; Williams, A.D.; Febbraio, M.A. Skeletal muscle phenotype is associated with exercise tolerance in patients with peripheral arterial disease. J. Vasc. Surg. 2005, 41, 802–807. [Google Scholar] [CrossRef] [Green Version]
- Tuomisto, T.T.; Rissanen, T.T.; Vajanto, I.; Korkeela, A.; Rutanen, J.; Ylä-Herttuala, S. HIF-VEGF-VEGFR-2, TNF-α and IGF pathways are upregulated in critical human skeletal muscle ischemia as studied with DNA array. Atherosclerosis 2004, 174, 111–120. [Google Scholar] [CrossRef]
- Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; et al. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, V.; Wineinger, M.A.; Kappagoda, C.T.; Kilmer, D.D.; Pevec, W.C.; Rosen, W.S.; Rubner, D. SENSORY AXONOPATHY IN MILD TO MODERATE PERIPHERAL ARTERIAL DISEASE. Am. J. Phys. Med. Rehabil. 1998, 77, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Mäkitie, J.; Teräväinen, H. Histochemical changes in striated muscle in patients with intermittent claudication. Arch. Pathol. Lab. Med. 1977, 101, 658–663. [Google Scholar] [PubMed]
- Robbins, J.L.; Jones, W.S.; Duscha, B.D.; Allen, J.D.; Kraus, W.E.; Regensteiner, J.G.; Hiatt, W.R.; Annex, B.H. Relationship between leg muscle capillary density and peak hyperemic blood flow with endurance capacity in peripheral artery disease. J. Appl. Physiol. 2011, 111, 81–86. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, M.R.; Bronks, R.; Newton, R.U.; Sharman, M.J.; Graham, J.C.; Cody, D.V.; Kraemer, W.J. Muscle fiber characteristics in patients with peripheral arterial disease. Med. Sci. Sports Exerc. 2001, 33, 2016–2021. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Reid, B.A.; Ro, B.; Casey, C.A.; Song, Q.; Kuang, S.; Roseguini, B.T. Heat therapy improves soleus muscle force in a model of ischemia-induced muscle damage. J. Appl. Physiol. 2019, 127, 215–228. [Google Scholar] [CrossRef]
- Kim, K.; Ro, B.; Damen, F.W.; Gramling, D.P.; Lehr, T.D.; Song, Q.; Goergen, C.J.; Roseguini, B.T. Heat therapy improves body composition and muscle function, but does not affect capillary or collateral growth in a model of obesity and hindlimb ischemia. J. Appl. Physiol. 2020, LID. [Google Scholar] [CrossRef]
- McGuigan, M.R.M.; Bronks, R.; Newton, R.U.; Sharman, M.J.; Graham, J.C.; Cody, D.V.; Kraemer, W.J. Resistance Training in Patients With Peripheral Arterial Disease: Effects on Myosin Isoforms, Fiber Type Distribution, and Capillary Supply to Skeletal Muscle. J. Gerontol. Ser. A: Boil. Sci. Med. Sci. 2001, 56, B302–B310. [Google Scholar] [CrossRef]
- Wang, J.; Bronks, R.; Graham, J.; Myers, S. Effects of Supervised Treadmill Walking Training on Calf Muscle Capillarization in Patients with Intermittent Claudication. Angiology 2009, 60, 36–41. [Google Scholar] [CrossRef]
- Gardner, A.W.; Killewich, L.A. Lack of functional benefits following infrainguinal bypass in peripheral arterial occlusive disease patients. Vasc. Med. 2001, 6, 9–14. [Google Scholar] [CrossRef]
- West, A.M.; Anderson, J.D.; Epstein, F.H.; Meyer, C.H.; Hagspiel, K.D.; Berr, S.S.; Harthun, N.L.; Weltman, A.L.; Annex, B.H.; Kramer, C.M. Percutaneous intervention in peripheral artery disease improves calf muscle phosphocreatine recovery kinetics: A pilot study. Vasc. Med. 2012, 17, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Roseguini, B.T.; Hirai, D.M.; Alencar, M.C.; Ramos, R.P.; Silva, B.M.; Wolosker, N.; Neder, J.A.; Nery, L.E. Sildenafil improves skeletal muscle oxygenation during exercise in men with intermittent claudication. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R396–R404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brass, E.P.; Hiatt, W.R.; Gardner, A.W.; Hoppel, C.L. Decreased NADH dehydrogenase and ubiquinol-cytochrome c oxidoreductase in peripheral arterial disease. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H603–H609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hands, L.J.; Bore, P.J.; Egalloway, G.; Morris, P.J.; Radda, G.K. Muscle metabolism in patients with peripheral vascular disease investigated by 31P nuclear magnetic resonance spectroscopy. Clin. Sci. 1986, 71, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Zatina, M.A.; Berkowitz, H.D.; Gross, G.M.; Maris, J.M.; Chance, B. P-31 Nuclear-Magnetic-Resonance Spectroscopy-Noninvasive Biochemical-Analysis of the Ischemic Extremity. J. Vasc. Surg. 1986, 3, 411–420. [Google Scholar] [PubMed]
