Abstract
Due to a multiplicity of causes provoking traumatic brain injury (TBI), TBI is a highly heterogeneous pathology, characterized by high mortality and disability rates. TBI is an acute neurodegenerative event, potentially and unpredictably evolving into sub-chronic and chronic neurodegenerative events, with transient or permanent neurologic, cognitive, and motor deficits, for which no valid standardized therapies are available. A vast body of literature demonstrates that TBI-induced oxidative/nitrosative stress is involved in the development of both acute and chronic neurodegenerative disorders. Cellular defenses against this phenomenon are largely dependent on low molecular weight antioxidants, most of which are consumed with diet or as nutraceutical supplements. A large number of studies have evaluated the efficacy of antioxidant administration to decrease TBI-associated damage in various animal TBI models and in a limited number of clinical trials. Points of weakness of preclinical studies are represented by the large variability in the TBI model adopted, in the antioxidant tested, in the timing, dosages, and routes of administration used, and in the variety of molecular and/or neurocognitive parameters evaluated. The analysis of the very few clinical studies does not allow strong conclusions to be drawn on the real effectiveness of antioxidant administration to TBI patients. Standardizing TBI models and different experimental conditions, as well as testing the efficacy of administration of a cocktail of antioxidants rather than only one, should be mandatory. According to some promising clinical results, it appears that sports-related concussion is probably the best type of TBI to test the benefits of antioxidant administration.
1. Introduction
Traumatic brain injury (TBI) affects more than 10 million people worldwide each year, representing 30% to 40% of all injury-related mortalities and disabilities among all age groups, with enormous social and economic impacts [1,2]. Epidemiological previsions until 2030 indicate a 2–3-times higher incidence of patients suffering from TBI-related disabilities than those with neurological disabilities from Alzheimer’s disease or cerebrovascular disorders. Despite recent advances in trauma research and the ongoing efforts of collaborative multidisciplinary studies to tackle this problem and improve patients’ outcomes, TBI still represents a major global health burden and public health challenge among all ages in all countries regardless of the patient’s income level.
1.1. Definition/Classification
TBI results from the absorption by the cerebral tissue of part of the energy associated with an external mechanical force, not necessarily acting directly to the head. This amount of energy causes the derangement of a myriad of biochemical, metabolic, and molecular functions, deeply affecting brain cell homeostasis and leading to temporary or permanent impairment of consciousness, neurocognitive deficits, neuromotor disabilities, or psychological disturbances [3,4]. The extent to which these mechanisms are damaged in TBI depends on the severity of the impact. There are various systems and scales to assess the severity of TBI, the most commonly used is the Glasgow Coma Scale (GCS) which classifies TBI into mild (GCS range 13–15), moderate (GCS range 9–12) and severe (GCS range 3–8). The GCS is obtained by scoring specific clinical assessments, including eye opening, motor and verbal responses [5] (Table 1).

Table 1.
Traumatic brain injury (TBI) severity classification, according to the Glasgow Coma Scale (GCS). The score for each patient is calculated by summing the points obtained in each set of neurological examination. Therefore, the minimal value that a TBI patient may score is 3 (corresponding to comatose severe TBI patients) and the maximal is 15 (corresponding to the mildest group of mild TBI patients).
As shown in Figure 1, the majority of TBI patients (80%) sustain mild injuries [6], which have less mortality rates compared to both moderate and severe injuries (30–40%) [7].
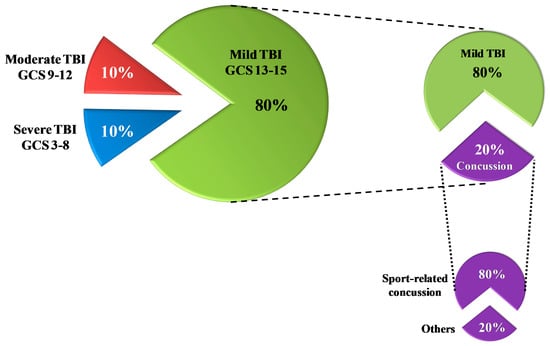
Figure 1.
Rates of traumatic brain injury (TBI) according to the severity classification based on the Glasgow Coma Scale (GCS) score. Of the total TBI, 80% are mild TBI. Of these, 20% are concussions; 80% of all concussions are sports-related concussions.
According to the available epidemiological data [1,6,8], men are more prone to sustain TBI, which is caused, more than 50% of the time, by falls and road traffic accidents (Figure 2).
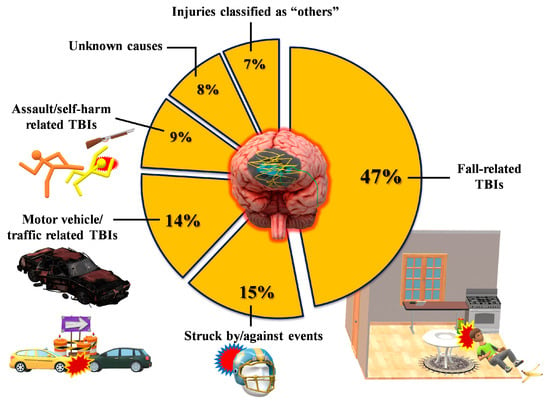
Figure 2.
Classification of TBI patients in terms of rates and types of the most frequent events causing a traumatic head injury. Data refer to USA epidemiological data, which are currently the most accurate worldwide.
Unfortunately, an increasing number of severe TBIs occur during brawls, and this is often due to gunshots, with very devastating consequences.
1.2. Triage
After assessing the injury severity, pre-hospital management at the scene of TBI occurrence is mainly conducted to maintain vital physiological functions of the patient (airway, breathing, circulation, spinal stability) until their transfer to an emergency department [9,10]. In the emergency department, clinicians assess the sustained injuries and continue the supportive treatment, often performing conventional neuroradiological exams to scan the head (and cervical spine on occurrence) by using computed tomography (CT) and/or magnetic resonance imaging (MRI). These initial interventions depend on the severity of TBI and the national/local guidelines. Neurosurgeons assess the surgical need and establish the treatment approach and management plan [9,10].
1.3. Therapy
Due to the heterogeneity of TBI and limited knowledge of the underlying pathophysiological mechanisms, there are no current standardized surgical and pharmacological treatments for TBI patients. Despite their promising preclinical outcomes, many intervention strategies have failed to demonstrate beneficial effects in randomized controlled trials so that TBI patients are still waiting for the discovery of drugs capable of decreasing mortality and disability associated with this pathology [11]. The primary insult resulting from the application of mechanical force in TBI occurs immediately and is inevitable. The amount of permanent damage due to sudden neuronal death and permanent loss of cerebral tissue functions is also inevitable, so that all the efforts at this stage are to prevent the primary injury from happening [12]. The secondary insult, involving biochemical and molecular processes described in the next paragraph, may last for days, weeks, or months, and its damaging activity might actively be decreased by proper therapies [12]. This secondary insult is characterized by the release of excitatory neurotransmitters (glutamate, aspartate), glucose dysmetabolism with mitochondrial dysfunction, and free radical overproduction. Final consequences are the activation of different molecular pathways and inflammatory cascades, leading to cellular apoptosis and damage of the blood–brain barrier (BBB) permeability [12].
As previously stated, treatments deeply vary because of the heterogeneity of TBI and the patient’s response to therapy and it is almost impossible to establish standard approaches to be used in all TBI patients. At present, the drugs most frequently administered are aimed at controlling intra-cranial pressure (ICP) within normal levels (<22 mm Hg) [13], maintaining cerebral blood flow and decreasing secondary injury associated damage [12]. A list of these drugs is summarized in Table 2 [10,11,12,13,14].
Table 2.
Summary of the main drug treatments administered to stabilize clinical conditions of TBI patients in emergency departments.
Acute neurosurgical intervention is often required in moderate to severe TBI patients to evacuate hematoma, causing compression of the brain within the closed skull and leading to the rise of intracranial pressure (ICP). TBI patients needing surgical intervention should be managed in centers with appropriate neurosurgery staff and a neurosurgical intensive care unit (NICU) [10]. A frequent medical intervention is aimed at avoiding the ICP raised by controlling ventilation in order to reduce partial CO2 pressure, a potent vasodilator. In the case of brain edema, the intravenous infusion of hyperosmolar agents is a common clinical practice targeted to improve the blood rheology and cerebral blood flow (CBF) [10,12]. Moderate to severe TBI patients often undergo therapy to induce pharmacological paralysis and sedation aimed at avoiding an increase in ICP and to reduce the metabolic energy requirements of the brain [10,12]. It is highly recommended that mildly injured TBI patients, particularly those who score <15 on the GCS, even in the absence of gross clinical symptoms, are admitted for neurological observation and conventional neuroradiological evaluation (CT scan, MRI), in order to exclude the presence of subdural and/or subarachnoid hematomas [10].
2. TBI and Oxidative/Nitrosative Stress: A Rationale for Antioxidant-Based Therapies
As mentioned above, primary TBI injury is the damage occurring to the tissue (cerebral cells, blood vessels) when part of the energy, associated with the mechanical force causing the injury, is discharged against nerve and blood vessel cells. Secondary TBI injury refers to a cascade of biochemical and molecular mechanisms triggered by the primary insult. These neurochemical changes start immediately after impact, last for hours, days, or weeks depending on the injury severity, and may culminate in cerebral cell death with a loss of neuronal functions. The secondary insult is characterized by the imbalance of ionic homeostasis, release of excitatory neurotransmitters (glutamate, aspartate), glucose dysmetabolism with mitochondrial dysfunction, and free radical overproduction. Final consequences are the activation of different molecular pathways and inflammatory cascades, leading to cellular apoptosis and damage of the BBB permeability [15]. The formation of reactive oxygen species (ROS) and free radicals in brain tissue following TBI is well documented and plays a crucial role in triggering molecular damaging processes (lipid peroxidation, DNA damage, protein oxidation) and in exacerbating glutamate excitotoxicity, mitochondrial dysfunction, ionic dysregulation, and activation of cellular proteases [16,17,18]. A schematic representation of secondary injury is illustrated in Figure 3.
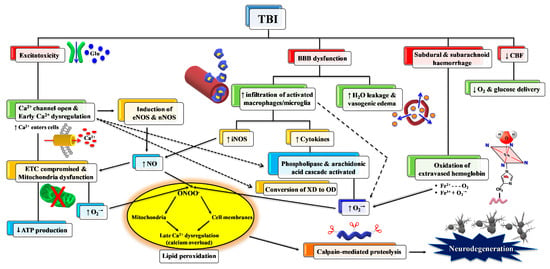
Figure 3.
Schematic representation of some of the main pathological processes characterizing the TBI-associated secondary insult. The force discharged and partly absorbed by the cerebral tissue at the time of impact (primary insult) induces immediate glutamate release by neurons, change in the blood–brain barrier (BBB) permeability, frequent hemorrhage, and decrease in the cerebral blood flow (CBF). Excitotoxic phenomena due to sustained glutamate (Glu) release deeply alter ionic homeostasis, particularly causing an increase in mitochondrial Ca2+. Malfunctioning of the mitochondrial electron transport chain (ETC) and oxidative phosphorylation (OXPHOS) is consequent to increased Ca2+ entry and decreased oxygen and glucose delivery (due to a decrease in CBF), and ultimately generating decreased ATP formation with an energy crisis. Ca2+ also activates endothelial (eNOS) and neuronal (nNOS) isoforms of nitric oxide (NO) synthase, promotes the conversion of xanthine dehydrogenase (XDH) into xanthine oxidase (XO), and triggers the arachidonic acid cascade activating phospholipases. The change in BBB permeability modifies water vascular permeability, causing vasogenic brain edema, and allows infiltration and activation of macrophages/microglia, that are responsible either for NO overproduction by the inducible NO synthase (iNOS) or for the release of pro-inflammatory cytokines. Hematomas generate the release of hemoglobin (Hb) from ruptured erythrocytes and the consequent oxidation of Fe2+ of Hb to Fe3+. This last process, together with XO activity, the arachidonic acid cascade, activated macrophages/microglia, and dysfunctional mitochondria, generates a flow of superoxide anion (O2•−) and gives rise to the reaction with NO and the formation of peroxynitrite (ONOO•−). The damaging action of ROS and RNS on polyunsaturated fatty acids of phospholipids of biological membranes triggers a lipid peroxidation reaction chain, culminating in neuronal cell death.
Glutamate release is one of the main processes activated after TBI [19] that causes an influx of Ca2+ into neuronal cells via activation of NMDA receptors [20] and leads to an “early” calcium dysregulation. This negatively impacts the main mitochondrial function, namely, the electron transport chain (ETC) coupled with oxidative phosphorylation (OXPHOS) for energy production (ATP), thus leading to energy imbalance [21,22] and contributing to increased ROS production [23] with documented post-traumatic membrane lipid peroxidation [24] (Figure 4).
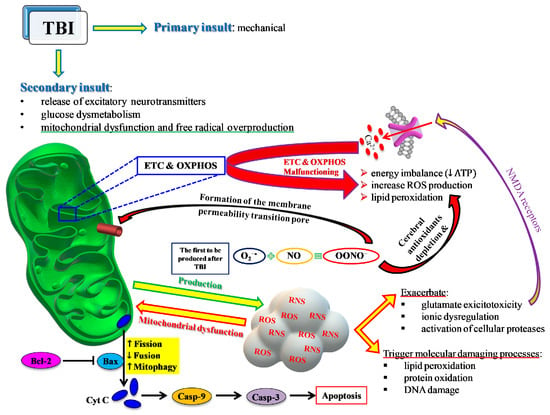
Figure 4.
Schematic representation of the central role played by mitochondria in most of the events characterizing the TBI-mediated secondary insult. ETC and OXPHOS are impaired by the increase in Ca2+ and the change in the mitochondrial quality control (MCQ) network. A decrease in fusion (OPA1, MFN1, MFN2) and an increase in fission (DRP1, FIS1) and mitophagy (PINK1, PARK2) greatly contribute to the reduced mitochondrial phosphorylating capacity, unbalanced ATP production and consumption, leading to an energy crisis. Concomitantly, an insurgence of oxidative/nitrosative stress takes place due to the overproduction of ROS and RNS exceeding the cell antioxidant defenses. The intrinsic pathway of apoptosis via cytochrome c release and caspase activation leads to increasing cerebral cell death.
Dysfunctional mitochondria are also characterized by an imbalance of the mitochondrial quality control network, regulating fusion, fission, and mitophagy. These processes, also known as mitochondrial dynamics, are tightly controlled by a set of proteins selectively promoting fusion (optic dominant atrophy 1, OPA1; mitofusin 1 and 2, MFN1 and MFN2), fission (mitochondrial fission protein 1, FIS1; dynamin-related protein 1, DRP1) or mitophagy (mitochondrial serine/threonine-protein kinase, PINK1; parkin, PARK2). Moderate to severe TBI inhibit fusion and activate fission and mitophagy, consequently decreasing both the total number of mitochondria and the number of those properly functioning. In this light, it has been shown that the peptide SS-31, specifically targeting the mitochondrial phospholipid cardiolipin and providing significant neuroprotection in a variety of neurological diseases, plays a significant role as a potential agent to reduce TBI-mediated mitochondrial dysfunction and oxidative/nitrosative stress [25]. Its pre-impact administration decreased ROS-mediated damage, inhibited apoptosis, and improved mitochondrial biogenesis, thus providing significant neuroprotection in experimental TBI in mice [25].
