Cannabidiol Protects Dopaminergic Neurons in Mesencephalic Cultures against the Complex I Inhibitor Rotenone Via Modulation of Heme Oxygenase Activity and Bilirubin

 ,
, 

Abstract
1. Introduction
1.1. Phytocannabinoids as Protectants in Neurodegenerative Diseases
1.2. Role of HO and the Biliverdin Reductase (BVR) System in Neurodegeneration and Neuroprotection
1.3. Phytocannabinoids and Interaction With the HO and BVR System
1.4. Experimental Models to Study Neurodegeneration and the Mechanisms Underlying Cytoprotection of CBs
2. Materials and Methods
2.1. Cell Culture
2.1.1. Primary Mesencephalic Cultures
2.1.2. Neuronal N18TG2 Adherent Cell Cultures and J774A.1 Macrophages Grown in Suspension
2.1.3. Determination of Cell Number and Viability
2.2. Determination of HO Activity
2.2.1. Preparation of Samples
2.2.2. HO Enzyme Assay
2.2.3. HO Enzyme Kinetics
2.3. Gene Expression Analysis
2.3.1. Preparation of Samples
2.3.2. RNA Extraction and cDNA Synthesis
2.3.3. Quantification of Target mRNA Expression by qPCR
2.4. Data Processing and Statistics
3. Results
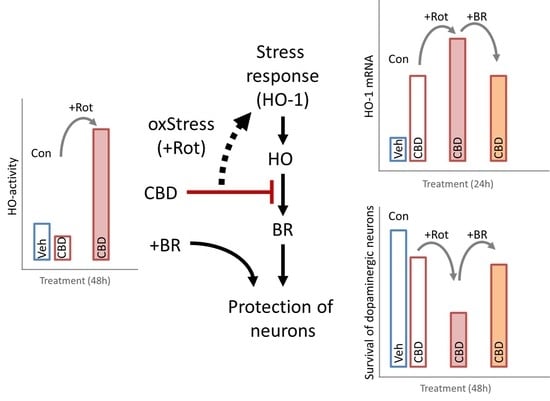
3.1. Modulation of HO Activity by CBs
3.2. Survival of Dopaminergic Neurons
3.3. Changes of Metabolic Activity of Neuroblastoma Cells upon Treatment
3.4. Determination of Treatment-induced Cell Stress Response Using Gene Expression
4. Discussion
4.1. Reduction of In-Vitro Heme Converting Capacity by CBs
4.2. Induction of Cell Stress Response by CBs
4.3. Acceleration of in Vitro Heme Converting Capacity by Treatment with CBs and Rotenone
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hill, A.J.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol. Ther. 2012, 133, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.J.; Martínez-Moreno, M.; Ortega, F.J.; Mahy, N. Targeting microglial K(ATP) channels to treat neurodegenerative diseases: A mitochondrial issue. Oxid. Med. Cell. Longev. 2013, 2013, 194546. [Google Scholar] [CrossRef] [PubMed]
- Mondragón-Rodríguez, S.; Perry, G.; Zhu, X.; Moreira, P.I.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: Implications for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 940603. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Zhang, J.; Butterfield, D.A. Oxidative stress and neurodegeneration. Brain Res. Bull. 2017, 133, 1–3. [Google Scholar] [CrossRef]
- Moldzio, R.; Pacher, T.; Krewenka, C.; Kranner, B.; Novak, J.; Duvigneau, J.C.; Rausch, W.D. Effects of cannabinoids Δ(9)-tetrahydrocannabinol, Δ(9)-tetrahydrocannabinolic acid and cannabidiol in MPP+ affected murine mesencephalic cultures. Phytomedicine 2012, 19, 819–824. [Google Scholar] [CrossRef]
- Hampson, A.J.; Grimaldi, M.; Axelrod, J.; Wink, D. Cannabidiol and (-) Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc. Natl. Acad. Sci. USA 1998, 95, 8268–8273. [Google Scholar] [CrossRef]
- Nguyen, C.H.; Krewenka, C.; Radad, K.; Kranner, B.; Huber, A.; Duvigneau, J.C.; Miller, I.; Moldzio, R. THC (Δ9-Tetrahydrocannabinol) exerts neuroprotective effect in glutamate-affected murine primary mesencephalic cultures through restoring mitochondrial membrane potential and anti-apoptosis involving CB1 receptor-dependent mechanism. Phytother. Res. 2016, 30, 2044–2052. [Google Scholar] [CrossRef]