- Pipinos, I.I.; Shepard, A.D.; Anagnostopoulos, P.V.; Katsamouris, A.; Boska, M.D. Phosphorus 31 nuclear magnetic resonance spectroscopy suggests a mitochondrial defect in claudicating skeletal muscle. J. Vasc. Surg. 2000, 31, 944–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.Y.; Green, S.; Askew, C.; Barker, G.; Green, A.; Walker, P. Skeletal muscle mitochondrial ATP production rate and walking performance in peripheral arterial disease. Clin. Physiol. Funct. Imaging 2002, 22, 226–232. [Google Scholar] [CrossRef]
- Pipinos, I.I.; Sharov, V.G.; Shepard, A.D.; Anagnostopoulos, P.V.; Katsamouris, A.; Todor, A.; Filis, K.A.; Sabbah, H.N. Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J. Vasc. Surg. 2003, 38, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Pipinos, I.I.; Judge, A.R.; Zhu, Z.; Selsby, J.T.; Swanson, S.A.; Johanning, J.M.; Baxter, B.T.; Lynch, T.G.; Dodd, S.L. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free. Radic. Biol. Med. 2006, 41, 262–269. [Google Scholar] [CrossRef]
- Tecilazich, F.; Dinh, T.; Lyons, T.E.; Guest, J.; Villafuerte, R.A.; Sampanis, C.; Gnardellis, C.; Zuo, C.S.; Veves, A. Postexercise phosphocreatine recovery, an index of mitochondrial oxidative phosphorylation, is reduced in diabetic patients with lower extremity complications. J. Vasc. Surg. 2013, 57, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.L.; Bækgaard, N.; Quistorff, B. Mitochondrial dysfunction in calf muscles of patients with combined peripheral arterial disease and diabetes type 2. Int. Angiol. 2017, 36, 482–495. [Google Scholar]
- Hart, C.R.; Layec, G.; Trinity, J.D.; Kwon, O.S.; Zhao, J.; Reese, V.R.; Gifford, J.R.; Richardson, R.S. Increased skeletal muscle mitochondrial free radical production in peripheral arterial disease despite preserved mitochondrial respiratory capacity. Exp. Physiol. 2018, 103, 838–850. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.R.; Layec, G.; Trinity, J.D.; Le Fur, Y.; Gifford, J.R.; Clifton, H.L.; Richardson, R.S. Oxygen availability and skeletal muscle oxidative capacity in patients with peripheral artery disease: Implications from in vivo and in vitro assessments. Am. J. Physiol. Circ. Physiol. 2018, 315, H897–H909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratl, A.; Frese, J.P.B.; Speichinger, F.; Pesta, D.; Frech, A.; Omran, S.; Greiner, A. Regeneration of Mitochondrial Function in Gastrocnemius Muscle in Peripheral Arterial Disease After Successful Revascularisation. Eur. J. Vasc. Endovasc. Surg. 2019, 59, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Groennebaek, T.; Billeskov, T.B.; Schytz, C.T.; Jespersen, N.R.; Bøtker, H.E.; Olsen, R.K.J.; Eldrup, N.; Nielsen, J.; Farup, J.; De Paoli, F.V.; et al. Mitochondrial Structure and Function in the Metabolic Myopathy Accompanying Patients with Critical Limb Ischemia. Cells 2020, 9, 570. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.E.; Yamaguchi, D.J.; Schmidt, C.A.; Zeczycki, T.N.; Shaikh, S.R.; Brophy, P.; Green, T.D.; Tarpey, M.D.; Karnekar, R.; Goldberg, E.J.; et al. Extensive skeletal muscle cell mitochondriopathy distinguishes critical limb ischemia patients from claudicants. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Isbell, D.C.; Berr, S.S.; Toledano, A.Y.; Epstein, F.H.; Meyer, C.H.; Rogers, W.J.; Harthun, N.L.; Hagspiel, K.D.; Weltman, A.; Kramer, C.M. Delayed Calf Muscle Phosphocreatine Recovery After Exercise Identifies Peripheral Arterial Disease. J. Am. Coll. Cardiol. 2006, 47, 2289–2295. [Google Scholar] [CrossRef] [Green Version]
- Keller, U.; Oberhänsli, R.; Huber, P.; Widmer, L.K.; Aue, W.P.; Hassink, R.I.; Muller, S.; Seelig, J. Phosphocreatine content and intracellular pH of calf muscle measured by phosphorus NMR spectroscopy in occlusive arterial disease of the legs. Eur. J. Clin. Investig. 1985, 15, 382–388. [Google Scholar] [CrossRef]