Among ROS, superoxide anion is the first to be produced after TBI by cerebral cells via different mechanisms, such as the activation of phospholipases and the arachidonic acid cascade (cycloxygenase, COX), the conversion of xanthine dehydrogenase to xanthine oxidase, and, mainly, the malfunctioning of mitochondrial ETC [26]. Activated microglia and infiltrating neutrophils and macrophages also provide additional sources of superoxide radical [27,28] at later time points after TBI, very frequently through the activity of NADPH-oxidase (NOX) [29]. Oxidation of Fe2+ of extravasated hemoglobin (Hb), as a result of the rupture of cerebral blood vessels, may also provide a further source of ROS. Physiologically, superoxide anion is rapidly and efficiently converted into H2O2 + O2 by the enzyme superoxide dismutase (SOD) [29], and H2O2 is then detoxified into O2 + H2O mainly by glutathione peroxidase and, partly, by catalase and peroxiredoxins. Superoxide anion overproduction by the aforementioned mechanisms, coupled with acidosis frequently occurring after TBI, favor H2O2 to react with reduced iron of extravasated Hb, producing hydroxyl radicals through the Fenton reaction. Hydroxyl radicals have no specific defense antioxidants, have a tremendously high oxido-reductive potential, and can easily attack nearly any biological molecule capable of donating one electron and causing its irreversible modification [26]. In addition, oxidized Fe3+ (either from extravasated Hb or released from ferritin) contributes to the hydroxyl radical generation through the reaction of Fe3+ with superoxide anions (the Haber–Weiss reaction) which regenerates Fe2+ for further Fenton reactions. The high oxido-reductive potential allows hydroxyl radicals to tear off one H• from double bonds of polyunsaturated membrane phospholipids, thus initiating the dangerous lipid peroxidation reaction chain disrupting the functions and integrities of biological membranes [30].
TBI also greatly affects the homeostasis of nitric oxide (NO). NO is a fundamental signaling gaseous molecule in the nervous, immune, and cardiovascular systems, transmitting both intracellular and intercellular signals crucial for cell and organ survival. It is produced by the activity of a family of enzymes named nitric oxide synthases (NOS). The 3 NOS isoforms (endothelial, neuronal, and inducible) are not equally upregulated after TBI [31,32,33]. Depending on which isoform is more upregulated, NO may or may not exert beneficial effects. For example, although increased NO synthesis occurs early after TBI, the amount generated by nNOS and eNOS [33,34] has a crucial role as a vasodilator in maintaining cerebral blood flow [35]. In contrast, NO formed intracellularly in cerebral cells by iNOS is deeply involved in the generation of reactive nitrogen species (RNS) and in the so-called nitrosative stress response [36]. The temporal coincidence of excess ROS and RNS production gives rise to the concomitant insurgence of oxidative/nitrosative stress [37]. In particular, the reaction between NO radical and superoxide anion (NO + O2•−) generates peroxynitrite (ONOO•−). Peroxynitrite decays into various unstable RNS, as well as into nitrite and nitrate, which are considered the stable NO end-products. Peroxynitrite and RNS actively take part in depleting cerebral antioxidant defences and in causing lipid peroxidation of mitochondrial membranes [38], further deteriorating Ca2+ homeostasis [39], and in the additional dysfunction of mitochondria through the formation of the membrane permeability transition pore [40], in delaying normalization of ionic homeostasis [41], and in mediating calpain-catalyzed proteolysis and neurodegeneration [42]. It is worth recalling that involvement of oxidative/nitrosative stress has been addressed as a fundamental pathobiological mechanism of cerebral tissue injuries occurring in numerous acute and chronic neurodegenerative disorders, affecting mitochondrial functions, and appearing particularly relevant in the progression of chronic neurodegenerative disorders (Alzheimer’s disease, Parkinson’s disease, multiple sclerosis) [43,44,45].
By definition, an insurgence of oxidative/nitrosative stress takes place anytime cells/tissues/organs undergo an imbalance between ROS and RNS formation and their respective antioxidant defenses. In addition to enzymes capable to scavenging ROS (SOD, catalase, glutathione peroxidase, heme oxygenase, thioredoxin), cells are protected from oxidative/nitrosative stress by low molecular weight antioxidants [46], particularly when considering protection from certain ROS (hydroxyl radicals) and RNS (peroxynitrite). A schematic representation of the main sources of ROS and RNS during oxidative/nitrosative stress occurring after TBI is shown in Figure 5.
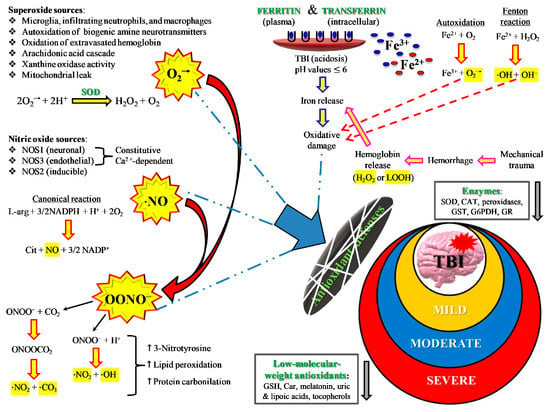
Figure 5.
Schematic representation of the main sources of ROS and RNS during oxidative/nitrosative stress occurring after TBI. The severity of injury is quite well correlated with high or low oxidative/nitrosative stress and with protraction of intra- and extracellular ROS and RNS generation from the various potential sources. For instance, mild TBI rarely causes hematomas with hemoglobin extravasation and mobilization of ferritin iron, thus strongly decreasing the amount of iron used in the Haber–Weiss-sustained Fenton reaction. Conversely, severe TBI induces long-lasting conditions of metabolic derangement due to mitochondrial dysfunction. A vicious cycle is formed between the increased energy requirement, either to satisfy repairing processes or to counteract dangerous phenomena (glutamate excitotoxicity, ionic homeostatic disequilibrium), and the damaging molecules originating from mitochondrial ETC unable to manage the tetravalent reduction of molecular oxygen to water minimizing superoxide formation.
In general, the common characteristic of these defense molecules is their remarkable reducing power that renders them highly “attractive” nearly for any type of oxidant, including ROS and RNS. A gross division into hydrophilic (water-soluble) and hydrophobic (fat-soluble) antioxidants clusters them into water-rich (cytoplasm, mitochondrial matrix) and fat-rich (biological membranes) cell compartments, thereby determining, and/or limiting, their possibility to interact with ROS and RNS (Figure 6).
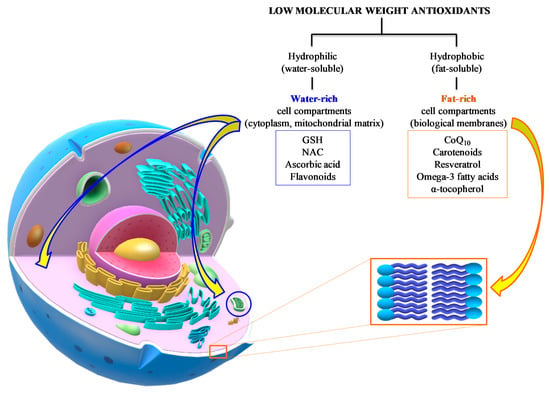
Figure 6.
Summary of the main water- and fat-soluble antioxidants potentially useful as an adjuvant therapy to TBI patients. Their respective chemical properties localize the components of the two categories into hydrophilic (cytoplasm, mitochondrial matrix) or hydrophobic compartments (biological membranes), therefore characterizing the potential antioxidant activities of the different compounds.
With the exclusion of reduced glutathione (GSH), uric acid, and coenzyme Q10 (these last two are secondary antioxidants since this is not their primary biological role), another common characteristic of low molecular weight antioxidants is that mammals are unable to perform their synthesis so that they depend on regular intake with diet to have adequate circulating and tissue concentrations of antioxidants [47]. Therefore, the quality of food consumed and/or supplementation of adjuvants and nutraceuticals is of fundamental importance in order to provide significant protection in the case of increased ROS and RNS formation [48,49]. Although the link between oxidative/nitrosative damage and TBI is evident and the awareness that deficiency in antioxidant-rich foods in the daily diet may further exacerbate TBI symptoms is highly plausible, there are still no clear evidence of successful antioxidant therapies in the clinical setting [50], despite a continuous growth of studies, reporting either preclinical or clinical data, using supplementation with natural or synthetic low molecular weight antioxidants, that have appeared in the literature in the last decades as potentially useful treatments in TBI [18,51]. This tendency is certainly due to different reasons, including the lack of standardized, clearly effective pharmacological approaches of TBI patients, the increased knowledge of the molecular mechanisms underlying cerebral damage after TBI, and the lack of unwanted side effects for the large majority of low molecular weight antioxidants.
In this review, we analyze data from the literature concerning the use of antioxidants in the post-injury period to alleviate the effects of TBI, considering either preclinical or clinical studies, with no limitations in terms of severity of TBI patients, experimental TBI models, animal species, routes of antioxidant administration, doses of antioxidant tested, parameters evaluated, but limiting our search only to those compounds having a well-established and direct antioxidant activity towards ROS and/or RNS.
3. Low Molecular Weight Antioxidants in TBI: Summary of Preclinical and Clinical Studies
3.1. Ascorbic Acid (Vitamin C)
Ascorbic acid (AA) is one of the most abundant water-soluble antioxidants within mammalian tissues, acting as a reducing cofactor in several enzymatic reactions [52]. Being a powerful reducing agent, it quickly reacts, through one or two electron reactions, with a wide range of ROS and RNS, including peroxynitrite and hydroxyl radicals. AA is involved in the recycling of oxidized -tocopherol (Vitamin E) contributing to the maintenance of -tocopherol redox state within biological membranes, and its deficiency is associated with impairments in connective tissue integrity. Since AA is not synthesized by most mammals (including human beings), the body depends on vitamin C-rich food ingestion to satisfy its AA need [53]. Efficient absorption of this molecule mainly occurs through the SVCT1 Na+-ascorbic acid transporter activity [54,55]. Although, circulating levels of AA are relatively low (30 to 70 mol/L serum in healthy humans) [47], abundant AA concentrations are found in peripheral tissues, particularly in the brain, where it is usually present in concentrations up to 3000 mol/L brain water [56,57,58]. The SVCT2 Na+-ascorbic acid transporter, ubiquitously distributed on the cell membrane of the different tissues, provides the electrogenic AA transport exploiting the favorable extracellular/intracellular sodium gradient [59,60]. Although AA is one of the most studied free radical scavengers and is particularly abundant in the brain, only one preclinical and one clinical study have examined the effects of AA in TBI, even though it has been shown that the level of AA in the cerebral tissue decreases rapidly following experimental TBI [56,61]. Its depletion is strictly dependent on the severity of the injury, remaining well below control values even longer after severe TBI and returning to pre-impact concentrations after 72 h in mildly injured animals [21,62]. In a model of closed-head TBI, animals received pre-treatment with different doses of AA (intraperitoneal administration of 45 or 60 mg/kg/die) for two weeks, alone or in combination with equal dosages of -tocopherol, that was administrated for an additional 2 weeks after impact [62,63]. Results of this study indicate that AA, even if administered alone, reduced mortality rate, decreased cerebral tissue and circulating levels of malondialdehyde (MDA), restored brain values of AA, and stimulated tissue superoxide dismutase levels [64].
In a double blind controlled clinical trial, Razmkon et al. [65] divided a cohort of 100 TBI patients into four groups receiving either a low dose of AA (500 mg/die i.v. for 7 days), or a high dose of AA (10 g i.v. on admission and 4 days after, followed by 4 g/die i.v. for the remaining 3 days), or vitamin E (400 IU/die i.m. for 7 days), or placebo. Beneficial effects of high dosage of this antioxidant were observed in AA-treated patients who showed decreased progression of perilesional edema on CT scan. The relatively modest effects of AA recorded in this study might be due to the malfunction of the SVCT2 transporter that normally ensures the electrogenic AA transport into cells. Potentially, the ionic imbalance typically encountered following TBI might perturb the functioning of the AA transporter, thus limiting the effectiveness of AA-based therapy. It might also be possible that decreased expression of SVCT2 occurs following TBI.
3.2. N-Acetyl-Cysteine
GSH is one of the low molecular weight antioxidants that mammals are able to synthesize [66]. Since the -SH group of GSH is supplied by cysteine, high levels of this amino acid are essential to ensure adequate GSH tissue levels. Due to the fundamental role of GSH in detoxifying reactions, both to scavenge ROS and RNS and to eliminate xenobiotics, increased cysteine availability is needed under conditions of oxidative/nitrosative stress. GSH functions as an essential antioxidant, as well as the most important reducing agent for protein –SH groups, and it has recently been demonstrated to be involved in the modulation of inflammatory processes [67]. It is the cofactor of the enzymes GSH peroxidases (GPx) and glutathione-S-transferases. The reduction of oxidized GSH (GSSG) is performed by the NADPH-dependent enzyme GSH reductase (GR), responsible for the maintenance of high values of the GSH/GSSG ratio. The brain is highly rich in GSH (3000 mol/L brain water), where it has been shown that GSH binds to the glutamate NMDA and AMPA receptors, possibly acting as an endogenous neuromodulators [68,69]. Additionally, GSH operates as an activating agent of ionotropic receptors, thus suggesting even a role of GSH in cerebral neurotransmission [70]. It is worth underlining that part of the total GSH cellular content is under the form of the stable S-nitrosoglutahione (GSNO) deriving from the interaction of NO with GSH. GSNO is considered a reservoir of intracellular NO, a transducer of the NO signaling, and an antioxidant due to its ability to break the lipid peroxidation reaction chain. GSNO levels are regulated by the NADH-dependent activity of GSNO-reductase, deeply involved in the regulation of protein nitrosylation and secondary formation of RNS [71]. Hence, maintaining high levels of GSH allows the formation of adequate concentrations of GSNO. Several experimental studies have observed a decrease in brain GSH following TBI. Among these, Ansari et al. showed a significant decrease of GSH/GSSG from 3 to 96 h post-TBI in rat hippocampus, which was mirrored by a time-dependent decline of GPx and GR activities [72]. It was reported that concentrations of rat brain GSH significantly declined depending on the severity of injury in a model of graded diffuse TBI [56]. Whilst in mild TBI (mTBI), a spontaneous recovery late post-injury was observed, severely injured animals had a permanent decrease of brain GSH up to a week following TBI [56]. In addition, a decrease of GSH in CSF of children who suffered a severe TBI has also been reported [73]. Similarly, lower levels of cysteine and glycine, both fundamental for the GSH biosynthesis, were also demonstrated after mild and severe TBI [74]. Interestingly, it has been demonstrated that the i.p. administration of 150 mg/kg γ-glutamylcysteine ethyl ester (GCEE), approximately 10 min postinjury, produced a significant decrease of oxidative/nitrosative damage to proteins following TBI in the rat [75], thus supporting the rationale of applying therapies aimed at increasing cerebral GSH of the post-injured brain.
The aforementioned results motivated the use of N-acetylcysteine (NAC) as a potential new treatment in TBI, either acting as a direct ROS and RNS scavenger, or operating by improving the availability of cysteine and ultimately enhancing GSH synthesis. Since these NAC effects have been well documented in both preclinical and clinical studies, various authors evaluated the benefits of NAC administration in experimental models of TBI, as well as its administration in TBI patients. The potential of GSH precursors as treatment options for TBI has been summarized in a recent review by Koza et al. [76]. In a model of contusion cortical impact, administration of a single dose of NAC (150 mg/kg) 15 min after injury decreased MDA formation, increased SOD and GPx, and protected morphology and the number of neurons of rats sacrificed at 2 and 12 h post-TBI [77]. NAC was also used in combination with minocycline (MINO) and administered 12 h after injury. NAC + MINO prevented the TBI-induced decrease in the expression of oligodendrocyte antigenic markers CC1, 2′,3′-cyclic-nucleotide 3′-phosphodiesterase, and phospholipid protein between 2 and 14 days post-CHI [78]. These results indicated an ample therapeutic window of NAC administration justifying the potential clinical application of NAC in the treatment of TBI patients. In a different study, 150 mg/kg NAC was orally administered (via gastric gavage) at 1, 24, 48, and 72 h after mild closed-head TBI. Results showed that NAC decreased plasma lipid peroxidation and IL-1b levels, increased brain cortex IL-4, GSH, TAS, vitamin A, AA and -tocopherol values, and increased erythrocyte GSHPx levels [79].