- Had-Aissouni, L.; Ré, D.B.; Nieoullon, A.; Kerkerian-Le Goff, L. Importance of astrocytic inactivation of synaptically released glutamate for cell survival in the central nervous system--are astrocytes vulnerable to low intracellular glutamate concentrations? J. Physiol. Paris 2002, 96, 317–322. [Google Scholar] [CrossRef]
- Xia, Y.; Xing, J.Z.; Krukoff, T.L. Neuroprotective effects of R, R-tetrahydrochrysene against glutamate-induced cell death through anti-excitotoxic and antioxidant actions involving estrogen receptor-dependent and -independent pathways. Neuroscience 2009, 162, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Cebeira, M.; de Ceballos, M.L.; Zeng, B.Y.; Jenner, P.; Ramos, J.A.; Fernández-Ruiz, J.J. Increased cannabinoid CB1 receptor binding and activation of GTP-binding proteins in the basal ganglia of patients with Parkinson’s syndrome and of MPTP-treated marmosets. Eur. J. Neurosci. 2001, 14, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. The pharmacology of cannabinoid receptors and their ligands: An overview. Int. J. Obes. 2006, 30 (Suppl. 1), S13–S18. [Google Scholar] [CrossRef] [PubMed]
- Zendulka, O.; Dovrtělová, G.; Nosková, K.; Turjap, M.; Šulcová, A.; Hanuš, L.; Juřica, J. Cannabinoids and cytochrome P450 interactions. Curr. Drug Metab. 2016, 17, 206–226. [Google Scholar] [CrossRef]
- Brann, D.W.; Bhat, G.K.; Lamar, C.A.; Mahesh, V.B. Gaseous transmitters and neuroendocrine regulation. Neuroendocrinology 1997, 65, 385–395. [Google Scholar] [CrossRef]
- Telezhkin, V.; Brazier, S.P.; Mears, R.; Müller, C.T.; Riccardi, D.; Kemp, P.J. Cysteine residue 911 in C-terminal tail of human BK(Ca)α channel subunit is crucial for its activation by carbon monoxide. Pflugers Arch. 2011, 461, 665–675. [Google Scholar] [CrossRef]
- Bernabeu, R.; Princ, F.; de Stein, M.L.; Fin, C.; Juknat, A.A.; Batile, A.; Izquierdo, I.; Medina, J.H. Evidence for the involvement of hippocampal CO production in the acquisition and consolidation of inhibitory avoidance learning. Neuroreport 1995, 6, 516–518. [Google Scholar] [CrossRef]
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Cell Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef]
- Chen, J.; Tu, Y.; Connolly, E.C.; Ronnett, G.V. Heme oxygenase-2 protects against glutathione depletion-induced neuronal apoptosis mediated by bilirubin and cyclic GMP. Curr. Neurovasc. Res. 2005, 2, 121–131. [Google Scholar] [CrossRef]
- Zeynalov, E.; Shah, Z.A.; Li, R.C.; Doré, S. Heme oxygenase 1 is associated with ischemic preconditioning-induced protection against brain ischemia. Neurobiol. Dis. 2009, 3, 264–269. [Google Scholar] [CrossRef]
- Ahmad, A.S.; Zhuang, H.; Doré, S. Heme oxygenase-1 protects brain from acute excitotoxicity. Neuroscience 2006, 141, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Doré, S.; Sampei, K.; Goto, S.; Alkayed, N.J.; Guastella, D.; Blackshaw, S.; Gallagher, M.; Traystman, R.J.; Hurn, P.D.; Koehler, R.C.; et al. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol. Med. 1999, 5, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Panahian, N.; Huang, T.; Maines, M.D. Enhanced neuronal expression of the oxidoreductase--biliverdin reductase--after permanent focal cerebral ischemia. Brain Res. 1999, 850, 1–13. [Google Scholar] [CrossRef]