- Williams, D.M.; Fencil, L.; Chenevert, T.L. Peripheral arterial occlusive disease: P-31 MR spectroscopy of calf muscle. Radiology 1990, 175, 381–385. [Google Scholar] [CrossRef]
- Schunk, K.; Romaneehsen, B.; Rieker, O.; Duber, C.; Kersjes, W.; Schadmand-Fischer, S.; Schmiedt, W.; Thelen, M. Dynamic phosphorus-31 magnetic resonance spectroscopy in arterial occlusive disease-Effects of vascular therapy on spectroscopic results. Invest. Radiol. 1998, 33, 329–335. [Google Scholar] [CrossRef]
- Bhat, H.K.; Hiatt, W.R.; Hoppel, C.L.; Brass, E.P. Skeletal Muscle Mitochondrial DNA Injury in Patients With Unilateral Peripheral Arterial Disease. Circulation 1999, 99, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Freire, M.; Moore, A.Z.; Peterson, C.A.; Kosmac, K.; McDermott, M.M.; Sufit, R.L.; Guralnik, J.M.; Polonsky, T.; Tian, L.; Kibbe, M.R.; et al. Associations of Peripheral Artery Disease With Calf Skeletal Muscle Mitochondrial DNA Heteroplasmy. J. Am. Hear. Assoc. 2020, 9, e015197. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2008, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Fisher-Wellman, K.H.; Lin, C.T.; Ryan, T.E.; Reese, L.R.; Gilliam, L.A.A.; Cathey, B.L.; Lark, D.S.; Smith, C.D.; Muoio, D.M.; Neufer, P.D. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem. J. 2015, 467, 271–280. [Google Scholar] [CrossRef]
- Go, Y.M.; Roede, J.R.; Walker, D.I.; Duong, D.M.; Seyfried, N.T.; Orr, M.; Liang, Y.; Pennell, K.D.; Jones, D. Selective Targeting of the Cysteine Proteome by Thioredoxin and Glutathione Redox Systems. Mol. Cell. Proteom. 2013, 12, 3285–3296. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. Jama 2002, 287, 2570–2581. [Google Scholar] [CrossRef]
- Malmstedt, J.; Leander, K.; Wahlberg, E.; Karlström, L.; Alfredsson, L.; Swedenborg, J. Outcome After Leg Bypass Surgery for Critical Limb Ischemia Is Poor in Patients With Diabetes: A population-based cohort study. Diabetes Care 2008, 31, 887–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Spangenburg, E.E.; Neufer, P.D.; McClung, J.M. Targeted Expression of Catalase to Mitochondria Protects Against Ischemic Myopathy in High-Fat Diet–Fed Mice. Diabetes 2016, 65, 2553–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, D.J.; Casale, G.P.; Koutakis, P.; Nella, A.A.; Swanson, S.A.; Zhu, Z.; Miserlis, D.; Johanning, J.M.; Pipinos, I.I. Oxidative damage and myofiber degeneration in the gastrocnemius of patients with peripheral arterial disease. J. Transl. Med. 2013, 11, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutakis, P.; Weiss, D.J.; Miserlis, D.; Shostrom, V.K.; Papoutsi, E.; Ha, D.M.; Carpenter, L.A.; McComb, R.D.; Casale, G.P.; Pipinos, I.I. Oxidative damage in the gastrocnemius of patients with peripheral artery disease is myofiber type selective. Redox Biol. 2014, 2, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive Oxygen Species: Impact on Skeletal Muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Morton, A.B.; Ahn, B.; Smuder, A.J. Redox control of skeletal muscle atrophy. Free. Radic. Biol. Med. 2016, 98, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottecher, J.; Kindo, M.; Chamaraux-Tran, T.-N.; Charles, A.-L.; Lejay, A.; Kemmel, V.; Vogel, T.; Chakfé, N.; Zoll, J.; Diemunsch, P.; et al. Skeletal muscle ischemia-reperfusion injury and cyclosporine A in the aging rat. Fundam. Clin. Pharmacol. 2016, 30, 216–225. [Google Scholar] [CrossRef]
- Charles, A.L.; Guilbert, A.S.; Guillot, M.; Talha, S.; Lejay, A.; Meyer, A.; Kindo, M.; Wolff, V.; Bouitbir, J.; Zoll, J.; et al. Muscles Susceptibility to Ischemia-Reperfusion Injuries Depends on Fiber Type Specific Antioxidant Level. Front. Physiol. 2017, 8, 52. [Google Scholar] [CrossRef] [Green Version]