3.3. Flavonoids
Flavonoids are a class of more than 5000 naturally occurring compounds, largely present in plants and fungi [80]. Their common structure is formed by one phenyl ring fused with an oxygen-containing heterocyclic ring to which is linked a second phenyl ring. Since, in the different flavonoids, the number of –OH groups is higher than one, flavonoids are more generally termed as polyhydroxy polyphenols, or simply polyphenols. Chemically, they are divided into three groups: flavonoids or bioflavonoids, isoflavonoids, and neoflavonoids. Flavonoids are divided into five sub-groups (anthocyanidins, anthoxanthins, flavanones, flavanonols, and flavans), isoflavonoids into five sub-groups (isoflavones, isoflavonones, isoflavans, pterocarpans, rotenoid), and neoflavonoids into two sub-groups (neoflavones and neoflavenes). The large majority of all the aforementioned compounds are water-soluble, and, it is assumed, they are consumed in variable proportions, with foods of vegetal origin, according to the different dietary habits. The common characteristic of flavonoids is that they possess a very negative oxido-reductive potential, therefore acting as powerful antioxidants towards any type of ROS and RNS [81]. A growing number of preclinical studies have investigated the effects of flavonoid administration in reducing TBI-associated damage, the results of which have been condensed into Table A1 of Appendix A.
Given the wide interest in the assessment of natural compounds (including flavonoids) obtained from a variety of vegetable sources and the large number of flavonoids, it is difficult to find two studies examining the same flavonoid, and it is therefore difficult to directly compare studies investigating the effectiveness of flavonoid-based therapies.
In a small pilot clinical trial, 60 patients who experienced mild TBI and showing symptoms from 3 to 12 months after injury, received oral enzogenol (1 g/die) for 6 weeks. Cognitive functioning before and after treatment was investigated. Compliance, side-effects, cognitive failures, working and episodic memory, post-concussive symptoms, and mood were assessed at baseline, 6, 12, and 16 weeks. Patients receiving enzogenol were found to have significantly fewer self-reported cognitive failures than those in the placebo group [82].
3.4. Resveratrol
Resveratrol is an antioxidant with modest, but significant, solubility in water and good solubility in organic solvents, thus indicating its prevalent hydrophobic nature. It is formed by two 6-carbon aromatic rings (one with two and the other with one -OH groups) linked by a C=C bridge and allowing the existence of cis- and trans-resveratrol. This form of polyphenol is abundantly present in the skin of red grapes. However, red wines are the food source with the highest resveratrol levels. High resveratrol intake, due to high red wine ingestion, was originally suggested as one of the possible explanation of the so-called “French paradox” concerning the apparently low occurrence of cardiovascular diseases in the French population, in spite of a high fat (mostly saturated) diet consumption [83]. Apart from its intrinsic antioxidant activity, resveratrol seems to modulate several cell functions, including defense mechanisms, mitochondrial functions, and inflammatory processes [84].
A preclinical study in rats evaluated the effects of a single dose of resveratrol (100 mg/kg) administered intraperitoneally immediately after TBI induced by the weight-drop model. Brain water content and levels of MDA, GSH, NO, and xanthine oxidase (XO) were evaluated at 24 h after impact. In the resveratrol-treated group, significantly lower values of MDA, XO and NO, and increased level of GSH were observed. Additionally, resveratrol treatment also attenuated tissue lesion area [85]. Another study analyzed the protective effect of resveratrol in TBI in vivo and in vitro. Results showed that cell death after TBI is induced through the ROS/GSK-3β/mitochondria signaling pathway and that administration of resveratrol (100 mg/kg) immediately after controlled cortical impact (CCI)–TBI can increase cell survival by suppressing GSK-3β-mediated autophagy and apoptosis [86]. In one additional study, mice were treated with either placebo or resveratrol (100 mg/kg) at 5 min and 12 h after induction of mild TBI using CCI. In comparison to placebo-receiving animals, results demonstrated a reduction of microglial activation in the brain regions of the cortex, corpus callosum, and dentate gyrus induced by mild TBI in resveratrol treated rats. In addition, resveratrol decreased IL-6 and IL-12 expressions suggesting a general decrease of neuroinflammation caused by TBI [87].
3.5. α-Tocopherol (Vitamin E)
Tocopherols are a family of fat-soluble compounds, found in high concentrations in various vegetal oils, having a remarkable antioxidant capacity. Tocopherol congeners having vitamin E activity are four tocopherols and four tocotrienols, the former having a saturated hydrocarbon harbored side chain, the latter having an unsaturated hydrocarbon harbored side chain composed by three isoprene units [88,89]. The chromane ring, to which the hydrocarbon chain is bound, is the oxido-reductive center common to all the eight compounds. Its single OH-group can easily lose one electron, in the form of a hydrogen radical, under the action of highly oxidizing ROS and RNS and forming the corresponding, relatively stable, tocopheryl- or tocotrienyl-radical. The hydrophobic side chain of tocopherols allows these compounds to localize within the phospholipid bilayer of biological membranes, where they act as interrupters of lipid peroxidation chain reactions. When oxidized by ROS and RNS, tocopheryl-radicals strictly depend on AA availability to get recycled into their respective fully reduced forms. Among the eight congeners, -tocopherol is the most abundant in the European diet and -tocopherol in the American diet. Albeit the brain is rich in easily oxidizable fat-soluble compounds, mainly found in the biological membrane structures of the nerve cells, it is one of the mammalian tissues with the lowest content of tocopherols. This renders the brain potentially more exposed to lipid peroxidation phenomena mediated by oxidative/nitrosative stress.
As in the case of AA, several animal studies using different TBI models at different severities have been conducted, but only one clinical study evaluated the effects of -tocopherol administration to TBI patients. In guinea pigs experiencing mild or severe TBI, Inci et al. found that -tocopherol (100 mg/kg), intraperitoneally administered before graded TBI, decreased brain levels of MDA derived from oxidative/nitrosative stress-mediated lipid peroxidation. The authors concluded that -tocopherol administered early post-injury is beneficial to decrease tissue damage associated with TBI [90]. In a model of focal moderate TBI, Sprague–Dawley rats received a daily intraperitoneal injection of -tocopherol (600 mg/kg), subsequently to TBI induction. At different times post-impact, neurocognitive tasks, histochemical evaluation of tissue damage, and expressions of Nogo-A and NgR, two protein factors inhibiting nervous cell regeneration, were evaluated in all animals. Results showed that rats treated with -tocopherol performed better on neurocognitive tests, had less evident histological signs of edema, inflammation, and necrosis, as well as decreased expression of Nogo-A and NgR. The authors proposed that -tocopherol may reduce ROS-mediated tissue damage and promote cerebral tissue regeneration following TBI [91]. In a model of focal moderate TBI, the effects of administration of tocopheryl-succinate (100 mg/kg) + polyethylene glycol (PEG) (2 mL/kg), injected 30 min before or 5 min after injury, were evaluated in rats subjected to fluid percussion. Although PEG alone improved the parameters under evaluation, the combination tocopheryl-succinate (100 mg/kg) + PEG (2mL/kg) showed the best increase in the survival rates and the best improvement of neurocognitive tasks and motor function, suggesting that this type of infusion might be effective in the clinical setting to decrease TBI-associated damage [92]. Aiguo et al. fed rats using a regular diet with or without 500 IU/kg of -tocopherol for 4 weeks, at the end of which animals received focal mild TBI, according to the fluid percussion injury model [93]. It was shown that the -tocopherol-rich diet prevented TBI-increased oxidative damage to proteins, the decrease in SOD, Sir2, and BDNF caused by TBI, and improved TBI-associated motor function impairments. The authors’ conclusions were that dietary -tocopherol supplementation could decrease the damaging effects of mild TBI on synaptic plasticity and cognitive functions.
Despite the beneficial effects of -tocopherol in preclinical studies, translation to the clinical setting was only performed in the previously cited Razmkon study [65]. Among the four groups of TBI patients, those who were administered -tocopherol (400 IU/die intramuscularly for 7 days) showed a significant reduction of mortality rates and improvement of long-term functional outcomes.
3.6. Coenzyme Q10
Coenzyme Q (ubiquinone) is a fat-soluble compound ubiquitously found as a component of the mitochondrial electron transport chain in oxygen-dependent living organisms. It is characterized by a quinone ring which, through oxido-reductive reactions exchanging one or two electrons, allows coenzyme Q to exist in a fully oxidized, fully reduced, or semi-reduced (semi-quinone radical) form. Depending on the organism considered, the quinone ring is bound to a hydrophobic chain, made up of isoprene units, repeated from a minimum of 3 to a maximum of 10 units (as it occurs in humans). This last form of coenzyme Q, named coenzyme Q10 (CoQ10), is found in the mitochondrial inner membrane actively participating in the transfer of electrons from Complex I and Complex II to Complex III, where the so-called CoQ10 cycle takes place [94].
Several studies were performed, testing the effects of exogenously administered CoQ10 under various pathological conditions to counteract oxidative/nitrosative stress, ameliorate mitochondrial dysfunction, and decrease inflammatory processes [95]. The rationale was attributed to the CoQ10 oxido-reductive potential, by its ability to affect one-electron transfer reactions by forming the semi-quinone radical, by the relatively long-living, low reactivity of its semi-quinone radical and by its high hydrophobicity clustering CoQ10 within biological membranes. It should, however, be recalled that the main CoQ10 biological role is that of ensuring the electron transfer through ETC and not that of acting as an antioxidant [96]. However, very recently, it has been shown that CoQ10 interacts with FSP1 acting as a novel plasma membrane oxidoreductase and protecting cells from glutathione-independent ferroptosis [97].
At present, few animal studies have evaluated the role of CoQ10 administration in the treatment of head trauma, with no clinical studies conducted to date. In the Marmarou model of moderate diffused closed-head TBI, CoQ10 was administered at a dose of 10 mg/kg immediately after trauma and after 24 h by gavage. Compared to trauma only, this treatment decreased MDA levels, vascular congestion, neuronal loss, nuclear pyknosis, nuclear hyperchromasia, cytoplasmic eosinophilia and axonal edema, and increased cerebral SOD [98]. In rats receiving TBI according to CCI, administration of CoQ10 immediately after impact was found to significantly affect the cerebral expression of genes involved in mechanisms of bioenergetics and oxidative/nitrosative stress [99]. The same research group tested the effects of intra-arterial CoQ10 infusion before or 30 min after TBI induced by CCI [100]. Results indicated a decrease in brain mitochondrial damage and apoptosis, as well as lower circulating values of two markers of TBI severity, namely, serum glial fibrillary acidic protein (GFAP) and ubiquitin C-terminal hydrolase-L1 (UCH-L1). However, in this study, in vivo MRS (magnetic resonance spectroscopy) failed to show any significant amelioration of neurometabolism produced by CoQ10 treatment [100].
3.7. Carotenoids
Carotenoids are a class of pigments synthesized by plants, algae, and photosynthetic bacteria [101], composed of 8 isoprene units with a total of 40 carbon atoms. There are over 1100 known carotenoids, which are categorized into a class of carotenoids containing oxygen (xanthophylls) and a class of oxygen-free, purely hydrocarbon carotenoids (carotenes). In the human brain, various carotenoids, including -carotene, -carotene, -carotene, -cryptoxanthin, lutein, and zeaxanthin, can generally be measured [102]. Some of them (-carotene, -carotene, -carotene, -cryptoxanthin) can be converted into vitamin A and are therefore directly involved in the mechanism of vision, ensuring adequate retinol formation. Since the different carotenoids are distributed into a variety of foods mainly of vegetal origin (fishes and crustaceans, such as salmon and red shrimps, derive their high carotenoid level on the high intake of carotenoid-containing algae), their daily ingestion, and therefore their relative amount within tissues, is strictly dependent on the individual dietary regimen, as well as on the supplementation of carotenoid-containing adjuvants or nutraceuticals. In general, carotenoids exhibit antioxidants and anti-inflammatory properties, as well as possess modulatory activities of autophagy. The use of various carotenoids as potential treatments for several pathologies, including neurodegenerative diseases, have been extensively studied [102].
In a stretch injury model of TBI using astrocyte cultures, astaxanthin attenuated apoptosis by inhibiting NKCC1 expression, and reduced the expression of NF-κB-mediated pro-inflammatory factors [103]. In mice, intraperitoneal injection of astaxanthin (10, 25, 50, or 100 mg/kg body weight) administrated 30 min after impact, decreased TBI-related brain tissue injury (induced by CCI) by dose-dependently ameliorating AQP4/NKCC1-mediated cerebral edema. In addition, the highest dose examined was shown to improve neurologic deficits and the BBB permeability [104]. In a different study, astaxanthin (25 or 75 mg/kg) was administered to mice via oral gavage beginning 30 min post-trauma and followed by six additional daily oral gavages. Animals received a left frontal TBI using a weight-drop device. Results showed that astaxanthin administration improved sensorimotor performance and enhanced cognitive recovery. In addition, reduction of lesion size in the cortex and expressions comparable to controls of brain-derived neurotrophic factor (BDNF), growth-associated protein-43 (GAP-43), synapsin, and synaptophysin (SYP), indicating the induction of neuronal survival and plasticity, were recorded [105].
Many other carotenoids have been investigated in TBI animal models, and molecular mechanisms have also been described. Among these, fucoxanthin showed neuroprotective effects via regulation of the Nrf2-ARE and Nrf2-autophagy pathways [106]. Bexarotene inhibited apoptosis and inflammation by upregulating the lncRNA Neat1 [107], and improved the neurological functions of mice after TBI, partially through apolipoprotein E [108]. Administration of crocin has shown neuroprotective effects partially dependent on the activation of Notch signalling [109]. Lastly, lutein, another strong antioxidant with anti-inflammatory and anti-apoptotic effects, and capable of crossing the BBB, showed protective effects by suppressing interleukin IL 1β, IL 6, and monocyte chemoattractant protein 1 expressions [110].
3.8. Omega-3 Fatty Acids
Omega-3 fatty acids are a group of polyunsaturated fatty acids (PUFA) that have three (linolenic acid) to six (docosohexaenoic acid) double bonds in their carbon chain of 18 (linolenic acid) to 22 (docosohexaenoic acid) carbon atoms in length. Considering their last carbon atom as the -atom, this group of PUFA has the common characteristic of one double bond positioned on the third carbon atom far from the -atom. Omega-3 fatty acids are either essential (linolenic acid) or semi-essential (eicosapentaenoic acid, docosahexaenoic acid) for humans. Our body is unable to synthesize linolenic acid (the precursor of the other omega-3 fatty acids) that should be ingested in adequate amounts with diet. However, humans are capable of providing little but significant amounts of docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) from the elongation and desaturation of linolenic acid, through a specific metabolic pathway. Omega-3 fatty acids are not only fundamental components of membrane phospholipids but also precursors of other important biologically active molecules (prostaglandins). Particularly DHA and EPA are abundantly found in the brain phospholipids, with the former representing about 50% of the weight of the neural tissue cell membranes [111]. Although real benefits in patients with neurodegenerative disorders are still controversial, the supplementation of omega-3 fatty acids has been tested in various pathologies characterized by increased ROS and RNS production. The rationale is either in the pure antioxidant activity of omega-3 fatty acids or in the possibility that an aliquot of those exogenously administered might help in restoring biological membranes damaged by oxidative/nitrosative stress-mediated lipid peroxidation.