- Huang, E.; Ong, W.Y.; Go, M.L.; Garey, L.J. Heme oxygenase-1 activity after excitotoxic injury: Immunohistochemical localization of bilirubin in neurons and astrocytes and deleterious effects of heme oxygenase inhibition on neuronal survival after kainate treatment. J. Neurosci. Res. 2005, 80, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kitamura, Y.; Kakimura, J.; Shibagaki, K.; Taniguchi, T.; Gebicke-Haerter, P.J.; Smith, M.A.; Perry, G.; Shimohama, S. Possible protective mechanisms of heme oxygenase-1 in the brain. Ann. N. Y. Acad. Sci. 2002, 977, 501–506. [Google Scholar] [CrossRef]
- Yamamoto, N.; Izumi, Y.; Matsuo, T.; Wakita, S.; Kume, T.; Takada-Takatori, Y.; Sawada, H.; Akaike, A. Elevation of heme oxygenase-1 by proteasome inhibition affords dopaminergic neuroprotection. J. Neurosci. Res. 2010, 88, 1934–1942. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Orozco-Ibarra, M.; Chánez-Cárdenas, M.E.; Rangel-López, E.; Santamaría, A.; Pedraza-Chaverri, J.; Barrera-Oviedo, D.; Maldonado, P.D. Heme oxygenase-1 (HO-1) upregulation delays morphological and oxidative damage induced in an excitotoxic/pro-oxidant model in the rat striatum. Neuroscience 2013, 231, 91–101. [Google Scholar] [CrossRef]
- Sharp, F.R.; Zhan, X.; Liu, D.Z. Heat shock proteins in the brain: Role of Hsp70, Hsp 27, and HO-1 (Hsp32) and their therapeutic potential. Transl. Stroke Res. 2013, 4, 685–692. [Google Scholar] [CrossRef]
- Mancuso, C. Heme oxygenase and its products in the nervous system. Antioxid. Redox. Signal. 2004, 6, 878–887. [Google Scholar]
- Mueller, C.; Zhou, W.; Vanmeter, A.; Heiby, M.; Magaki, S.; Ross, M.M.; Espina, V.; Schrag, M.; Dickson, C.; Liotta, L.A.; et al. The heme degradation pathway is a promising serum biomarker source for the early detection of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1081–1091. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Cini, C.; Preziosi, P.; Perluigi, M.; Mancuso, C.; Butterfield, D.A. Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim. Biophys. Acta 2011, 1812, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Barone, E.; Mancuso, C.; Perluigi, M.; Cocciolo, A.; Mecocci, P.; Butterfield, D.A.; Coccia, R. HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: A potential biochemical marker for the prediction of the disease. J. Alzheimers Dis. 2012, 32, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Kimpara, T.; Takeda, A.; Yamaguchi, T.; Arai, H.; Okita, N.; Takase, S.; Sasaki, H.; Itoyama, Y. Increased bilirubins and their derivatives in cerebrospinal fluid in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 551–554. [Google Scholar] [CrossRef]
- Macías-García, D.; Méndez-Del Barrio, C.; Jesús, S.; Labrador, M.A.; Adarmes-Gómez, A.; Vargas-González, L.; Carrillo, F.; Gómez-Garre, P.; Mir, P. Increased bilirubin levels in Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 63, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Nidecker, A.; Kocher, M.; Maeder, M.; Gratzl, O.; Zäch, G.A.; Benz, U.F.; Burckhardt, B. MR-imaging of chronic spinal cord injury. Assoc. Neurol. Funct. Neurosurg. Rev. 1991, 14, 169–179. [Google Scholar]
- Silva, S.L.; Vaz, A.R.; Diógenes, M.J.; van Rooijen, N.; Sebastião, A.M.; Fernandes, A.; Silva, R.F.; Brites, D. Neuritic growth impairment and cell death by unconjugated bilirubin is mediated by NO and glutamate, modulated by microglia, and prevented by glycoursodeoxycholic acid and interleukin-10. Neuropharmacology 2012, 62, 2398–2408. [Google Scholar] [CrossRef]