- Thaveau, F.; Zoll, J.; Rouyer, O.; Chafke, N.; Kretz, J.G.; Piquard, F.; Geny, B. Ischemic preconditioning specifically restores complexes I and II activities of the mitochondrial respiratory chain in ischemic skeletal muscle. J. Vasc. Surg. 2007, 46, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Pottecher, J.; Guillot, M.; Belaidi, E.; Charles, A.-L.; Lejay, A.; Gharib, A.; Diemunsch, P.; Geny, B. Cyclosporine A normalizes mitochondrial coupling, reactive oxygen species production, and inflammation and partially restores skeletal muscle maximal oxidative capacity in experimental aortic cross-clamping. J. Vasc. Surg. 2013, 57, 1100–1108.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, S.; Charles, A.-L.; Georg, I.; Goupilleau, F.; Meyer, A.; Kindo, M.; Laverny, G.; Metzger, D.; Geny, B. Aging Exacerbates Ischemia-Reperfusion-Induced Mitochondrial Respiration Impairment in Skeletal Muscle. Antioxidants 2019, 8, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottecher, J.; Adamopoulos, C.; Lejay, A.; Bouitbir, J.; Charles, A.L.; Meyer, A.; Singer, M.; Wolff, V.; Diemunsch, P.; Laverny, G.; et al. Diabetes Worsens Skeletal Muscle Mitochondrial Function, Oxidative Stress, and Apoptosis After Lower-Limb Ischemia-Reperfusion: Implication of the RISK and SAFE Pathways? Front. Physiol. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novelli, G.P.; Adembri, C.; Gandini, E.; Orlandini, S.Z.; Papucci, L.; Formigli, L.; Manneschi, L.I.; Quattrone, A.; Pratesi, C.; Capaccioli, S. Vitamin E protects human skeletal muscle from damage during surgical ischemia-reperfusion. Am. J. Surg. 1997, 173, 206–209. [Google Scholar] [CrossRef]
- Ostman, B.; Michaelsson, K.; Rahme, H.; Hillered, L. Tourniquet-Induced Ischemia and Reperfusion in Human Skeletal Muscle. Clin. Orthop. Relat. Res. 2004, 418, 260–265. [Google Scholar] [CrossRef]
- Fuglestad, M.A.; Hernandez, H.; Gao, Y.; Ybay, H.; Schieber, M.N.; Brunette, K.E.; Myers, S.A.; Casale, G.P.; Pipinos, I.I. A low-cost, wireless near-infrared spectroscopy device detects the presence of lower extremity atherosclerosis as measured by computed tomographic angiography and characterizes walking impairment in peripheral artery disease. J. Vasc. Surg. 2020, 71, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Manfredini, F.; Malagoni, A.M.; Felisatti, M.; Mandini, S.; Mascoli, F.; Manfredini, R.; Basaglia, N.; Zamboni, P. A Dynamic Objective Evaluation of Peripheral Arterial Disease by Near-Infrared Spectroscopy. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Mofarrahi, M.; Kristof, A.S.; Nkengfac, B.; Harel, S.; Hussain, S.N.A. Reactive Oxygen Species Regulation of Autophagy in Skeletal Muscles. Antioxidants Redox Signal. 2014, 20, 443–459. [Google Scholar] [CrossRef]
- Berru, F.N.; Gray, S.E.; Thome, T.; Kumar, R.A.; Salyers, Z.R.; Coleman, M.; Le, D.; O’Malley, K.; Ferreira, L.F.; Berceli, S.A.; et al. Chronic kidney disease exacerbates ischemic limb myopathy in mice via altered mitochondrial energetics. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- McClung, J.M.; Mccord, T.J.; Southerland, K.; Schmidt, C.A.; Padgett, M.E.; Ryan, T.E.; Kontos, C.D. Subacute limb ischemia induces skeletal muscle injury in genetically susceptible mice independent of vascular density. J. Vasc. Surg. 2016, 64, 1101–1111.e2. [Google Scholar] [CrossRef] [Green Version]
- McClung, J.M.; Mccord, T.J.; Ryan, T.E.; Schmidt, C.A.; Green, T.D.; Southerland, K.W.; Reinardy, J.L.; Mueller, S.B.; Venkatraman, T.N.; Lascola, C.D.; et al. BAG3 (Bcl-2–Associated Athanogene-3) Coding Variant in Mice Determines Susceptibility to Ischemic Limb Muscle Myopathy by Directing Autophagy. Circulation 2017, 136, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.A.; Ryan, T.E.; Lin, C.-T.; Inigo, M.M.; Green, T.D.; Brault, J.J.; Spangenburg, E.E.; McClung, J.M. Diminished force production and mitochondrial respiratory deficits are strain-dependent myopathies of subacute limb ischemia. J. Vasc. Surg. 2017, 65, 1504–1514.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.A.; Amorese, A.J.; Ryan, T.E.; Goldberg, E.J.; Tarpey, M.D.; Green, T.D.; Karnekar, R.R.; Yamaguchi, D.J.; Spangenburg, E.E.; McClung, J.M. Strain-Dependent Variation in Acute Ischemic Muscle Injury. Am. J. Pathol. 2018, 188, 1246–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokun, A.O.; Keum, S.; Hazarika, S.; Li, Y.; LaMonte, G.M.; Wheeler, F.; Marchuk, D.A.; Annex, B.H. A Quantitative Trait Locus (LSq-1) on Mouse Chromosome 7 Is Linked to the Absence of Tissue Loss After Surgical Hindlimb Ischemia. Circulation 2008, 117, 1207–1215. [Google Scholar] [CrossRef]