In Table A2 of Appendix A, we summarize key preclinical studies evaluating the role of omega-3 fatty acids and DHA in TBI and examined more detailed clinical studies carried out in TBI patients. It is worth underlining that the three clinical studies available in the literature do not support the results reported in experimental TBI.
Matsouoka et al. [112] evaluated the effects of the combined daily administration for 12 weeks of 1470 mg DHA and 147 mg EPA to a group of 53 traumatized patients, 52% of which suffered from mild TBI. A control group of 20/57 traumatized subjects sustained mild TBI and was administered with placebos. Outcome measures were posttraumatic stress disorder (PTSD) symptoms, assessed by the Clinician-Administered PTSD Scale, and depressive symptoms, assessed by the Montgomery–Åsberg Depression Rating Scale, both evaluated at baseline and after 12-weeks, together with the serum levels of mature BDNF and precursor pro-BDNF. Results showed no effect of DHA + EPA on PTSD, depressive symptoms, or serum BDNF levels, thus confuting the efficacy of these omega-3 in ameliorating mental status of patients sustaining mild TBI [109]. Same authors reported no effects of the combined daily administration for 12 weeks of 1470 mg DHA and 147 mg EPA, even if enriched with 0.3% a-tocopherol, when the same patients were fully evaluated for the post-injury quality of life. Also in this case, the authors concluded that the administration of omega-3, specifically DHA and EPA, is not justified by their clinical results to treat TBI patients [113]. The last study showed a clinical case of an elderly patient taking warfarin and fish oil supplementation (rich in omega-3 fatty acids) after suffering blunt head trauma and who manifested an irreversible fatal warfarin-induced coagulopathy. This study, although reporting only one case, reveals the risks of omega-3 fatty acid supplementation in patients taking anticoagulant medications, because of the synergistic effect of the two treatments (warfarin and omega-3 fatty acids) in inhibiting platelet aggregation promoting untreatable bleeding [114].
4. Discussion
Concussion is a traumatically induced transient perturbation of brain functions occurring at any time impacts of different intensities, not necessarily hitting the head, cause an acceleration–deceleration phenomena of the skull [115,116]. The most dangerous impacts are those inducing rotational and translational movements of the head. Very commonly, and erroneously, the term concussion is interchangeably used with mild TBI. Therefore, it is certainly true that all concussions are mTBIs, and it is equally true that not all mTBIs are concussions [117]. Concussions are characterized by a rapid onset of spontaneously resolving clinical symptoms, typically represented by neurological impairment and cognitive deficits. Since during a concussive event part of the energy associated to the impact is also discharged towards the cerebral tissue, concussion is known to trigger a plethora of biochemical, metabolic, and molecular changes transiently, but deeply, altering cerebral cell functions and homeostasis. This complex pathobiological process is also known as the neurometabolic cascade of concussion [117,118,119]. The concussed brain is characterized by transient alteration of ionic homeostasis, imbalanced cellular calcium exchange and storage, which leads to mitochondrial dysfunction with an energy crisis. Glucose dysmetabolism [120], alteration of the mitochondrial quality control [121,122], impairment of cell energy state [121,122,123], and increase of ROS and RNS production [124] characterize a temporal window following concussion during which the brain is “metabolically vulnerable” It has been clearly demonstrated that alterations of brain metabolism last much longer than symptom disappearance and return of neurocognitive functions to pre-impact levels. A second concussive event of even lesser magnitude than the first, falling within this period of vulnerability, may have catastrophic consequences causing a clinical condition termed as second impact syndrome (SIS). When occurring, SIS is fatally characterized by untreatable malignant cerebral edema associated with high disability and mortality rates. Repeat concussions, taking place before complete restoration of neurometabolism, may be the basis of the degeneration into chronic traumatic encephalopathy (CTE). CTE has been described in retired athletes who sustained a high number of concussions (mainly unreported) in their career [125], and it is a progressive neurodegenerative disorder causing a dramatic loss of neurocognitive functions and evolving into dementia [125,126].
Concussions represent about 20% of all mTBI. Of all concussions, 80% are sports-related concussions. Athletes, particularly those practicing sports characterized by frequent physical contact (such as boxing, martial arts, American football, ice hockey), are at much higher risks of concussions than non-athletes. Amateur and non-athletes suffer the influence of post-concussive symptoms in their everyday life as the main consequences connected to concussions. In contrast, the main problems for semi-professional and professional athletes refer to the time they are forced to stay out of the competitions, before their return to play following a concussion. According to what is stated above, sports-related concussions are a type of TBI in which prevention might effectively be applied either by modifying rules of those sports disciplines at higher risk of concussion, or in preventively treating athletes with drugs capable of inhibiting specific molecular pathways activated by concussions. It should also be taken into account that drug treatments might be helpful in allowing safer return of athletes to play. In this light, few studies have been carried out to evaluate the effects of the administration of antioxidants prior to concussion in reducing molecular changes and symptoms associated with concussion [127].
Antioxidant Therapies in Sports-Related Concussion
In a clinical study evaluating the effect of enzogenol administration, 42 athletes with a history of sports-related concussion in the chronic phase of injury were enrolled. Athletes sustained concussions from 0.5 to 3 years prior to the start of the study and 28/42 of them experienced repeat concussions. They were blindly and randomly divided into two groups, one receiving placebos and the other 1 g/die of enzogenol. Both treatments were administrated for six weeks. Athletes were assessed on recruitment and at the end of drug administration, using virtual reality (VR), electroencephalography (EEG), and neuropsychological (NP) tasks. The enzogenol-treated group showed enhanced frontal-midline theta and decreased parietal theta power, indicating reduced mental fatigue compared to the placebo group. Subjects enrolled in the enzogenol group also self-reported reduced mental fatigue and sleep problems. The authors concluded that enzogenol has the potential to improve brain functioning in the chronic phase of concussion [128].
In a study evaluating the prophylactic effects of DHA administration, 81 American football athletes were recruited, and blindly and randomly administered with 2, 4, and 6 g/die of DHA or placebo. During the 189 days of the study duration, blood of athletes was analyzed at various times (coincident with changes in intensity, hours of contact, and likely changes in head impacts) neurofilament light (NFL) serum levels, as an indicator of axonal injury. The main finding was that DHA likely attenuates the increase in serum NFL, suggesting a neuroprotective effect of DHA towards a central pathogenic mechanism of concussion (axonal injury). Interestingly, the lowest dose (2 g/die) appeared to produce the most marked decrease in serum NFL levels [127].
Although it did not refer to sports-related concussions, one clinical trial (NCT00822263), carried out to test the efficacy of NAC on symptoms associated with blast exposure mild TBI (causing the same symptoms as concussion) in a Southwest Asia combat setting, is certainly worth being mentioned. In this prospective study, 81 symptomatic U.S. service members who were exposed to a blast wave or who were in a vehicle that was damaged by a blast wave, and who met the criteria for concussive mild TBI (balance dysfunction, confusion, headache, sensorineural hearing loss, impaired memory, and sleep disturbances), accepted to participate in the study. Randomly assigned patients received either placebo or 4-g loading dose of NAC followed by 4 g daily (in two divided doses of 2 gm morning and night) for 4 days and then by 3 gm daily (in two divided doses of 1.5 gm morning and night). Treatments started in a variable time ranging 24–72 h from blast exposure. When compared to placebo, results showed beneficial effects of oral NAC administration on neuropsychological test results, number of TBI symptoms, and full symptom resolution on day seven, following the beginning of treatment. Early initiation of NAC administration was also found to have additional benefit on the neurological but not on neuropsychological outcome measures [126].
5. Conclusions
The analysis of the literature concerning the beneficial effects of antioxidant-based therapies to reduce TBI-associated damage leads to the following indications: (i) there are no sufficient clinical data to determine whether adding the administration of antioxidants to that of drugs commonly used in TBI patients improves outcome in terms of decreased mortality and disability; (ii) for the majority of both water- and fat-soluble antioxidants, clinical studies are completely lacking; (iii) data from experimental TBI in laboratory animals are supportive of beneficial effects of administration of antioxidants in TBI of any severity; (iv) clear effectiveness of antioxidants, dosages, timing and route of administration, biochemical and molecular parameters, clinical and neurological parameters have not yet been established, rendering less strong the indications reported in these preclinical studies; (v) there are no data in the literature, either clinical or preclinical, for natural antioxidants (lycopene, anthocyanins) commonly found in a balanced diet, such as the Mediterranean diet.
It is our opinion that it will be very difficult to obtain conclusive evidence showing that antioxidant administration to TBI patients is truly beneficial. In the past, preclinical studies demonstrated that ROS generation [129] and ROS-mediated damage [130,131] take place in the very early phase following TBI. Furthermore, a clinical study, in which TBI patients were analyzed for CSF content of antioxidants and ROS-mediated lipid peroxidation biomarkers within 3 h from injury, clearly showed that all TBI patients reached the NICUs, having dramatic ascorbate decrease and evident MDA increase in their CSF samples [132], i.e., most of ROS- and RNS-mediated damage was already completed at the time of potential antioxidant administration. Altogether, the aforementioned preclinical and clinical findings indicate a very narrow therapeutic window to allow antioxidant therapies to have a reasonable probability of having positive effects. It is also our opinion that it should be much more effective to test, either in animals or in human beings, the administration of a cocktail of antioxidants, distributing in different cell compartments, having differential ROS and RNS scavenging activity and performing secondary activations of different pathways beneficial for the post-injured brain, rather than expecting resounding ameliorations using the administration of one antioxidant only.
One further crucial observation is that, in the case of TBI, it is not feasible to attempt preventive medicine based on the administration of antioxidants, since there is no chance to know whether one will sustain TBI during his life span. Differently, preventive administration of antioxidants might be performed in sports-related concussion. By initially selecting those athletes practicing sports at high risk of concussion (any contact and some non-contact sports), it might certainly be easy to demonstrate whether athletes treated with antioxidants and who experienced a concussion have less concussion-associated symptoms, cognitive deficits, and neurometabolic alterations, as well as having shorter recovery periods than control, placebo-treated, concussed athletes. Therefore, it appears quite evident that sports-related concussion and athletes have the best chance of representing the preferential TBI type and patient population to evaluate future antioxidant therapies in humans.
Author Contributions
Conceptualization, V.D.P. and K.M.Y.; reviewing of the papers, G.C., G.L. (Giacomo Lazzarino)., and S.S.; writing the first draft of the manuscript V.D.P., B.T., G.L. (Giuseppe Lazzarino), and A.M.A.; editing the final text, A.K.B. and A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AA | Ascorbic Acid |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AQP4 | Aquaporin-4 |
| BBB | Blood–Brain Barrier |
| BDNF | Brain-Derived Neurotrophic Factor |
| CBF | Cerebral Blood Flow |
| CBV | Cerebral Blood Volume |
| CCI | Controlled Cortical Impact |
| COX | Cyclooxygenase |
| CSF | Cerebro-Spinal Fluid |
| CT | Computed Tomography |
| CTE | Chronic Traumatic Encephalopathy |
| DHA | Docosohexaenoic Acid |
| DHE | Dihydroethidium Staining |
| DRP1 | Dynamin-Related Protein 1 |
| EEG | Electroencephalography |
| eNOS | Endothelial Nitric Oxide Synthase |
| EPA | Eicosapentaenoic Acid |
| ETC | Electron Transfer Chain |
| FIS1 | Fission Protein 1, mitochondrial |
| FJB/C | Fluoro Jade B or C staining |
| FST | Forced Swimming Test |
| FPI | Fluid Percussion Injury |
| GAP-43 | Growth-Associated Protein-43 |
| GCS | Glasgow Coma Scale |
| GFAP | Glial Fibrillary Acidic Protein |
| GPx | Glutathione Peroxidases |
| GSH | Reduced Glutathione |
| GSK-3β | Glycogen Synthase Kinase 3 Beta |
| GSNA | Greater Splanchnic Nerve Activity |
| GSNO | Nitrosoglutathione |
| GR | GSH Reductase |
| GSSG | Oxidized Glutathione |
| Hb | Haemoglobin |
| H&E | Hematoxylin and Eosin staining rotocol |
| ICH | Immunohistochemistry |
| ICP | Intracranial Pressure |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| i.m. | Intramuscular |
| i.v. | Intravenous |
| lncRNA Neat1 | Long noncoding RNA nuclear enriched abundant transcript 1 |
| MDA | Malondialdehyde |
| MFN1 or 2 | Mitofusin 1 or 2 |
| MINO | Minocycline |
| mNSS | Modified Neurological Severity Score |
| mTBI | Mild Traumatic Brain Injury |
| MWM | Morris Water Maze |
| NAC | N-Acetylcystein |
| NFL | Neurofilament Light |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NICUs | Neurosurgical Intensive Care Units |
| NKCC | Sodium–potassium–chloride cotransporter |
| NMDA | N-Methyl-D-Aspartate |
| nNOS | Neuronal Nitric Oxide Synthase |
| NO | Nitric oxide |
| NOR | Novel Object Recognition |
| NOS | Nitric Oxide Synthases |
| NOX | NADPH Oxidase |
| NP | Neuropsychological |
| Nrf2-ARE | Nuclear factor E2-related factor 2 Antioxidant Response Elements |
| NSS | Neurological Soft Signs |
| OPA1 | Optic dominant Atrophy 1 |
| OXPHOS | Oxidative Phosphorylation |
| PARK2 | Parkin |
| PAT | Passive Avoidance Task |
| PEG | Polyethylene Glycol |
| PINK1 | mitochondrial serine/threonine-protein kinase 1 |
| PTSD | Posttraumatic Stress Disorder |
| PUFA | Polyunsaturated Fatty Acids |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| SIS | Second Impact Syndrome |
| SOD | Superoxide Dismutase |
| SVCT1 or 2 | Sodium dependent vitamin C transporter 1 or 2 |
| SYP | Synaptophysin |
| TAC | Total Antioxidant Capacity |
| TBARS | Thiobarbituric Acid Reactive Substance |
| TBI | Traumatic Brain Injury |
| TEM | Transmission Electron Microscopy |
| TST | Tail Suspension Test |
| TUNEL | Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling assay |
| UCH-L1 | Ubiquitin Carboxy-terminal Hydrolase-L1 |
| VCS | Veterinary Coma Scale |
| VR | Virtual Reality |
| WB | Western Blot |
| WD | Weight Drop |
| XO | Xanthine Oxidase |
Appendix A
Table A1.
Summary of preclinical studies on flavonoid administration in different models of TBI.
Table A1.