- Van Bergen, P.; Rauhala, P.; Spooner, C.M.; Chiueh, C.C. Hemoglobin and iron-evoked oxidative stress in the brain: Protection by bile pigments, manganese and S-nitrosoglutathione. Free Radic. Res. 1999, 31, 631–640. [Google Scholar] [CrossRef]
- Falcão, A.S.; Silva, R.F.; Vaz, A.R.; Silva, S.L.; Fernandes, A.; Brites, D. Cross-talk between neurons and astrocytes in response to bilirubin: Early beneficial effects. Neurochem. Res. 2013, 38, 644–659. [Google Scholar] [CrossRef]
- Doré, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef]
- Stocker, R. Antioxidant activities of bile pigments. Antioxid. Redox. Signal. 2004, 6, 841–849. [Google Scholar]
- Jansen, T.; Daiber, A. Direct antioxidant properties of bilirubin and biliverdin. Is there a role for biliverdin reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Maghzal, G.J.; Leck, M.C.; Collinson, E.; Li, C.; Stocker, R. Limited role for the bilirubin-biliverdin redox amplification cycle in the cellular antioxidant protection by biliverdin reductase. J. Biol. Chem. 2009, 284, 29251–29259. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Otterbein, L.; Robson, S. The molecular basis for the immunomodulatory activities of unconjugated bilirubin. Int. J. Biochem. Cell Biol. 2013, 45, 2843–2851. [Google Scholar] [CrossRef]
- Carcolé, M.; Castany, S.; Leánez, S.; Pol, O. Treatment with a heme oxygenase 1 inducer enhances the antinociceptive effects of µ-opioid, δ-opioid, and cannabinoid 2 receptors during inflammatory pain. J. Pharmacol. Exp. Ther. 2014, 351, 224–232. [Google Scholar] [CrossRef]
- Steib, C.J.; Gmelin, L.; Pfeiler, S.; Schewe, J.; Brand, S.; Göke, B.; Gerbes, A.L. Functional relevance of the cannabinoid receptor 2—Heme oxygenase pathway: A novel target for the attenuation of portal hypertension. Life Sci. 2013, 93, 543–551. [Google Scholar] [CrossRef]
- Juknat, A.; Pietr, M.; Kozela, E.; Rimmerman, N.; Levy, R.; Coppola, G.; Geschwind, D.; Vogel, Z. Differential transcriptional profiles mediated by exposure to the cannabinoids cannabidiol and Δ9-tetrahydrocannabinol in BV-2 microglial cells. Br. J. Pharmacol. 2012, 165, 2512–2528. [Google Scholar] [CrossRef]
- Schwartz, M.; Böckmann, S.; Hinz, B. Up-regulation of heme oxygenase-1 expression and inhibition of disease-associated features by cannabidiol in vascular smooth muscle cells. Oncotarget 2018, 9, 34595–34616. [Google Scholar] [CrossRef]
- Fontanellas, A.; Manzanares, J.; García-Bravo, M.; Buzaleh, A.M.; Méndez, M.; Oliva, J.M.; Batlle, A.; Palomo, T.; Enríquez de Salamanca, R. Effects of repeated administration with CP-55,940, a cannabinoid CB1 receptor agonist on the metabolism of the hepatic heme. Int. J. Biochem. Cell Biol. 2005, 37, 1620–1625. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S26–S36. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.F.; Geihs, M.A.; França, T.F.A.; Moreira, D.C.; Hermes-Lima, M. Is preparation for oxidative stress a case of physiological conditioning hormesis? Front. Physiol. 2018, 9, 945. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Molina-Holgado, F.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity In Vivo and In Vitro: Relevance to Parkinson´s disease. Neurobiol. Dis. 2005, 19, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Doré, S.; Goto, S.; Sampei, K.; Blackshaw, S.; Hester, L.D.; Ingi, T.; Sawa, A.; Traystman, R.J.; Koehler, R.C.; Snyder, S.H. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 2000, 99, 587–592. [Google Scholar] [CrossRef]