- McClung, J.M.; Mccord, T.J.; Keum, S.; Johnson, S.; Annex, B.H.; Marchuk, D.A.; Kontos, C.D. Skeletal Muscle–Specific Genetic Determinants Contribute to the Differential Strain-Dependent Effects of Hindlimb Ischemia in Mice. Am. J. Pathol. 2012, 180, 2156–2169. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Tarpey, M.D.; Amorese, A.J.; Yamaguchi, D.J.; Goldberg, E.J.; Iñigo, M.M.; Karnekar, R.; O’Rourke, A.; Ervasti, J.M.; et al. PFKFB3-mediated glycolysis rescues myopathic outcomes in the ischemic limb. JCI Insight 2020, 5, 139628. [Google Scholar] [CrossRef]
- Miura, S.; Saitoh, S.-I.; Kokubun, T.; Owada, T.; Yamauchi, H.; Machii, H.; Takeishi, Y. Mitochondrial-Targeted Antioxidant Maintains Blood Flow, Mitochondrial Function, and Redox Balance in Old Mice Following Prolonged Limb Ischemia. Int. J. Mol. Sci. 2017, 18, 1897. [Google Scholar] [CrossRef] [Green Version]
- Lejay, A.; Paradis, S.; Lambert, A.; Charles, A.-L.; Talha, S.; Enache, I.; Thaveau, F.; Chakfe, N.; Geny, B. N-Acetyl Cysteine Restores Limb Function, Improves Mitochondrial Respiration, and Reduces Oxidative Stress in a Murine Model of Critical Limb Ischaemia. Eur. J. Vasc. Endovasc. Surg. 2018, 56, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Orfany, A.; Arriola, C.G.; Doulamis, I.P.; Guariento, A.; Ramirez-Barbieri, G.; Moskowitzova, K.; Shin, B.; Blitzer, D.; Rogers, C.; Del Nido, P.J.; et al. Mitochondrial transplantation ameliorates acute limb ischemia. J. Vasc. Surg. 2020, 71, 1014–1026. [Google Scholar] [CrossRef]
- Bertero, E.; O’Rourke, B.; Maack, C. Response by Bertero et al. to Letter Regarding Article, “Mitochondria Do Not Survive Calcium Overload”. Circ. Res. 2020, 126, e58–e59. [Google Scholar] [CrossRef]
- Liu, X.; Sun, X.; Liao, H.; Dong, Z.; Zhao, J.; Zhu, H.; Wang, P.; Shen, L.; Xu, L.; Ma, X.; et al. Mitochondrial Aldehyde Dehydrogenase 2 Regulates Revascularization in Chronic Ischemia. Arter. Thromb. Vasc. Biol. 2015, 35, 2196–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowell, B.B. Mitochondrial Dysfunction and Type 2 Diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hey-Mogensen, M.; Sahlin, K.; Fernström, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Højlund, K. Mitochondrial Respiration Is Decreased in Skeletal Muscle of Patients With Type 2 Diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshanravan, B.; Kestenbaum, B.; Gamboa, J.; Jubrias, S.A.; Ayers, E.; Curtin, L.; Himmelfarb, J.; De Boer, I.H.; Conley, K.E. CKD and Muscle Mitochondrial Energetics. Am. J. Kidney Dis. 2016, 68, 658–659. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Klein, J.D.; Du, J.; Franch, H.A.; Zhang, L.; Hassounah, F.; Hudson, M.B.; Wang, X.H. Chronic kidney disease induces autophagy leading to dysfunction of mitochondria in skeletal muscle. Am. J. Physiol. Physiol. 2017, 312, F1128–F1140. [Google Scholar] [CrossRef]
- Kestenbaum, B.R.; Gamboa, J.; Liu, S.; Ali, A.S.; Shankland, E.; Jue, T.; Giulivi, C.; Smith, L.R.; Himmelfarb, J.; De Boer, I.H.; et al. Impaired skeletal muscle mitochondrial bioenergetics and physical performance in chronic kidney disease. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Örlander, J.; Kiessling, K.; Larsson, L. Skeletal muscle metabolism, morphology and function in sedentary smokers and nonsmokers. Acta Physiol. Scand. 1979, 107, 39–46. [Google Scholar] [CrossRef]
- Degens, H.; Gayan-Ramirez, G.; Van Hees, H.W.H. Smoking-induced Skeletal Muscle Dysfunction. From Evidence to Mechanisms. Am. J. Respir. Crit. Care Med. 2015, 191, 620–625. [Google Scholar] [CrossRef]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.R.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Bonnard, C.; Durand, A.; Peyrol, S.; Chanseaume, E.; Chauvin, M.-A.; Morio, B.; Vidal, H.; Rieusset, J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Investig. 2008, 118, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Wattanakit, K.; Folsom, A.R.; Selvin, E.; Coresh, J.; Hirsch, A.T.; Weatherley, B.D. Kidney Function and Risk of Peripheral Arterial Disease: Results from the Atherosclerosis Risk in Communities (ARIC) Study. J. Am. Soc. Nephrol. 2007, 18, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hare, A.M.; Glidden, D.V.; Fox, C.S.; Hsu, C.-Y. High Prevalence of Peripheral Arterial Disease in Persons With Renal Insufficiency. Circulation 2004, 109, 320–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdi, P.G.; Moradi, H.; Yang, J.-Y.; Wang, P.H.; Vaziri, N. Skeletal muscle mitochondrial depletion and dysfunction in chronic kidney disease. Int. J. Clin. Exp. Med. 2013, 6, 532–539. [Google Scholar] [PubMed]