Summary of preclinical studies on flavonoid administration in different models of TBI.
| Name of Flavonoid | Dosage and Route of Administration | Time Points | Tests and Assays | TBI Model | Main Findings | Ref |
|---|---|---|---|---|---|---|
| Chrysin | 25, 50, or 100 mg/kg orally, started immediately post-injury and continued for up to 3 or 14 days | 1 day before injury; 0, 1, 4 h, and 1, 2, 3, 4, 7, 13, 14, 28 days post-injury | VCS, rotarod test, PAT, biochemical assay, histochemical staining, TUNEL, IHC | WD | Chrysin enhanced post-injury motor function, cognitive status and neuronal loss via reduction of oxidative stress (increased concentrations of SOD, CAT, GPx, GSH, and decreased MDA level) and inhibition of the apoptotic proteins (decreased Bax and increased Bcl-2) in the cerebral cortex and CA3 hippocampal neurons | [133] |
| Wogonin | 1, 2.5, 5 mg/kg. (i.v.) before and after moderate TBI; Alternatively, 2 µL (10 mM) injected into the lateral cerebral ventricle (i.c.v.) before and after moderate TBI | Before injury and 0.5, 1, 2, 4 h post-injury | Arterial pressure, heart rate, baroreflex and GSNA, histochemical staining of hippocampus | FPI | Wogonin administered before or after TBI significantly improved the cardiovascular changes (MAP, HR, and baroreflex) occurring after FPI. | [134] |
| Isoliquiritigenin (ILG) | 20 mg/kg (i.p.) 1 h post-TBI | 1 and 7 days after TBI | Sensorimotor Garcia test, brain water content, BBB permeability assay, histochemical staining, cell viability assay, WB, RT–qPCR, fluorescence immunoassays | CCI | ILG improved neurologic functions by reducing brain edema, BBB permeability, apoptosis (decrease in expression of cleaved caspase 3), and oxidative stress (translocation of Nrf2 into the nucleus with activation of downstream proteins). | [135] |
| Hesperidin | 50 mg/kg orally from day 10 to 24 post-injury | Day 21 and 24 post-injury | Sucrose preference test, FST, suppressed feeding test, TST, biochemical analysis | WD | Hesperidin reduced depression symptoms, levels of IL-1β, TNF-α and MDA, and increased BDNF levels in the hippocampus | [136] |
| Icariin | Oral administration of 3, 10, 30 mg/kg on injury induction and daily for 7 days post-injury | 0, 3, 7, 8 days post-TBI | Rotarod test, balance beam test, Y-maze, WB, IHC, histochemical staining, protein expression | Modified CCI | Icariin improved sensory motor and cognitive functions, and upregulated BDNF, SYP, and PSD-95 after injury. However, no improvement of brain histology and neuronal death were found. | [137] |
| Baicalin | 50, 100, 150 mg/kg (i.p.) 30 min after TBI | 2 h, and 1 and 3 days post-TBI | NSS, brain water content, TUNEL assay, WB Immunostaining, RT-qPCR | WD | Baicalin improved neurological function, brain edema, apoptosis, and oxidative stress via activating the Akt/Nrf2 pathways | [138] |
| Formononetin | Intragastrical administration of 10, 30 mg/kg/die for up to 7 days after TBI | 7 days post-injury | ELISA of tissue and serum cytokines, Cytohistologic stains, RT-qPCR, WB | WD | Formononetin counteracted TBI-induced neuroinflammation by decreasing tissue and serum levels of IL-6 and BDNF, and increasing tissue and serum levels IL-10 | [139] |
| Troxerutin | 1.5 mL/kg (i.p.) for 5 days before TBI | 3 days post-injury | NSS, MRI, ELISA, Nissl, TEM, WB and Immunostaining | WD | Troxerutin improved neurovascular function and integrity post-injury through action on eNOS and NO level, with consequent decrease in peroxynitrite formation | [140] |
| Quercetin | 50 mg/kg (i.p.) at 30 min, 12 h and 24 h post-TBI | 1, 3, 5 days post-injury | Brain water content, NSS, H&E staining and neuron count, IHC, WB | WD | Reduced brain edema, neural apoptosis and improved motor function (inhibition of extracellular signal-regulated kinase 1/2 phosphorylation and activated Akt serine/threonine protein kinase phosphorylation) | [141] |
| Resveratrol | 0.05, 0.1 mg/kg via oral gavage for 10 consecutive days, 7 days after TBI | 7 days post-injury | MWM, WB, TAC, Apoptosis, DHE staining | CCI | Reduced cognitive deficits, ROS generation, and apoptosis after TBI via recovered activation of p38/Nrf2/HO1 signaling pathway | [142] |
| Silymarin | 50 mg/kg via gavage for 20 days before injury | 8–10, 11, 18–20, 21 post-injury | Open field, elevated plus-maze, light-dark box, elevated zero-maze, sucrose preference, FST, TST, TNF-α | WD | Decreased anxiety and depression-like behaviours after injury due to reduced TNF-α levels in the prefrontal cortex and hippocampus | [143] |
| Diosmin | 100 mg/kg (p.o.) for 7 days before TBI | –1 h, 1 h, 1,2,15days post-injury | VCS, passive avoidance memory, BBB permeability, brain edema, ELISA | WD | Protective effects against TBI-induced memory and long-term potentiation impairment through reduction of TNF-α concentration in hippocampus | [144] |
| Catechin | 1, 5, 10, 20 or 30 mg/kg daily via gavage up to 28 days post-TBI | 3, 5, 7, 14, 21, 28 days pot-injury | Brain infarct volume edema, foot-fault test, MWM, BBB permeability, RT-qPCR, WB | CCI | Catechin prevented tight junction disruption and preserved BBB integrity, reducing post-injury inflammatory reaction | [145] |
| 7,8-dihydroxyflavone | 5 mg/kg (i.p) post-TBI either had access to voluntary wheel running for 7 days after injury or were sedentary | 7, 14 days post-injury | Barnes maze, voluntary running wheel exercise, WB, rsfMRI | FPI | 7,8-DHF enhanced the levels of cell energy metabolism (COII, PGC-1α, AMPK) and hippocampal functional connectivity | [146] |
| Pycnogenol | 50, 100 mg/kg (i.p.) 15 min, 3 h, 6 h post-TBI | 2, 5, 7, 12 days post-injury | MWM, FJB, cortical tissue sparing | CCI | No improvement of cognitive ability post-injury in MWM maze task. Pycnogenol suppressed NO through the inhibition of iNOS and also the NF-kB/AP-1 pathway. | [147] |
| Breviscapine | 50 mg/kg (i.v.) post-TBI | 1, 4, 7, 14, 21 days post-injury | NSS, RT-PCR, WB, IHC, Immunostaining, TUNEL | WD | Neurobehavioral function improved after treatment due to GSK3β signaling pathway inhibition | [148] |
| Hydroxysafflor yellow A | 10, 30 mg/kg orally post-TBI | 6 h, 12 h, 24 h post-TBI | Detection of HSYA, SOD, MDA, CAT, GSH/ GSSG | CCI | HSYA reduced oxidative stress by improving the activities of SOD and CAT, the level of GSH, and the GSH/GSSG ratio. Additionally, it decreased the levels of MDA and GSSG | [149] |
| Genistein | 15 mg/kg (i.p.) 30 min and again 24 h after TBI | –1, 1, 2 days post-TBI | Brain oedema, BBB permeability, ICP, VCS, beam-walk task | WD | Genistein inhibited brain edema, BBB permeability, and improved ICP after TBI. It also improved neurobehavioral performance and motor disorder | [150] |
| Epicatechin | 5, 15, 45 mg/kg by gavage at 3 h after TBI and once daily for 3 days or 15 mg/kg EC at 3 h after TBI and then once daily for 7 days | 1, 2, 3, 7, 14, 21, 28 days post-TBI | Neurologic deficit score, forelimb placing test, wire-hanging test, rotarod test, TST, FST, sucrose preference test, IHC, brain water content, Hb, WB, Immunostaining | CCI | EC significantly reduced lesion volume, edema, and cell death and improved neurologic function on days 3 and 28. Cognitive performance and depression-like behaviors were also improved by activating the Nrf2 pathway, inhibiting heme oxygenase-1 protein expression, and reducing iron deposition | [151] |
| Procyanidins | 100 mg/kg (i.v.) PC within 30 min post-TBI | 24 h, and 11, 12, 13, 14 days post-injury | MWM, MDA, GSH, SOD, ELISA, WB | CCI | Procyanidins improved cognitive performance by reducing the level of MDA, increasing GSH and activity of SOD, elevating the levels of BDNF, phosphorylation-cAMP-response element-binding protein (pCREB), total CREB, and cyclic AMP (cAMP) | [152] |
| Proanthocyanidin | Not mentioned | 72 h post-injury | Cerebral water content, TBARS, nitrite and nitrate, Thiols | Cold injury | Proanthocyanidin attenuated oxidative and nitrosative stress and decreased brain edema | [153] |
| (–)-epigallocatechin gallate (EGCG) | 0.1% (w/v) EGCG pre- and post-TBI. Solution was prepared by dissolving the drug in drinking water | 1, 3, and 7 days post-TBI | MWM, IHC, immunostaining for ssDNA and NeuN, lipid peroxidation | CCI | EGCG treatments improved cognitive impairment through inhibiting free radical-mediated neuronal degeneration and apoptotic cell death around the area damaged by TBI. | [154] |
| Puerarin | 200 mg/kg (i.p.) before injury | 24 h post-injury | WB, MDA, GSH, Naþ-Kþ-ATPase activity, Myeloperoxidase activity, FJC | WD | Puerarin ameliorated oxidative neurodegeneration after TBI through the activation of PI3K-Akt pathway | [155] |
| Luteolin | In vivo 10, 30, 50 (i.p.) mg/kg post-TBI In vitro 5, 10, and 25 µM post-TBI | 1, 3, 7 days post-TBI | Grip test, brain water content, MDA, GPx activity, TUNEL, IHC, WB, nuclear extraction and electrophoresis mobility shift assay, RT-qPCR, cell viability | WD | Luteolin enhanced the translocation of Nrf2 to the nucleus both in vivo and in vitro, upregulation of heme oxygenase 1 (HO1) and NAD(P)H:quinone oxidoreductase 1 (NQO1). Luteolin neuroprotective effects are possibly mediated by the activation of the Nrf2–ARE pathway | [156] |
| Naringin | 100 mg/kg orally 7 days before and 7 days after the TBI | 7 days post-injury | NSS, brain water content, serum and tissue biochemical analysis, WB | WD | Naringin improved behavioral dysfunction by attenuating the increases in MDA and NO; enhancing the activation of SOD; decreasing the over-activation of iNOS; down-regulating the overexpression of IL-1b; and reducing the brain edema. | [157] |
| Baicalein | 30 mg/kg (i.p.) immediately following injury or daily for 4 dayspost-injury | Before injury, 1, 4, 7, 14, 21 days after injury | Rotarod test, y test, mNSS, beam walk test, RT-PCR, WB, IHC, ELISA, FJB | CCI | Baicalein attenuated the contusion’s site expression of TNF-α, IL-1β and IL-6 mRNA and cytokine protein. | [158] |
Table A2.
Summary of preclinical studies on omega-3 fatty acids administration in different models of TBI.
Table A2.
Summary of preclinical studies on omega-3 fatty acids administration in different models of TBI.
| Dosage and Route of Administration | Time Points | Tests and Assays | TBI Model | Main Findings | Ref |
|---|---|---|---|---|---|
| DHA: 500 nmol/kg (i.v.) 30 min post-injury | 7, 28 days post-injury | mNSS, MRI, ICH | CCI | Reduced lesion size, microglia, and astrocytic reactivity, decreased the accumulation of beta-amyloid precursor protein (APP) at 7 days post-TBI. Reduced the neurofilament light (NFL) levels in plasma at 28 days. | [159] |
| DHA: 7.5 mg/100 mL (i.p.) 30 min after TBI | 1, 3, 7, 14 days post-injury | Brain nitrates and nitrites (NOx), NOR, markers of microglial activation, astrocyte marker, mRNA of inflammation-related genes | CCI | Decreased oxidative stress at day 1 and pro-inflammatory microglial activation at day 3. Decreased oxidative stress, histologic damage, and mRNA markers of microglial pro-inflammatory activation. Improved short term cognitive function. | [160] |
| NPD 1:50 ng intra-lesionally, immediately following TBI | 1,3 days post-injury | FJB, TUNEL, Immuno-staining and lesion size analyses | PBI | NPD1 decreased the lesion area at 72 h | [161] |
| DHA: 370, 550, 740 mg/kg/day, intragastric administration 30 min after TBI. | 1, 4, 7, 14, 21 days post-injury | NSS, MWM TUNEL, Nrf2-ARE pathway-related genes | FPI | Improved neuromotor and cognitive functions, increased anti-apoptotic protein expression, SOD and GPx activity, translocation of Nrf2 to the nucleus. Increased the expression of the downstream factors NAD(P)H:quinone oxidoreductase (NQO-1) and HO-1. Neuroprotective potentially mediated through activating the Nrf2- ARE pathway. | [162] |
| DHA: 16 mg/kg (i.p) at 30 min, 1, 3, 5 days after TBI | 1, 2, 3, 4, 5 days post-injury | NSS, beam walking test and rotarod test, qRT-PCR, WB, TUNEL, IHC | CCI | Improved neurological and cognitive functions. Decreased apoptosis, TLR4 expression, and the expression of inflammatory mediator NF-Kappa B. | [163] |
| DHA: 370, 740 mg/kg/day (i.v.) 30 min after TBI and daily for 15 days | 2, 7 and 15 days post-injury | Beam-walking, MWM, RT-qPCR | FPI | Protection against motor deficits. Inhibition of caspase-3 upregulating the Bcl-2:Bax ratio. | [164] |
| Resveratrol 50 mg/L via drinking water; Omega3 fatty acids and prebiotics were administered via powdered food (100 g of prebiotic, 300 g of DHA, and 600 g of standard diet per 10 kg of food) for 43 days before TBI. | 43–60 days | Neurological test battery, expression of Aqp4, Gfap, Igf1, Nfl, Sirt1, and Tau genes | Focal closed- head | Treatment altered the behavioral performance and prevented injury-related deficits in the longer-term behavior measures, Decreased expression of Aqp4, Gfap, Igf1, Nfl, and Sirt1in the prefrontal cortex. | [165] |
| DHA: 16 mg/kg (i.p.) at 5 min, 3 to 21 days, after TBI | 3, 7, 21 days post-injury | Activation of microglia or macrophages, inflammatory response, neurons expression of the endoplasmic reticulum (ER) stress marker CHOP | CCI | DHA administration reduced neuronal ER stress and subsequent association with microglial or macrophage polarization after TBI. Potential amelioration of TBI-induced cellular pathology | [166] |
| DHA: 0.1% diet. 1 d before TBI and for 50 days after TBI | 1, 2, 3, 12, 28, 41, 47, 50 days post-injury | Inflammatory cytokines, nitrates, and nitrites, MWM, T2-weighted MRI, diffusion tensor imaging, histological exams | CCI | Decreased cognitive impairment, oxidative stress, and white matter injury in adult rats | [167] |
| ω-3 PUFA: 2 mL/kg (i.p.) 30 min after TBI and daily for 7 days | 1, 3, 7 days post-injury | mNSS, brain water content, NISSl staining, microglial activation, immunofluorescent staining, WB, TLR4/NF-κB | Feeney DM TBI model | ω-3 PUFA supplementation inhibited TBI-induced microglial activation and the subsequent inflammatory response by regulating HMGB1 nuclear translocation and secretion and also HMGB1-mediated activation of the TLR4/NF-κB signaling pathway, leading to neuroprotective effects. | [168] |
| DHA: 16 mg/kg (i.p.) 15 min after the injury and daily for 3 or 7 days. | 3, 7 days post-injury. | RT–qPCR, immunoblotting, immunostaining, DTI, MWM | CCI | DHA administration restored hippocampal lysosomal biogenesis and function, demonstrating its therapeutic potential. | [169] |
| Diet containing 10% corn oil with a fatty acid profile high in ω-6 PUFAs and low in ω-3 PUFAs for 3 days before injury | 3, 6, 12 h | Two-photon laser scanning microscopy (2PLSM), parenchymal cell death and reactive oxidative species (ROS), (GSH), neuroprotectin D1 (NPD1) | Closed-head | Neuroprotection by decreasing cell death, ROS formation, and preserving GSH and NPD1 expression. | [170] |
| DHA (1.2%) enriched diets and/or curcumin (500 ppm) for 2 weeks post-injury | 1, 2, 3, 4, 5 days and 2 weeks | Cognitive tests, WB, markers of lipid peroxidation | FPI | DHA alone or in combination improved cognitive functions, decreased oxidative stress and damage to membrane phospholipids. Curcumin complemented the action of DHA on TBI pathology | [171] |
| DHA = 1.2% of diet coupled with exercise for 12 days | 12 days | Expression of acyl-CoA oxidase 1 (Acox1), 17β-hydroxysteroid dehydrogenase type 4 (17β-HSD4), calcium-independent phospholipases A2 (iPLA2), syntaxin-3 (STX-3), and BDNF | FPI | DHA activity was synergistic with exercise. The effects were evident on restoration of membrane homeostasis after TBI, thus supporting synaptic plasticity and cognition. | [172] |
| Control diet: unhydrogenated soybean oil (70 g/kg). Deficient diet: safflower oil (66.5 g/kg) and soybean oil (3.5 g/kg). | 1, 7, 14, 21, and 28 days after TBI | Ccl2, Gfap, and Mmp 9 mRNA levels, MMP-2 and -9 enzymatic activities, lesion volume | CCI | Decreased brain DHA content in TBI rats fed with deficient diet contributed to poorer sensorimotor outcomes after TBI through a mechanism involving modulation of Timp1 expression. | [173] |
| DHA: 3, 12, 40 mg/kg diet for 30 days prior TBI | 1 week after injury | WMW, IHC | WD | Dietary supplementation with DHA increased DHA serum levels, reduced TBI-associated tissue damage (axonal injury counts, markers for cellular injury and apoptosis), and improved memory assessment by the water maze testing. | [174] |
References
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Buki, A.; Chesnut, R.M.; et al. InTBIR Participants and Investigators. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef]
- World Health Oraganization. Neurological Disorders: Public Health Challenges. 2006. Available online: https://www.who.int/mental_health/neurology/chapter_3_b_neuro_disorders_public_h_challenges.pdf?ua=1 (accessed on 19 July 2019).