- Ryan, D.; Drysdale, A.J.; Lafourcade, C.; Pertwee, R.G.; Platt, B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J. Neurosci. 2009, 29, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; De Filippis, D.; Maiuri, M.C.; De Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef]
- Borges, R.S.; Batista, J., Jr.; Viana, R.B.; Baetas, A.C.; Orestes, E.; Andrade, M.A.; Honório, K.M.; da Silva, A.B. Understanding the molecular aspects of tetrahydrocannabinol and cannabidiol as antioxidants. Molecules 2013, 18, 12663–12674. [Google Scholar] [CrossRef]
- Müllebner, A.; Moldzio, R.; Redl, H.; Kozlov, A.V.; Duvigneau, J.C. Heme degradation by heme oxygenase protects mitochondria but induces ER stress via formed bilirubin. Biomolecules 2015, 5, 679–701. [Google Scholar] [CrossRef]
- Zhao, H.; Wong, R.J.; Nguyen, X.; Kalish, F.; Mizobuchi, M.; Vreman, H.J.; Stevenson, D.K.; Contag, C.H. Expression and regulation of heme oxygenase isozymes in the developing mouse cortex. Pediatr. Res. 2006, 60, 518–523. [Google Scholar] [CrossRef]
- Giulietti, A.; Overbergh, L.; Valckx, D.; Decallonne, B.; Bouillon, R.; Mathieu, C. An overview of real-time quantitative PCR: Applications to quantify cytokine gene expression. Methods 2001, 25, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Korematsu, K.; Horie, N.; Suezawa, T.; Okuma, Y.; Nomura, Y.; Ozawa, K. Inhibition of casein kinase 2 modulates XBP1-GRP78 arm of unfolded protein responses in cultured glial cells. PLoS ONE 2012, 7, e40144. [Google Scholar] [CrossRef] [PubMed]
- Wey, S.; Luo, B.; Lee, A.S. Acute inducible ablation of GRP78 reveals its role in hematopoietic stem cell survival, lymphogenesis and regulation of stress signaling. PLoS ONE 2012, 7, e39047. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenbach, K.T.; Pong, M.; Morgan, T.E.; Wang, H.; Ott, K.; Zhou, B.; Longo, V.D.; Lee, A.S. GRP78/BiP is a novel downstream target of IGF-1 receptor mediated signaling. J. Cell Physiol. 2012, 227, 3803–3811. [Google Scholar] [CrossRef]
- Weidinger, A.; Müllebner, A.; Paier-Pourani, J.; Banerjee, A.; Miller, I.; Lauterböck, L.; Duvigneau, J.C.; Skulachev, V.P.; Redl, H.; Kozlov, A.V. Vicious inducible nitric oxide synthase-mitochondrial reactive oxygen species cycle accelerates inflammatory response and causes liver injury in rats. Antioxid. Redox. Signal. 2015, 22, 572–586. [Google Scholar] [CrossRef]
- Amrhein, V.; Greenland, S.; McShane, B. Scientists rise up against statistical significance. Nature 2019, 567, 305–307. [Google Scholar] [CrossRef]
- Jiang, R.; Yamaori, S.; Okamoto, Y.; Yamamoto, I.; Watanabe, K. Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab. Pharmacokinet. 2013, 28, 332–338. [Google Scholar] [CrossRef]
- Qian, Y.; Wang, X.; Markowitz, J.S. In vitro inhibition of carboxylesterase 1 by major cannabinoids and selected metabolites. Drug Metab. Dispos. 2019, 47, 465–472. [Google Scholar] [CrossRef]
- Yamaori, S.; Koeda, K.; Kushihara, M.; Hada, Y.; Yamamoto, I.; Watanabe, K. Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab. Pharmacokinet. 2012, 27, 294–300. [Google Scholar] [CrossRef]