- Gamboa, J.L.; Billings, F.T.; Bojanowski, M.T.; Gilliam, L.A.; Yu, C.; Roshanravan, B.; Roberts, L.J.; Himmelfarb, J.; Ikizler, T.A.; Brown, N.J. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiol. Rep. 2016, 4, e12780. [Google Scholar] [CrossRef] [PubMed]
- Criqui, M.H.; Ho, L.A.; Denenberg, J.O.; Ridker, P.M.; Wassel, C.L.; McDermott, M.M. Biomarkers in peripheral arterial disease patients and near- and longer-term mortality. J. Vasc. Surg. 2010, 52, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Fiessler, C.; Stavroulakis, K.; Torsello, G.; Bisdas, T.; Lang, W.; Adili, F.; Balzer, K.; Billing, A.; Böckler, D.; et al. Outcomes of dialysis patients with critical limb ischemia after revascularization compared with patients with normal renal function. J. Vasc. Surg. 2018, 68, 822–829.e1. [Google Scholar] [CrossRef]
- Aboyans, V.; Criqui, M.H.; Denenberg, J.O.; Knoke, J.D.; Ridker, P.M.; Fronek, A. Risk Factors for Progression of Peripheral Arterial Disease in Large and Small Vessels. Circulation 2006, 113, 2623–2629. [Google Scholar] [CrossRef] [Green Version]
- O’Hare, A.M.; Bertenthal, D.; Shlipak, M.G.; Sen, S.; Chren, M.-M. Impact of Renal Insufficiency on Mortality in Advanced Lower Extremity Peripheral Arterial Disease. J. Am. Soc. Nephrol. 2004, 16, 514–519. [Google Scholar] [CrossRef]
- Liew, Y.P.; Bartholomew, J.R.; Demirjian, S.; Michaels, J.; Schreiber, M.J. Combined Effect of Chronic Kidney Disease and Peripheral Arterial Disease on All-Cause Mortality in a High-Risk Population. Clin. J. Am. Soc. Nephrol. 2008, 3, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.I.; Mukhopadhyay, S.; Guest, J.M.; Conrad, M.F.; Watkins, M.T.; Kwolek, C.J.; Lamuraglia, G.M.; Cambria, R.P. Impact of severe chronic kidney disease on outcomes of infrainguinal peripheral arterial intervention. J. Vasc. Surg. 2014, 59, 368–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvataneni, K.C.; Piyaskulkaew, C.; Szpunar, S.; Sharma, T.; Patel, V.C.; Patel, S.; Davis, T.; LaLonde, T.; Yamasaki, H.; Rosman, H.S.; et al. Relation of Baseline Renal Dysfunction With Outcomes in Patients Undergoing Popliteal and Infrapopliteal Percutaneous Peripheral Arterial Interventions. Am. J. Cardiol. 2016, 118, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Smith, R.A. Drug delivery to mitochondria: The key to mitochondrial medicine. Adv. Drug Deliv. Rev. 2000, 41, 235–250. [Google Scholar] [CrossRef]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial Energetics and Therapeutics. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 297–348. [Google Scholar] [CrossRef] [Green Version]
- Lang, C.J.G.; Sittl, H.; Erbguth, F. Coenzyme Q10 Treatment in Mitochondrial Encephalomyopathies. Eur. Neurol. 1997, 37, 212–218. [Google Scholar] [CrossRef]
- Rötig, A.; Appelkvist, E.-L.; Geromel, V.; Chretien, D.; Kadhom, N.; Edery, P.; Lebideau, M.; Dallner, G.; Munnich, A.; Ernster, L.; et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000, 356, 391–395. [Google Scholar] [CrossRef]
- Lerman-Sagie, T.; Rustin, P.; Lev, D.; Yanoov, M.; Leshinsky-Silver, E.; Sagie, A.; Ben-Gal, T.; Munnich, A. Dramatic improvement in mitochondrial cardiomyopathy following treatment with idebenone. J. Inherit. Metab. Dis. 2001, 24, 28–34. [Google Scholar] [CrossRef]
- Liet, J.M.; Pelletier, V.; Robinson, B.H.; Laryea, M.D.; Wendel, U.; Morneau, S.; Morin, C.; Mitchell, G.; Lacroix, J. The effect of short-term dimethylglycine treatment on oxygen consumption in cytochrome oxidase deficiency: A double-blind randomized crossover clinical trial. J. Pediatr. 2003, 142, 62–66. [Google Scholar] [CrossRef]