- Carroll, L.J.; Cassidy, J.D.; Holm, L.; Kraus, J.; Coronado, V.G. WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. Methodological issues and research recommendations for mild traumatic brain injury: The WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J. Rehabil. Med. 2004, 43, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974, 2, 81–84. [Google Scholar] [CrossRef]
- Gardner, A.J.; Zafonte, R. Neuroepidemiology of traumatic brain injury. Handb. Clin. Neurol. 2016, 138, 207–223. [Google Scholar] [PubMed]
- Rosenfeld, J.V.; Maas, A.I.; Bragge, P.; Morganti-Kossmann, M.C.; Manley, G.T.; Gruen, R.L. Early management of severe traumatic brain injury. Lancet 2012, 380, 1088–1098. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Web-based Injury Statistics Query and Reporting System (WISQARS) (2003). National Center for Injury Prevention and Control, Centers for Disease Control and Prevention. Available online: www.cdc.gov/injury/wisqars (accessed on 19 July 2019).
- Greenberg, M.S. Handbook of Neurosurgery, 8th ed.; Thieme: New York, NY, USA, 2016; pp. 825–830. [Google Scholar]
- Kolias, A.G.; Guilfoyle, M.R.; Helmy, A.; Allanson, J.; Hutchinson, P.J. Traumatic brain injury in adults. Pract. Neurol. 2013, 13, 228–235. [Google Scholar] [CrossRef]
- Kochanek, P.M.; Dixon, C.E.; Mondello, S.; Wang, K.K.K.; Lafrenaye, A.; Bramlett, H.M.; Dietrich, W.D.; Hayes, R.L.; Shear, D.A.; Gilsdorf, J.S.; et al. Multi-Center Pre-clinical Consortia to Enhance Translation of Therapies and Biomarkers for Traumatic Brain Injury: Operation Brain Trauma Therapy and Beyond. Front. Neurol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.R. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef]
- Carney, N.; Totten, A.M.; O’Reilly, C.; Ullman, J.S.; Hawryluk, G.W.; Bell, M.J.; Bratton, S.L.; Chesnut, R.; Harris, O.A.; Kissoon, N.; et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery 2017, 80, 6–15. [Google Scholar] [CrossRef]
- Kim, S.; Mortera, M.; Hu, X.; Krishnan, S.; Hoffecker, L.; Herrold, A.; Terhorst, L.; King, L.; Machtinger, J.; Zumsteg, J.M.; et al. Overview of pharmacological interventions after traumatic brain injuries: Impact on selected outcomes. Brain Inj. 2019, 33, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Kumar Sahel, D.; Kaira, M.; Raj, K.; Sharma, S.; Singh, S. Mitochondrial dysfunctioning and neuroinflammation: Recent highlights on the possible mechanisms involved in Traumatic Brain Injury. Neurosci. Lett. 2019, 710, 134347. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Fujisawa, H. The role of glutamate antagonists for the treatment of CNS injury. J. Neurotrauma 1992, 9 (Suppl. 2), S443–S462. [Google Scholar]
- Tymianski, M.; Tator, C.H. Normal and abnormal calcium homeostasis in neurons: A basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery 1996, 38, 1176–1195. [Google Scholar] [PubMed]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 2010, 7, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Severity of experimental traumatic brain injury modulates changes in concentrations of cerebral free amino acids. J. Cell. Mol. Med. 2017, 21, 530–542. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci. 2004, 61, 657–668. [Google Scholar] [CrossRef]
- Tavazzi, B.; Signoretti, S.; Lazzarino, G.; Amorini, A.M.; Delfini, R.; Cimatti, M.; Marmarou, A.; Vagnozzi, R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery 2005, 56, 582–589. [Google Scholar] [CrossRef]
- Cristofori, L.; Tavazzi, B.; Gambin, R.; Vagnozzi, R.; Signoretti, S.; Amorini, A.M.; Fazzina, G.; Lazzarino, G. Biochemical analysis of the cerebrospinal fluid: Evidence for catastrophic energy failure and oxidative damage preceding brain death in severe head injury: A case report. Clin. Biochem. 2005, 38, 97–100. [Google Scholar] [CrossRef]
- Hall, E.D.; Springer, J.E. Neuroprotection and acute spinal cord injury: A reappraisal. NeuroRx 2004, 1, 80–100. [Google Scholar] [CrossRef]
- Ozsuer, H.; Gorgulu, A.; Kiris, T.; Cobanoglu, S. The effects of memantine on lipid peroxidation following closed-head trauma in rats. Neurosurg. Rev. 2005, 28, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, H.; Fang, J.; Dai, W.; Zhou, J.; Wang, X.; Zhou, M. SS-31 Provides Neuroprotection by Reversing Mitochondrial Dysfunction After Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2018, 2018, 4783602. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Narabayashi, H.; Sata, T.; Takeshige, K. Kinetics of superoxide formation by respiratory chain NADH- dehydrogenase of bovine heart mitochondria. J. Biochem. 1983, 94, 1301–1306. [Google Scholar] [CrossRef]
- De Campos, R.P.; Siegel, J.M.; Fresta, C.G.; Caruso, G.; da Silva, J.A.; Lunte, S.M. Indirect detection of superoxide in RAW 264.7 macrophage cells using microchip electrophoresis coupled to laser-induced fluorescence. Anal. Bioanal. Chem. 2015, 407, 7003–7012. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Hogard, M.L.; Caruso, G.; Melo Costa, E.E.; Lazzarino, G.; Lunte, S.M. Monitoring carnosine uptake by RAW 264.7 macrophage cells using microchip electrophoresis with fluorescence detection. Anal. Methods 2017, 9, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.G.; Alshboul, O.A. Pathophysiology of traumatic brain injury. Neurosciences 2013, 18, 222–234. [Google Scholar]
- Gutteridge, J.M. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem. 1995, 41, 1819–1828. [Google Scholar] [CrossRef]
- Cobbs, C.S.; Fenoy, A.; Bredt, D.S.; Noble, L.J. Expression of nitric oxide synthase in the cerebral microvasculature after traumatic brain injury in the rat. Brain Res. 1997, 751, 336–338. [Google Scholar] [CrossRef]
- Rao, V.L.; Dogan, A.; Bowen, K.K.; Dempsey, R.J. Traumatic injury to rat brain upregulates neuronal nitric oxide synthase expression and L-[3H]nitroarginine binding. J. Neurotrauma 1999, 16, 865–877. [Google Scholar] [CrossRef]
- Gahm, C.; Holmin, S.; Mathiesen, T. Temporal profiles and cellular sources of three nitric oxide synthase isoforms in the brain after experimental contusion. Neurosurgery 2000, 46, 169–177. [Google Scholar] [CrossRef]
- Cherian, L.; Goodman, J.C.; Robertson, C.S. Brain nitric oxide changes after controlled cortical impact injury in rats. J. Neurophysiol. 2000, 83, 2171–2178. [Google Scholar] [CrossRef] [PubMed]
- Hlatky, R.; Lui, H.; Cherian, L.; Goodman, J.C.; O’Brien, W.E.; Contant, C.F.; Robertson, C.S. The role of endothelial nitric oxide synthase in the cerebral hemodynamics after controlled cortical impact injury in mice. J. Neurotrauma 2003, 20, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Fresta, C.G.; Chakraborty, A.; Wijesinghe, M.B.; Amorini, A.M.; Lazzarino, G.; Lazzarino, G.; Tavazzi, B.; Lunte, S.M.; Caraci, F.; Dhar, P.; et al. Non-toxic engineered carbon nanodiamond concentrations induce oxidative/nitrosative stress, imbalance of energy metabolism, and mitochondrial dysfunction in microglial and alveolar basal epithelial cells. Cell Death Dis. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Fresta, C.G.; Siegel, J.M.; Wijesinghe, M.B.; Lunte, S.M. Microchip electrophoresis with laser-induced fluorescence detection for the determination of the ratio of nitric oxide to superoxide production in macrophages during inflammation. Anal. Bioanal. Chem. 2017, 409, 4529–4538. [Google Scholar] [CrossRef]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 2012, 1822, 675–684. [Google Scholar] [CrossRef]
- Hall, E.D.; Andrus, P.K.; Oostveen, J.A.; Fleck, T.J.; Gurney, M.E. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J. Neurosci. Res. 1998, 53, 66–77. [Google Scholar] [CrossRef]
- Castilho, R.F.; Kowaltowski, A.J.; Meinicke, A.R.; Bechara, E.J.; Vercesi, A.E. Permeabilization of the inner mitochondrial membrane by Ca2+ ions is stimulated by t-butyl hydroperoxide and mediated by reactive oxygen species generated by mitochondria. Free Radic. Biol. Med. 1995, 18, 479–486. [Google Scholar] [CrossRef]
- Jacquard, C.; Trioulier, Y.; Cosker, F.; Escartin, C.; Bizat, N.; Hantraye, P.; Cancela, J.M.; Bonvento, G.; Brouillet, E. Brain mitochondrial defects amplify intracellular [Ca2+] rise and neurodegeneration but not Ca2+ entry during NMDA receptor activation. FASEB J. 2006, 20, 1021–1023. [Google Scholar] [CrossRef]
- Khan, M.; Dhammu, T.S.; Matsuda, F.; Annamalai, B.; Dhindsa, T.S.; Singh, I.; Singh, A.K. Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI. Brain Res. 2016, 1630, 159–170. [Google Scholar] [CrossRef][Green Version]
- Nakamura, T.; Lipton, S.A. ‘SNO’-Storms Compromise Protein Activity and Mitochondrial Metabolism in Neurodegenerative Disorders. Trends Endocrinol. Metab. 2017, 28, 879–892. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Petzold, A.; Gasperini, C.; Ruggieri, S.; Quartuccio, M.E.; Lazzarino, G.; Di Stasio, E.; Tavazzi, B. Serum Compounds of Energy Metabolism Impairment Are Related to Disability, Disease Course and Neuroimaging in Multiple Sclerosis. Mol. Neurobiol. 2017, 54, 7520–7533. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Rotilio, G.; Ciriolo, M.R. Role of nitric oxide synthases in Parkinson’s disease: A review on the antioxidant and anti-inflammatory activity of polyphenols. Neurochem. Res. 2008, 33, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Lazzarino, G.; Listorti, I.; Muzii, L.; Amorini, A.M.; Longo, S.; Di Stasio, E.; Caruso, G.; D’Urso, S.; Puglia, I.; Pisani, G.; et al. Low-molecular weight compounds in human seminal plasma as potential biomarkers of male infertility. Hum. Reprod. 2018, 33, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Lazzarino, G.; Listorti, I.; Bilotta, G.; Capozzolo, T.; Amorini, A.M.; Longo, S.; Caruso, G.; Lazzarino, G.; Tavazzi, B.; Bilotta, P. Water- and Fat-Soluble Antioxidants in Human Seminal Plasma and Serum of Fertile Males. Antioxidants 2019, 8, 96. [Google Scholar] [CrossRef]
- Perez-Gregorio, R.; Simal-Gandara, J. A Critical Review of Bioactive Food Components, and of their Functional Mechanisms, Biological Effects and Health Outcomes. Curr. Pharm. Des. 2017, 23, 2731–2741. [Google Scholar] [CrossRef]
- Hasadsri, L.; Wang, B.H.; Lee, J.V.; Erdman, J.W.; Llano, D.A.; Barbey, A.K.; Wszalek, T.; Sharrock, M.F.; Wang, H.J. Omega-3 fatty acids as a putative treatment for traumatic brain injury. J. Neurotrauma 2013, 30, 897–906. [Google Scholar] [CrossRef]
- Rigg, J.L.; Elovic, E.P.; Greenwald, B.D. A review of the effectiveness of antioxidant therapy to reduce neuronal damage in acute traumatic brain injury. J. Head Trauma Rehabil. 2005, 20, 389–391. [Google Scholar] [CrossRef]
- Vonder Haar, C.; Peterson, T.C.; Martens, K.M.; Hoane, M.R. Vitamins and nutrients as primary treatments in experimental brain injury: Clinical implications for nutraceutical therapies. Brain Res. 2016, 1640, 114–129. [Google Scholar] [CrossRef]
- Granger, M.; Eck, P. Dietary Vitamin C in Human Health. Adv. Food Nutr. Res. 2018, 83, 281–310. [Google Scholar]
- Castiglione, D.; Platania, A.; Conti, A.; Falla, M.; D’Urso, M.; Marranzano, M. Dietary Micronutrient and Mineral Intake in the Mediterranean Healthy Eating, Ageing, and Lifestyle (MEAL) Study. Antioxidants 2018, 7, 79. [Google Scholar] [CrossRef]
- Burzle, M.; Suzuki, Y.; Ackermann, D.; Miyazaki, H.; Maeda, N.; Clemencon, B.; Burrier, R.; Hediger, M.A. The sodium-dependent ascorbic acid transporter family SLC23. Mol. Asp. Med. 2013, 34, 436–454. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M.V. SVCT1 and SVCT2: Key proteins for vitamin C uptake. Amino Acids 2008, 34, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Tavazzi, B.; D’Urso, S.; Longo, S.; Vagnozzi, R.; Signoretti, S.; Clementi, E.; Giardina, B.; et al. Neuroglobin expression and oxidant/antioxidant balance after graded traumatic brain injury in the rat. Free Radic. Biol. Med. 2014, 69, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, R.A. Ascorbic acid in the brain. Brain Res. Rev. 1993, 18, 123–133. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Parker, W.H.; Qu, Z.C.; May, J.M. Ascorbic acid transport in brain microvascular pericytes. Biochem. Biophys. Res. Commun. 2015, 458, 262–267. [Google Scholar] [CrossRef]
- Angulo, C.; Castro, M.A.; Rivas, C.I.; Segretain, D.; Maldonado, R.; Yanez, A.J.; Slebe, J.C.; Vera, J.C.; Concha, I.I. Molecular identification and functional characterization of the vitamin C transporters expressed by Sertoli cells. J. Cell Physiol. 2008, 217, 708–716. [Google Scholar] [CrossRef]
- Awasthi, D.; Church, D.F.; Torbati, D.; Carey, M.E.; Pryor, W.A. Oxidative stress following traumatic brain injury in rats. Surg. Neurol. 1997, 47, 575–581. [Google Scholar] [CrossRef]
- Tyurin, V.A.; Tyurina, Y.Y.; Borisenko, G.G.; Sokolova, T.V.; Ritov, V.B.; Quinn, P.J.; Rose, M.; Kochanek, P.; Graham, S.H.; Kagan, V.E. Oxidative stress following traumatic brain injury in rats: Quantitation of biomarkers and detection of free radical intermediates. J. Neurochem. 2000, 75, 2178–2189. [Google Scholar] [CrossRef]
- Ishaq, G.M.; Saidu, Y.; Bilbis, L.S.; Muhammad, S.A.; Jinjir, N.; Shehu, B.B. Effects of alpha-tocopherol and ascorbic acid in the severity and management of traumatic brain injury in albino rats. J. Neurosci. Rural Pract. 2013, 4, 292–297. [Google Scholar]