- Duvigneau, J.C.; Kozlov, A.V.; Zifko, C.; Postl, A.; Hartl, R.T.; Miller, I.; Gille, L.; Staniek, K.; Moldzio, R.; Gregor, W.; et al. Reperfusion does not induce oxidative stress but sustained endoplasmic reticulum stress in livers of rats subjected to traumatic-hemorrhagic shock. Shock 2010, 33, 289–298. [Google Scholar] [CrossRef]
- Weidinger, A.; Dungel, P.; Perlinger, M.; Singer, K.; Ghebes, C.; Duvigneau, J.C.; Müllebner, A.; Schäfer, U.; Redl, H.; Kozlov, A.V. Experimental data suggesting that inflammation mediated rat liver mitochondrial dysfunction results from secondary hypoxia rather than from direct effects of inflammatory mediators. Front. Physiol. 2013, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- Burstein, S. Cannabidiol (CBD) and its analogs: A review of their effects on inflammation. Bioorg. Med. Chem. 2015, 23, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.; Horgan, D.J.; Tisdale, H.; Singer, T.P.; Beinert, H. Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. XIV. Location of the sites of inhibition of rotenone, barbiturates, and piericidin by means of electron paramagnetic resonance spectroscopy. J. Biol. Chem. 1968, 243, 844–847. [Google Scholar] [PubMed]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Bartova, A.; Birmingham, M.K. Effect of delta9-tetrahydrocannabinol on mitochondrial NADH-oxidase activity. J. Biol. Chem. 1976, 251, 5002–5006. [Google Scholar]
- Poulos, T.L. Heme enzyme structure and function. Chem Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef]
- Fang, J.; Sawa, T.; Akaike, T.; Akuta, T.; Sahoo, S.K.; Khaled, G.; Hamada, A.; Maeda, H. In vivo antitumor activity of pegylated zinc protoporphyrin: Targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003, 63, 3567–3574. [Google Scholar]
- Fang, J.; Tsukigawa, K.; Liao, L.; Yin, H.; Eguchi, K.; Maeda, H. Styrene-maleic acid-copolymer conjugated zinc protoporphyrin as a candidate drug for tumor-targeted therapy and imaging. J. Drug Target. 2016, 24, 399–407. [Google Scholar] [CrossRef]
- Salerno, L.; Pittala, V.; Romeo, G.; Modica, M.N.; Marrazzo, A.; Siracusa, M.A.; Sorrenti, V.; Di, G.C.; Vanella, L.; Parayath, N.N.; et al. Novel imidazole derivatives as heme oxygenase-1 (HO-1) and heme oxygenase-2 (HO-2) inhibitors and their cytotoxic activity in human-derived cancer cell lines. Eur. J. Med. Chem. 2015, 96, 162–172. [Google Scholar] [CrossRef]
- Greish, K.F.; Salerno, L.; Al, Z.R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel structural insight into inhibitors of heme oxygenase-1 (HO-1) by new imidazole-based compounds: Biochemical and in vitro anticancer activity evaluation. Molecules 2018, 23, 1209. [Google Scholar] [CrossRef] [PubMed]
- Dariš, B.; Verboten, M.T.; Knez, Ž.; Ferk, P. Cannabinoids in cancer treatment: Therapeutic potential and legislation. Bosn. J. Basic Med. Sci. 2019, 19, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Ochs, R.J. Understanding enzyme inhibition. Chem. Educ. 2000, 77, 1453. [Google Scholar] [CrossRef]
- Sardana, M.K.; Kappas, A. Dual control mechanism for heme oxygenase: Tin (IV)-protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc. Natl. Acad. Sci. USA 1987, 84, 2464–2468. [Google Scholar] [CrossRef]
- Chang, E.F.; Wong, R.J.; Vreman, H.J.; Igarashi, T.; Galo, E.; Sharp, F.R.; Stevenson, D.K.; Noble-Haeusslein, L.J. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J. Neurosci. 2003, 23, 3689–3696. [Google Scholar] [CrossRef]