- Park, M.; Nishimura, T.; Baeza-Garza, C.D.; Caldwell, S.T.; Pun, P.B.L.; Prag, H.A.; Young, T.; Sauchanka, O.; Logan, A.; Forkink, M.; et al. Confirmation of the Cardioprotective Effect of MitoGamide in the Diabetic Heart. Cardiovasc. Drugs Ther. 2020, 34, 823–834. [Google Scholar] [CrossRef]
- Lodi, R.; Hart, P.E.; Rajagopalan, B.; Taylor, D.J.; Crilley, J.G.; Bradley, J.L.; Blamire, A.M.; Manners, D.; Styles, P.; Schapira, A.H.; et al. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich’s ataxia. Ann. Neurol. 2001, 49, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Babizhayev, M.A.; Seguin, M.C.; Gueyne, J.; Evstigneeva, R.P.; Ageyeva, E.A.; Zheltukhina, G.A. l-carnosine (β-alanyl-l-histidine) and carcinine (β-alanylhistamine) act as natural antioxidants with hydroxyl-radical-scavenging and lipid-peroxidase activities. Biochem. J. 1994, 304, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.M.; Decker, E.A.; Chow, C.K.; Boissonneault, G.A. Effect of dietary carnosine on plasma and tissue antioxidant concentrations and on lipid oxidation in rat skeletal muscle. Lipids 1994, 29, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Methner, C.; Chouchani, E.T.; Buonincontri, G.; Pell, V.R.; Sawiak, S.J.; Murphy, M.P.; Krieg, T. Mitochondria selective S -nitrosation by mitochondria-targeted S -nitrosothiol protects against post-infarct heart failure in mouse hearts. Eur. J. Hear. Fail. 2014, 16, 712–717. [Google Scholar] [CrossRef] [Green Version]
- Lejay, A.; Charles, A.L.; Georg, I.; Goupilleau, F.; DeLay, C.; Talha, S.; Thaveau, F.; Chakfé, N.; Geny, B. Critical Limb Ischaemia Exacerbates Mitochondrial Dysfunction in ApoE–/– Mice Compared with ApoE+/+ Mice, but N-acetyl Cysteine still Confers Protection. Eur. J. Vasc. Endovasc. Surg. 2019, 58, 576–582. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochromec/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.E.; Schmidt, C.A.; Alleman, R.J.; Tsang, A.M.; Green, T.D.; Neufer, P.D.; Brown, D.A.; McClung, J.M. Mitochondrial therapy improves limb perfusion and myopathy following hindlimb ischemia. J. Mol. Cell. Cardiol. 2016, 97, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.J.; Drake, J.C.; Cui, D.; Lewellen, B.M.; Fisher, C.C.; Zhang, M.; Kashatus, D.F.; Palmer, L.A.; Murphy, M.P.; Yan, Z. Mitochondrial protein S- nitrosation protects against ischemia reperfusion-induced denervation at neuromuscular junction in skeletal muscle. Free Radic. Biol. Med. 2018, 117, 180–190. [Google Scholar] [CrossRef]
- Powers, S.K.; Nelson, W.B.; Hudson, M.B. Exercise-induced oxidative stress in humans: Cause and consequences. Free Radic. Biol. Med. 2011, 51, 942–950. [Google Scholar] [CrossRef]
- Brevetti, G.; Perna, S.; Sabba, C.; Rossini, A.; Di Uccio, V.S.; Berardi, E.; Godi, L. Superiority of L-propionylcarnitine vs L-carnitine in improving walking capacity in patients with peripheral vascular disease: An acute, intravenous, double-blind, cross-over study. Eur. Hear. J. 1992, 13, 251–255. [Google Scholar] [CrossRef]
- Brevetti, G.; di Lisa, F.; Perna, S.; Menabo, R.; Barbato, R.; Martone, V.D.; Siliprandi, N. Carnitine-related alterations in patients with intermittent claudication: Indication for a focused carnitine therapy. Circulation 1996, 93, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Brevetti, G.; Chiariello, M.; Ferulano, G.; Policicchio, A.; Nevola, E.; Rossini, A.; Attisano, T.; Ambrosio, G.; Siliprandi, N.; Angelini, C. Increases in walking distance in patients with peripheral vascular disease treated with L-carnitine: A double-blind, cross-over study. Circulation 1988, 77, 767–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brevetti, G.; Diehm, C.; Lambert, D. European multicenter study on propionyl-L-carnitine in intermittent claudication. J. Am. Coll. Cardiol. 1999, 34, 1618–1624. [Google Scholar] [CrossRef] [Green Version]
- Hiatt, W.R.; Regensteiner, J.G.; Creager, M.A.; Hirsch, A.T.; Cooke, J.P.; Olin, J.W.; Gorbunov, G.N.; Isner, J.; Lukjanov, Y.V.; Tsitsiashvili, M.S.; et al. Propionyl-L-carnitine improves exercise performance and functional status in patients with claudication. Am. J. Med. 2001, 110, 616–622. [Google Scholar] [CrossRef]