- Razmkon, A.; Sadidi, A.; Sherafat-Kazemzadeh, E.; Mehrafshan, A.; Jamali, M.; Malekpour, B.; Saghafinia, M. Administration of vitamin C and vitamin E in severe head injury: A randomized double-blind controlled trial. Clin. Neurosurg. 2011, 58, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhen, C.; Liu, J.; Yang, P.; Hu, L.; Shang, P. Unraveling the Potential Role of Glutathione in Multiple Forms of Cell Death in Cancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 3150145. [Google Scholar] [CrossRef] [PubMed]
- Limongi, D.; Baldelli, S.; Checconi, P.; Marcocci, M.E.; De Chiara, G.; Fraternale, A.; Magnani, M.; Ciriolo, M.R.; Palamara, A.T. GSH-C4 Acts as Anti-inflammatory Drug in Different Models of Canonical and Cell Autonomous Inflammation Through NFκB Inhibition. Front. Immunol. 2019, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Steullet, P.; Neijt, H.C.; Cuenod, M.; Do, K.Q. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: Relevance to schizophrenia. Neuroscience 2006, 137, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Varga, V.; Jenei, Z.; Janaky, R.; Saransaari, P.; Oja, S.S. Glutathione is an endogenous ligand of rat brain N-methyl-D-aspartate (NMDA) and 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors. Neurochem. Res. 1997, 22, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Oja, S.S.; Janaky, R.; Varga, V.; Saransaari, P. Modulation of glutamate receptor functions by glutathione. Neurochem. Int. 2000, 37, 299–306. [Google Scholar] [CrossRef]
- Barnett, S.D.; Buxton, I.L.O. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 340–354. [Google Scholar] [CrossRef]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef]
- Bayir, H.; Kagan, V.E.; Tyurina, Y.Y.; Tyurin, V.; Ruppel, R.A.; Adelson, P.D.; Graham, S.H.; Janesko, K.; Clark, R.S.; Kochanek, P.M. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr. Res. 2002, 51, 571–578. [Google Scholar] [CrossRef]
- Dash, P.K.; Hergenroeder, G.W.; Jeter, C.B.; Choi, H.A.; Kobori, N.; Moore, A.N. Traumatic Brain Injury Alters Methionine Metabolism: Implications for Pathophysiology. Front. Syst. Neurosci. 2016, 10, 36. [Google Scholar] [CrossRef]
- Koza, L.; Linseman, D.A. Glutathione precursors shield the brain from trauma. Neural Regen. Res. 2019, 14, 1701–1702. [Google Scholar] [PubMed]
- Reed, T.T.; Owen, J.; Pierce, W.M.; Sebastian, A.; Sullivan, P.G.; Butterfield, D.A. Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gamma-glutamylcysteine ethyl ester: Insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. J. Neurosci. Res. 2009, 87, 408–417. [Google Scholar] [PubMed]
- Hicdonmez, T.; Kanter, M.; Tiryaki, M.; Parsak, T.; Cobanoglu, S. Neuroprotective effects of N-acetylcysteine on experimental closed head trauma in rats. Neurochem. Res. 2006, 31, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Sangobowale, M.; Nikulina, E.; Bergold, P.J. Minocycline plus N-acetylcysteine protect oligodendrocytes when first dosed 12 h after closed head injury in mice. Neurosci. Lett. 2018, 682, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Senol, N.; Naziroglu, M.; Yuruker, V. N-acetylcysteine and selenium modulate oxidative stress, antioxidant vitamin and cytokine values in traumatic brain injury-induced rats. Neurochem. Res. 2014, 39, 685–692. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; Feliciano, A.S. Flavonoids: From Structure to Health Issues. Molecules 2017, 22, 477. [Google Scholar] [CrossRef] [PubMed]
- Izzi, V.; Masuelli, L.; Tresoldi, I.; Sacchetti, P.; Modesti, A.; Galvano, F.; Bei, R. The effects of dietary flavonoids on the regulation of redox inflammatory networks. Front. Biosci. (Landmark Ed.) 2012, 17, 2396–2418. [Google Scholar] [CrossRef]
- Theadom, A.; Mahon, S.; Barker-Collo, S.; McPherson, K.; Rush, E.; Vandal, A.C.; Feigin, V.L. Enzogenol for cognitive functioning in traumatic brain injury: A pilot placebo-controlled RCT. Eur. J. Neurol. 2013, 20, 1135–1144. [Google Scholar] [CrossRef]
- Yang, X.; Li, X.; Ren, J. From French Paradox to cancer treatment: Anti-cancer activities and mechanisms of resveratrol. Anticancer Agents Med. Chem. 2014, 14, 806–825. [Google Scholar] [CrossRef]
- Tsai, H.Y.; Ho, C.T.; Chen, Y.K. Biological actions and molecular effects of resveratrol, pterostilbene, and 3′-hydroxypterostilbene. J. Food Drug Anal. 2017, 25, 134–147. [Google Scholar] [CrossRef]
- Ates, O.; Cayli, S.; Altinoz, E.; Gurses, I.; Yucel, N.; Sener, M.; Kocak, A.; Yologlu, S. Neuroprotection by resveratrol against traumatic brain injury in rats. Mol. Cell. Biochem. 2007, 294, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chen, T.H.; Yang, L.Y.; Shih, C.M. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014, 5, e1147. [Google Scholar] [CrossRef] [PubMed]
- Gatson, J.W.; Liu, M.M.; Abdelfattah, K.; Wigginton, J.G.; Smith, S.; Wolf, S.; Minei, J.P. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J. Trauma Acute Care Surg. 2013, 74, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Irias-Mata, A.; Stuetz, W.; Sus, N.; Hammann, S.; Gralla, K.; Cordero-Solano, A.; Vetter, W.; Frank, J. Tocopherols, Tocomonoenols, and Tocotrienols in Oils of Costa Rican Palm Fruits: A Comparison between Six Varieties and Chemical versus Mechanical Extraction. J. Agric. Food Chem. 2017, 65, 7476–7482. [Google Scholar] [CrossRef] [PubMed]
- Comitato, R.; Ambra, R.; Virgili, F. Tocotrienols: A Family of Molecules with Specific Biological Activities. Antioxidants 2017, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Inci, S.; Ozcan, O.E.; Kilinc, K. Time-level relationship for lipid peroxidation and the protective effect of alpha-tocopherol in experimental mild and severe brain injury. Neurosurgery 1998, 43, 330–335. [Google Scholar] [CrossRef]
- Yang, J.; Han, Y.; Ye, W.; Liu, F.; Zhuang, K.; Wu, G. Alpha tocopherol treatment reduces the expression of Nogo-A and NgR in rat brain after traumatic brain injury. J. Surg. Res. 2013, 182, e69–e77. [Google Scholar] [CrossRef]
- Clifton, G.L.; Lyeth, B.G.; Jenkins, L.W.; Taft, W.C.; DeLorenzo, R.J.; Hayes, R.L. Effect of D, alpha-tocopheryl succinate and polyethylene glycol on performance tests after fluid percussion brain injury. J. Neurotrauma 1989, 6, 71–81. [Google Scholar] [CrossRef]
- Aiguo, W.; Zhe, Y.; Gomez-Pinilla, F. Vitamin E protects against oxidative damage and learning disability after mild traumatic brain injury in rats. Neurorehabil. Neural Repair 2010, 24, 290–298. [Google Scholar] [CrossRef]
- Artuch, R.; Salviati, L.; Jackson, S.; Hirano, M.; Navas, P. Coenzyme Q10 deficiencies in neuromuscular diseases. Adv. Exp. Med. Biol. 2009, 652, 117–128. [Google Scholar]
- Jorat, M.V.; Tabrizi, R.; Kolahdooz, F.; Akbari, M.; Salami, M.; Heydari, S.T.; Asemi, Z. The effects of coenzyme Q10 supplementation on biomarkers of inflammation and oxidative stress in among coronary artery disease: A systematic review and meta-analysis of randomized controlled trials. Inflammopharmacology 2019, 27, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, M.; Unal, M.M.; Gul, S.; Acikgoz, S.; Kandemir, N.; Hanci, V.; Edebali, N.; Acikgoz, B. Effect of coenzyme Q10 on ischemia and neuronal damage in an experimental traumatic brain-injury model in rats. BMC Neurosci. 2011, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M. The Antioxidant Role of Non-mitochondrial CoQ10: Mystery Solved! Cell Metab. 2020, 31, 13–15. [Google Scholar] [CrossRef]
- Pierce, J.D.; Shen, Q.; Peltzer, J.; Thimmesch, A.; Hiebert, J.B. A pilot study exploring the effects of ubiquinol on brain genomics after traumatic brain injury. Nurs. Outlook 2017, 65, S44–S52. [Google Scholar] [CrossRef]
- Pierce, J.D.; Gupte, R.; Thimmesch, A.; Shen, Q.; Hiebert, J.B.; Brooks, W.M.; Clancy, R.L.; Diaz, F.J.; Harris, J.L. Ubiquinol treatment for TBI in male rats: Effects on mitochondrial integrity, injury severity, and neurometabolism. J. Neurosci. Res. 2018, 96, 1080–1092. [Google Scholar] [CrossRef]
- Ross, A.C.; Caballero, B.H.; Cousins, R.J.; Tucker, K.L.; Ziegler, T.R. Modern Nutrition in Health and Disease, 11th ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2012. [Google Scholar]
- Cho, K.S.; Shin, M.; Kim, S.; Lee, S.B. Recent Advances in Studies on the Therapeutic Potential of Dietary Carotenoids in Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 4120458. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, Z.; Cui, H.; Wang, Y.; Zhong, C. Astaxanthin protects astrocytes against trauma-induced apoptosis through inhibition of NKCC1 expression via the NF-kappaB signaling pathway. BMC Neurosci. 2017, 18, 42. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, Z.; Cui, H.; Cao, Y.; Zhong, C.; Wang, Y. Astaxanthin alleviates cerebral edema by modulating NKCC1 and AQP4 expression after traumatic brain injury in mice. BMC Neurosci. 2016, 17, 60. [Google Scholar] [CrossRef]
- Ji, X.; Peng, D.; Zhang, Y.; Zhang, J.; Wang, Y.; Gao, Y.; Lu, N.; Tang, P. Astaxanthin improves cognitive performance in mice following mild traumatic brain injury. Brain Res. 2017, 1659, 88–95. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Fan, Y.; Gao, Y.; Li, X.; Hu, Z.; Ding, K.; Wang, Y.; Wang, X. Fucoxanthin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE and Nrf2-autophagy pathways. Sci. Rep. 2017, 7, 46763. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Jiang, L.; Huang, Z.; Zhang, H.; Cheng, C.; Liu, H.; He, J.; Wu, J.; Darwazeh, R.; Wu, Y.; et al. The long non-coding RNA Neat1 is an important mediator of the therapeutic effect of bexarotene on traumatic brain injury in mice. Brain Behav. Immun. 2017, 65, 183–194. [Google Scholar] [CrossRef]
- Zhong, J.; Cheng, C.; Liu, H.; Huang, Z.; Wu, Y.; Teng, Z.; He, J.; Zhang, H.; Wu, J.; Cao, F.; et al. Bexarotene protects against traumatic brain injury in mice partially through apolipoprotein E. Neuroscience 2017, 343, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, L.; Rao, W.; Su, N.; Hui, H.; Wang, L.; Peng, C.; Tu, Y.; Zhang, S.; Fei, Z. Neuroprotective effects of crocin against traumatic brain injury in mice: Involvement of notch signaling pathway. Neurosci. Lett. 2015, 591, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Yu, X.; Chen, M.; Chen, J.; Xu, J. Lutein protects against severe traumatic brain injury through antiinflammation and antioxidative effects via ICAM1/Nrf2. Mol. Med. Rep. 2017, 16, 4235–4240. [Google Scholar] [CrossRef]
- Lauritzen, L.; Hansen, H.S.; Jorgensen, M.H.; Michaelsen, K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Nishi, D.; Tanima, Y.; Itakura, M.; Kojima, M.; Hamazaki, K.; Noguchi, H.; Hamazaki, T. Serum pro-BDNF/BDNF as a treatment biomarker for response to docosahexaenoic acid in traumatized people vulnerable to developing psychological distress: A randomized controlled trial. Transl. Psychiatry 2015, 5, e596. [Google Scholar] [CrossRef]
- Noguchi, H.; Nishi, D.; Matsumura, K.; Hamazaki, K.; Hamazaki, T.; Matsuoka, Y.J. Limited effect of omega-3 fatty acids on the quality of life in survivors of traumatic injury: A randomized, placebo-controlled trial. Prostaglandins Leukot. Essent. Fatty Acids 2017, 127, 1–5. [Google Scholar] [CrossRef]
- Gross, B.W.; Gillio, M.; Rinehart, C.D.; Lynch, C.A.; Rogers, F.B. Omega-3 Fatty Acid Supplementation and Warfarin: A Lethal Combination in Traumatic Brain Injury. J. Trauma Nurs. 2017, 24, 15–18. [Google Scholar] [CrossRef]
- McCrory, P.; Feddermann-Demont, N.; Dvorak, J.; Cassidy, J.D.; McIntosh, A.; Vos, P.E.; Echemendia, R.J.; Meeuwisse, W.; Tarnutzer, A.A. What is the definition of sports-related concussion: A systematic review. Br. J. Sports Med. 2017, 51, 877–887. [Google Scholar] [CrossRef]
- McCrory, P.; Meeuwisse, W.; Dvorak, J.; Aubry, M.; Bailes, J.; Broglio, S.; Cantu, R.C.; Cassidy, D.; Echemendia, R.J.; Castellani, R.J.; et al. Consensus statement on concussion in sport-the 5(th) international conference on concussion in sport held in Berlin, October 2016. Br. J. Sports Med. 2017, 51, 838–847. [Google Scholar] [PubMed]
- Signoretti, S.; Tavazzi, B.; Lazzarino, G.; Vagnozzi, R. The Pathophysiology of Concussive Brain Injury. In Concussion and Traumatic Encephalopathy: Causes, Diagnosis and Management; Victoroff, J., Bigler, E.D., Eds.; Cambridge University Press: Cambridge, UK, 2019; pp. 138–152. [Google Scholar]
- Giza, C.C.; Hovda, D.A. The Neurometabolic Cascade of Concussion. J. Athl. Train. 2001, 36, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Giza, C.C.; Hovda, D.A. The new neurometabolic cascade of concussion. Neurosurgery 2014, 75 (Suppl. 4), S24–S33. [Google Scholar] [CrossRef]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Metabolic, enzymatic and gene involvement in cerebral glucose dysmetabolism after traumatic brain injury. Biochim. Biophys. Acta 2016, 1862, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Tavazzi, B.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Mitochondrial-related impairment—Part I. Neurosurgery 2007, 61, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Hill, L.J.; Porto, E.; Tavazzi, B.; Lazzarino, G.; Belli, A. Fusion or Fission: The Destiny of Mitochondria In Traumatic Brain Injury of Different Severities. Sci. Rep. 2017, 7, 9189. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Signoretti, S.; Floris, R.; Marziali, S.; Manara, M.; Amorini, A.M.; Belli, A.; Di Pietro, V.; D’Urso, S.; Pastore, F.S.; et al. Decrease in N-acetylaspartate following concussion may be coupled to decrease in creatine. J. Head Trauma Rehabil. 2013, 28, 284–292. [Google Scholar] [CrossRef]