- Piras, S.; Furfaro, A.L.; Brondolo, L.; Passalacqua, M.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. Differentiation impairs Bach1 dependent HO-1 activation and increases sensitivity to oxidative stress in SH-SY5Y neuroblastoma cells. Sci. Rep. 2017, 7, 7568. [Google Scholar] [CrossRef]
- McCarty, M.F. Serum bilirubin may serve as a marker for increased heme oxygenase activity and inducibility in tissues—A rationale for the versatile health protection associated with elevated plasma bilirubin. Med. Hypotheses 2013, 81, 607–610. [Google Scholar] [CrossRef]
- Schiavon, E.; Smalley, J.L.; Newton, S.; Greig, N.H.; Forsythe, I.D. Neuroinflammation and ER-stress are key mechanisms of acute bilirubin toxicity and hearing loss in a mouse model. PLoS ONE 2018, 13, e0201022. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Veh | CBs | CBs + Rot | CBs + Rot + BR | |
|---|---|---|---|---|
| CBD | 100.00 | 103.88 | 61.42 * | 76.07 * # |
| SEM | 0.41 | 3.05 | 2.30 | 1.61 |
| THC | 100.00 | 110.46 | 88.35 * | 90.95 * |
| SEM | 1.16 | 4.22 | 3.77 | 1.59 |
| Variable | Main Effect (p Value) | Main effect on Rotenone (p Value) | Interaction1 of BR on CBD (α Value) | Interaction 1 of BR on THC (α Value) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Rotenone | CBD | THC | BR | CBD | THC | BR | |||
| Survival | ↓<0.001 | 0.374 | 0.051 | 0.196 | ↑<0.001 | 0.392 | ↑0.024 | 0.137 | 0.575 |
| mRNA | |||||||||
| HO-1 | 0.072 | ↑<0.001 | ↑<0.001 | ↓0.006 | ↑<0.001 | ↑<0.001 | ↓0.006 | ↓0.007 | 0.951 |
| HO-2 | 0.060 | ↓0.01 | 0.188 | 0.442 | 0.562 | 0.922 | 0.281 | 0.264 | 0.271 |
| IL6 | ↓<0.001 | ↑<0.001 | ↑<0.001 | ↓0.001 | ↓<0.001 | ↓0.001 | ↓0.01 | ↓0.01 | 0.636 |
| CHOP | ↑<0.001 | ↑<0.001 | ↑0.001 | ↓<0.001 | ↑<0.001 | ↑0.037 | ↓0.003 | ↓0.01 | 0.926 |
| GRP78 | ↑<0.001 | ↑<0.001 | ↑<0.001 | ↓<0.001 | ↑<0.001 | ↑0.005 | ↓0.013 | ↓0.035 | 0.857 |
| XBP1 | 0.156 | ↑<0.001 | ↑0.004 | 0.2 | ↑<0.001 | 0.255 | ↓0.046 | 0.559 | 0.987 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duvigneau, J.C.; Trovato, A.; Müllebner, A.; Miller, I.; Krewenka, C.; Krenn, K.; Zich, W.; Moldzio, R. Cannabidiol Protects Dopaminergic Neurons in Mesencephalic Cultures against the Complex I Inhibitor Rotenone Via Modulation of Heme Oxygenase Activity and Bilirubin. Antioxidants 2020, 9, 135. https://doi.org/10.3390/antiox9020135
Duvigneau JC, Trovato A, Müllebner A, Miller I, Krewenka C, Krenn K, Zich W, Moldzio R. Cannabidiol Protects Dopaminergic Neurons in Mesencephalic Cultures against the Complex I Inhibitor Rotenone Via Modulation of Heme Oxygenase Activity and Bilirubin. Antioxidants. 2020; 9(2):135. https://doi.org/10.3390/antiox9020135
Chicago/Turabian StyleDuvigneau, Johanna Catharina, Alice Trovato, Andrea Müllebner, Ingrid Miller, Christopher Krewenka, Kristina Krenn, Wilhelm Zich, and Rudolf Moldzio. 2020. "Cannabidiol Protects Dopaminergic Neurons in Mesencephalic Cultures against the Complex I Inhibitor Rotenone Via Modulation of Heme Oxygenase Activity and Bilirubin" Antioxidants 9, no. 2: 135. https://doi.org/10.3390/antiox9020135
APA StyleDuvigneau, J. C., Trovato, A., Müllebner, A., Miller, I., Krewenka, C., Krenn, K., Zich, W., & Moldzio, R. (2020). Cannabidiol Protects Dopaminergic Neurons in Mesencephalic Cultures against the Complex I Inhibitor Rotenone Via Modulation of Heme Oxygenase Activity and Bilirubin. Antioxidants, 9(2), 135. https://doi.org/10.3390/antiox9020135