- McDermott, M.M.; Criqui, M.H.; Domanchuk, K.; Ferrucci, L.; Guralnik, J.M.; Kibbe, M.R.; Kosmac, K.; Kramer, C.M.; Leeuwenburgh, C.; Li, L.; et al. Cocoa to Improve Walking Performance in Older People With Peripheral Artery Disease. Circ. Res. 2020, 126, 589–599. [Google Scholar] [CrossRef]
- Park, S.Y.; Pekas, E.J.; Headid, R.J.; Son, W.M.; Wooden, T.K.; Song, J.; Layec, G.; Yadav, S.K.; Mishra, P.K.; Pipinos, I.I. Acute mitochondrial antioxidant intake improves endothelial function, antioxidant enzyme activity, and exercise tolerance in patients with peripheral artery disease. Am. J. Physiol. Circ. Physiol. 2020, 319, H456–H467. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preclinical Studies in Rodents | |||
|---|---|---|---|
| Reference | Species and PAD Model | Treatment | Main Results |
| Lejay et al. [128] | Swiss mice—femoral artery ligation 40 days post-surgery | N-acetylcysteine | -decrease tissue damage -improved mitochondrial respiration and calcium retention -decreased ROS levels |
| Lejay et al. [165] | Apolipoprotein E deficient mice—femoral artery ligation 40 days post-surgery | N-acetylcysteine | -improved mitochondrial respiration and calcium retention -decreased ROS production |
| Miura et al. [127] | Mice—femoral artery ligation 21 days post-surgery | MitoTEMPO | -improved limb perfusion recovery -decreased ROS production -decreased mtDNA damage |
| Pottecher et al. [111] | Wistar rats (young)—acute ischemia (3 h) and reperfusion (2 h) | Cyclosporin A | -improved mitochondrial respiration -decreased ROS production |
| Pottecher et al. [108] | Wistar rats (old)—acute ischemia (3 h) and reperfusion (2 h) | Cyclosporin A | -no rescue of mitochondrial respiration -increased ROS production |
| Ryan et al. [102] | C57BL6J mice—femoral artery ligation 7 days post-surgery | Transgenic overexpression of mitochondrial-targeted catalase | -reduced ischemic muscle injury and limb necrosis -improved ischemic muscle contractile function -improved mitochondrial respiration -decreased ROS levels |
| Ryan et al. [167] | BALB/c mice—femoral artery ligation 7 days post-surgery | Elampretide | -decreased limb necrosis -Improved mitochondrial respiration -increase limb perfusion recovery and capillary density |
| Wilson et al. [168] | Mice—acute ischemia (1 h) and 7–14 days post-ischemia | MitoSNO | -increased muscle contractile function -decreased muscle denervation |
| Clinical Studies in Human PAD Patients | |||
| Brevetti et al. [170,171,172] | IC patients (n = 8) IC patients (n = 10) IC patients (n = 30) | l-carnitine | -did not change ABI -improved walking distance |
| Brevetti et al. [173] | IC Patients (n = 485) | Propionyl-l-carnitine | -improved walking distance in severe claudicants but not mild |
| McDermott et al. [175] | IC Patients (n = 44) | Epicatechin | -improved 6-min walk performance-increase cytochrome c oxidase activity -improved muscle perfusion and capillary density |
| Park et al. [176] | IC Patients (n = 22) | mitoQ | -improved endothelial function -improved walking distance and time -delayed onset of claudication |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Anderson, E.M.; Scali, S.T.; Ryan, T.E. Skeletal Muscle Mitochondrial Dysfunction and Oxidative Stress in Peripheral Arterial Disease: A Unifying Mechanism and Therapeutic Target. Antioxidants 2020, 9, 1304. https://doi.org/10.3390/antiox9121304
Kim K, Anderson EM, Scali ST, Ryan TE. Skeletal Muscle Mitochondrial Dysfunction and Oxidative Stress in Peripheral Arterial Disease: A Unifying Mechanism and Therapeutic Target. Antioxidants. 2020; 9(12):1304. https://doi.org/10.3390/antiox9121304
Chicago/Turabian StyleKim, Kyoungrae, Erik M. Anderson, Salvatore T. Scali, and Terence E. Ryan. 2020. "Skeletal Muscle Mitochondrial Dysfunction and Oxidative Stress in Peripheral Arterial Disease: A Unifying Mechanism and Therapeutic Target" Antioxidants 9, no. 12: 1304. https://doi.org/10.3390/antiox9121304
APA StyleKim, K., Anderson, E. M., Scali, S. T., & Ryan, T. E. (2020). Skeletal Muscle Mitochondrial Dysfunction and Oxidative Stress in Peripheral Arterial Disease: A Unifying Mechanism and Therapeutic Target. Antioxidants, 9(12), 1304. https://doi.org/10.3390/antiox9121304