- Vagnozzi, R.; Signoretti, S.; Tavazzi, B.; Floris, R.; Ludovici, A.; Marziali, S.; Tarascio, G.; Amorini, A.M.; Di Pietro, V.; Delfini, R.; et al. Temporal window of metabolic brain vulnerability to concussion: A pilot 1H-magnetic resonance spectroscopic study in concussed athletes—Part III. Neurosurgery 2008, 62, 1286–1295. [Google Scholar] [CrossRef]
- Tavazzi, B.; Vagnozzi, R.; Signoretti, S.; Amorini, A.M.; Belli, A.; Cimatti, M.; Delfini, R.; Di Pietro, V.; Finocchiaro, A.; Lazzarino, G. Temporal window of metabolic brain vulnerability to concussions: Oxidative and nitrosative stresses—Part II. Neurosurgery 2007, 61, 390–395. [Google Scholar] [CrossRef]
- Mez, J.; Daneshvar, D.H.; Kiernan, P.T.; Abdolmohammadi, B.; Alvarez, V.E.; Huber, B.R.; Alosco, M.L.; Solomon, T.M.; Nowinski, C.J.; McHale, L.; et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 2017, 318, 360–370. [Google Scholar] [CrossRef]
- Hoffer, M.E.; Balaban, C.; Slade, M.D.; Tsao, J.W.; Hoffer, B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: A double-blind, placebo controlled study. PLoS ONE 2013, 8, e54163. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.M.; Jones, M.T.; Kirk, K.M.; Gable, D.A.; Repshas, J.T.; Johnson, T.A.; Andreasson, U.; Norgren, N.; Blennow, K.; Zetterberg, H. Effect of Docosahexaenoic Acid on a Biomarker of Head Trauma in American Football. Med. Sci. Sports Exerc. 2016, 48, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Finelli, K.; Bai, X.; Arnett, P.; Bream, T.; Seidenberg, P.; Lynch, S.; Johnson, B.; Slobounov, S. Effect of Enzogenol® Supplementation on Cognitive, Executive, and Vestibular/Balance Functioning in Chronic Phase of Concussion. Dev. Neuropsychol. 2017, 42, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Andrus, P.K.; Zhang, J.R.; Hall, E.D. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J. Neurotrauma 1994, 11, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Vagnozzi, R.; Marmarou, A.; Tavazzi, B.; Signoretti, S.; Di Pierro, D.; del Bolgia, F.; Amorini, A.M.; Fazzina, G.; Sherkat, S.; Lazzarino, G. Changes of cerebral energy metabolism and lipid peroxidation in rats leading to mitochondrial dysfunction after diffuse brain injury. J. Neurotrauma 1999, 16, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.N.; Sullivan, P.G.; Deng, Y.; Mbye, L.H.; Hall, E.D. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: Implications for neuroprotective therapy. J. Cereb. Blood Flow Metab. 2006, 26, 1407–1418. [Google Scholar] [CrossRef]
- Cristofori, L.; Tavazzi, B.; Gambin, R.; Vagnozzi, R.; Vivenza, C.; Amorini, A.M.; Di Pierro, D.; Fazzina, G.; Lazzarino, G. Early onset of lipid peroxidation after human traumatic brain injury: A fatal limitation for the free radical scavenger pharmacological therapy? J. Investig. Med. 2001, 49, 450–458. [Google Scholar] [CrossRef]
- Rashno, M.; Sarkaki, A.; Farbood, Y.; Rashno, M.; Khorsandi, L.; Naseri, M.K.G.; Dianat, M. Therapeutic effects of chrysin in a rat model of traumatic brain injury: A behavioral, biochemical, and histological study. Life Sci. 2019, 228, 285–294. [Google Scholar] [CrossRef]
- Umemoto, Y.; Patel, A.; Huynh, T.; Chitravanshi, V.C. Wogonin attenuates the deleterious effects of traumatic brain injury in anesthetized Wistar rats. Eur. J. Pharmacol. 2019, 848, 121–130. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, L.L.; Teng, C.H.; Wu, F.F.; Ge, L.Y.; Shi, Y.J.; He, Z.L.; Liu, L.; Jiang, C.J.; Hou, R.N.; et al. Isoliquiritigenin Provides Protection and Attenuates Oxidative Stress-Induced Injuries via the Nrf2-ARE Signaling Pathway After Traumatic Brain Injury. Neurochem. Res. 2018, 43, 2435–2445. [Google Scholar] [CrossRef]
- Kosari-Nasab, M.; Shokouhi, G.; Ghorbanihaghjo, A.; Abbasi, M.M.; Salari, A.A. Hesperidin attenuates depression-related symptoms in mice with mild traumatic brain injury. Life Sci. 2018, 213, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.; Bae, J.; Lee, J.S.; Bang, Y.; Lee, B.J.; Park, J.W.; Lee, K.; Cho, J.H.; Bu, Y. Icariin Improves Functional Behavior in a Mouse Model of Traumatic Brain Injury and Promotes Synaptic Plasticity Markers. Planta Med. 2019, 85, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wang, H.; Zhou, J.; Dai, W.; Zhu, Y.; Zhou, Y.; Wang, X.; Zhou, M. Baicalin provides neuroprotection in traumatic brain injury mice model through Akt/Nrf2 pathway. Drug Des. Dev. Ther. 2018, 12, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zeng, G.; Zheng, X.; Wang, W.; Ling, Y.; Tang, H.; Zhang, J. Neuroprotective effect of formononetin against TBI in rats via suppressing inflammatory reaction in cortical neurons. Biomed. Pharmacother. 2018, 106, 349–354. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, Y.; Zeng, J.; Li, D.; Huang, Y. Troxerutin cerebroprotein hydrolysate injection ameliorates neurovascular injury induced by traumatic brain injury—Via endothelial nitric oxide synthase pathway regulation. Int. J. Neurosci. 2018, 128, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Zhao, Z.; Chen, Y.; Li, Z.; Tian, Y.; Liu, Z.; Liu, B.; Song, J. Quercetin protects rat cortical neurons against traumatic brain injury. Mol. Med. Rep. 2018, 17, 7859–7865. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Qiu, W.; Xiao, G.; Cheng, J.; Zhang, N. Resveratrol Attenuates Cognitive Deficits of Traumatic Brain Injury by Activating p38 Signaling in the Brain. Med. Sci. Monit. 2018, 24, 1097–1103. [Google Scholar] [CrossRef]
- Kosari-Nasab, M.; Shokouhi, G.; Ghorbanihaghjo, A.; Abbasi, M.M.; Salari, A.A. Anxiolytic- and antidepressant-like effects of Silymarin compared to diazepam and fluoxetine in a mouse model of mild traumatic brain injury. Toxicol. Appl. Pharmacol. 2018, 338, 159–173. [Google Scholar] [CrossRef]
- Mirshekar, M.A.; Fanaei, H.; Keikhaei, F.; Javan, F.S. Diosmin improved cognitive deficit and amplified brain electrical activity in the rat model of traumatic brain injury. Biomed. Pharmacother. 2017, 93, 1220–1229. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, J.; Cai, Y.; Huang, J.; You, L. Catechin attenuates traumatic brain injury-induced blood-brain barrier damage and improves longer-term neurological outcomes in rats. Exp. Physiol. 2017, 102, 1269–1277. [Google Scholar] [CrossRef]
- Krishna, G.; Agrawal, R.; Zhuang, Y.; Ying, Z.; Paydar, A.; Harris, N.G.; Royes, L.F.; Gomez-Pinilla, F. 7,8-Dihydroxyflavone facilitates the action exercise to restore plasticity and functionality: Implications for early brain trauma recovery. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Roberts, K.N. Cognitive assessment of pycnogenol therapy following traumatic brain injury. Neurosci. Lett. 2016, 634, 126–131. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, L.; Xia, Q.J.; Dong, X.J.; Hu, Y.; Chen, Z.W.; Chen, K.; Wang, K.H.; Liu, J.; Wang, T.H. Neuroprotective effect of breviscapine on traumatic brain injury in rats associated with the inhibition of GSK3beta signaling pathway. Brain Res. 2017, 1660, 1–9. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Peng, W.; Xia, Z.; Gan, P.; Huang, W.; Shi, Y.; Fan, R. Hydroxysaf fl or yellow A exerts antioxidant effects in a rat model of traumatic brain injury. Mol. Med. Rep. 2016, 14, 3690–3696. [Google Scholar] [CrossRef] [PubMed]
- Soltani, Z.; Khaksari, M.; Jafari, E.; Iranpour, M.; Shahrokhi, N. Is genistein neuroprotective in traumatic brain injury? Physiol. Behav. 2015, 152, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Wang, W.; Li, Q.; Han, X.; Xing, J.; Qi, C.; Lan, X.; Wan, J.; Potts, A.; Guan, F.; et al. Cerebroprotection of flavanol (-)-epicatechin after traumatic brain injury via Nrf2-dependent and -independent pathways. Free Radic. Biol. Med. 2016, 92, 15–28. [Google Scholar] [CrossRef]
- Mao, X.; Hao, S.; Zhu, Z.; Zhang, H.; Wu, W.; Xu, F.; Liu, B. Procyanidins protects against oxidative damage and cognitive deficits after traumatic brain injury. Brain Inj. 2015, 29, 86–92. [Google Scholar] [CrossRef]
- Erdem, Y.; Tekiner, A.; Erkoc, Y.S.; Yilmaz, M.B.; Celik, H.; Yildirim, A.E.; Tekiner, A.S.; Bayar, M.A. Antiedema effects of proanthocyanidin on experimental traumatic brain edema. Turk. Neurosurg. 2015, 25, 85–89. [Google Scholar] [CrossRef][Green Version]
- Itoh, T.; Tabuchi, M.; Mizuguchi, N.; Imano, M.; Tsubaki, M.; Nishida, S.; Hashimoto, S.; Matsuo, K.; Nakayama, T.; Ito, A.; et al. Neuroprotective effect of (-)-epigallocatechin-3-gallate in rats when administered pre- or post-traumatic brain injury. J. Neural Transm. 2013, 120, 767–783. [Google Scholar] [CrossRef]
- Wang, J.W.; Wang, H.D.; Cong, Z.X.; Zhou, X.M.; Xu, J.G.; Jia, Y.; Ding, Y. Puerarin ameliorates oxidative stress in a rodent model of traumatic brain injury. J. Surg. Res. 2014, 186, 328–337. [Google Scholar] [CrossRef]
- Xu, J.; Wang, H.; Ding, K.; Zhang, L.; Wang, C.; Li, T.; Wei, W.; Lu, X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic. Biol. Med. 2014, 71, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.J.; Wang, L.Y.; Wei, Z.X.; Qu, W.S. Continual naringin treatment benefits the recovery of traumatic brain injury in rats through reducing oxidative and inflammatory alterations. Neurochem. Res. 2014, 39, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Hsu, C.W.; Huang, W.H.; Wang, J.Y. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br. J. Pharmacol. 2008, 155, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Thau-Zuchman, O.; Ingram, R.; Harvey, G.G.; Cooke, T.; Palmas, F.; Pallier, P.N.; Brook, J.; Priestley, J.V.; Dalli, J.; Lopez-Tremoleda, J.; et al. A single injection of docosahexaenoic acid induces a pro-resolving lipid mediator profile in the injured tissue and a long-lasting reduction in neurological deficit after traumatic brain injury in mice. J. Neurotrauma 2020, 37, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.E.; Requena, D.F.; Casper, T.C.; Velhorst, A.K.; Lolofie, A.; McFarlane, K.E.; Otto, T.E.; Terry, C.; Gensel, J.C. Docosahexaenoic acid decreased neuroinflammation in rat pups after controlled cortical impact. Exp. Neurol. 2019, 320, 112971. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.W.V.; Davidsson, J.; Lidin, E.; Angeria, M.; Risling, M.; Gunther, M. Brain tissue saving effects by single-dose intralesional administration of Neuroprotectin D1 on experimental focal penetrating brain injury in rats. J. Clin. Neurosci. 2019, 64, 227–233. [Google Scholar] [CrossRef]
- Zhu, W.; Ding, Y.; Kong, W.; Li, T.; Chen, H. Docosahexaenoic Acid (DHA) Provides Neuroprotection in Traumatic Brain Injury Models via Activating Nrf2-ARE Signaling. Inflammation 2018, 41, 1182–1193. [Google Scholar] [CrossRef]
- Tang, R.; Lin, Y.M.; Liu, H.X.; Wang, E.S. Neuroprotective effect of docosahexaenoic acid in rat traumatic brain injury model via regulation of TLR4/NF-Kappa B signaling pathway. Int. J. Biochem. Cell Biol. 2018, 99, 64–71. [Google Scholar] [CrossRef]
- Zhu, W.; Chi, N.; Zou, P.; Chen, H.; Tang, G.; Zhao, W. Effect of docosahexaenoic acid on traumatic brain injury in rats. Exp. Ther. Med. 2017, 14, 4411–4416. [Google Scholar] [CrossRef]
- Salberg, S.; Yamakawa, G.; Christensen, J.; Kolb, B.; Mychasiuk, R. Assessment of a nutritional supplement containing resveratrol, prebiotic fiber, and omega-3 fatty acids for the prevention and treatment of mild traumatic brain injury in rats. Neuroscience 2017, 365, 146–157. [Google Scholar] [CrossRef]
- Harvey, L.D.; Yin, Y.; Attarwala, I.Y.; Begum, G.; Deng, J.; Yan, H.Q.; Dixon, C.E.; Sun, D. Administration of DHA Reduces Endoplasmic Reticulum Stress-Associated Inflammation and Alters Microglial or Macrophage Activation in Traumatic Brain Injury. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.E.; Requena, D.F.; Abdullah, O.M.; Casper, T.C.; Beachy, J.; Malleske, D.; Pauly, J.R. Dietary Docosahexaenoic Acid Improves Cognitive Function, Tissue Sparing, and Magnetic Resonance Imaging Indices of Edema and White Matter Injury in the Immature Rat after Traumatic Brain Injury. J. Neurotrauma 2016, 33, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, S.; Chen, C.; Xie, B.; Fang, Z.; Hu, W.; Chen, J.; Fu, H.; He, H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-kappaB pathway following experimental traumatic brain injury. J. Neuroinflamm. 2017, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, E.; Sun, G.; Yan, H.Q.; Foley, L.M.; Andrzejczuk, L.A.; Attarwala, I.Y.; Hitchens, T.K.; Kiselyov, K.; Dixon, C.E.; et al. Effects of DHA on Hippocampal Autophagy and Lysosome Function After Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 2454–2470. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Yang, Z.; Luo, C.; Zeng, H.; Li, P.; Kang, J.X.; Wan, J.B.; He, C.; Su, H. Enriched Endogenous Omega-3 Fatty Acids in Mice Ameliorate Parenchymal Cell Death After Traumatic Brain Injury. Mol. Neurobiol. 2017, 54, 3317–3326. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Dietary strategy to repair plasma membrane after brain trauma: Implications for plasticity and cognition. Neurorehabil. Neural Repair 2014, 28, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience 2013, 248, 655–663. [Google Scholar] [CrossRef]
- Russell, K.L.; Berman, N.E.; Levant, B. Low brain DHA content worsens sensorimotor outcomes after TBI and decreases TBI-induced Timp1 expression in juvenile rats. Prostaglandins Leukot. Essent. Fatty Acids 2013, 89, 97–105. [Google Scholar] [CrossRef]
- Mills, J.D.; Hadley, K.; Bailes, J.E. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery 2011, 68, 474–481. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).