WIP Modulates Oxidative Stress through NRF2/KEAP1 in Glioblastoma Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Immunoblotting
2.3. Lentiviral Vector Production and Infection
2.4. Analysis of mRNA Levels
2.5. Immunofluorescence and Image Analysis
2.6. Flow Cytometry Determination of Reactive Oxygen Species
2.7. Statistical Analyses
3. Results
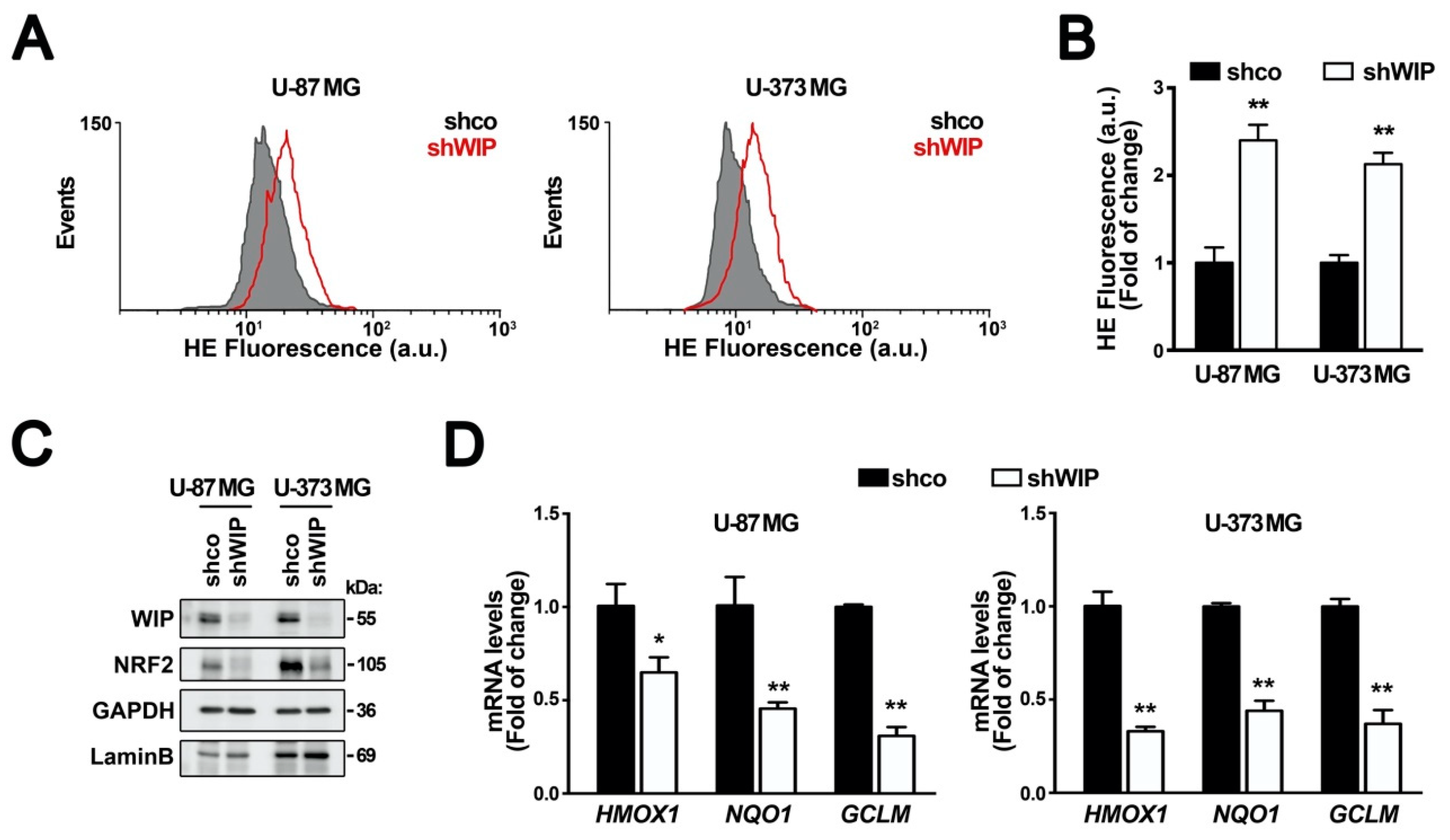
3.1. WIP Knocked-Down Cells Exhibit Increased ROS and Decreased NRF2 Levels
3.2. The Regulation of NRF2 by WIP is KEAP1-Dependent
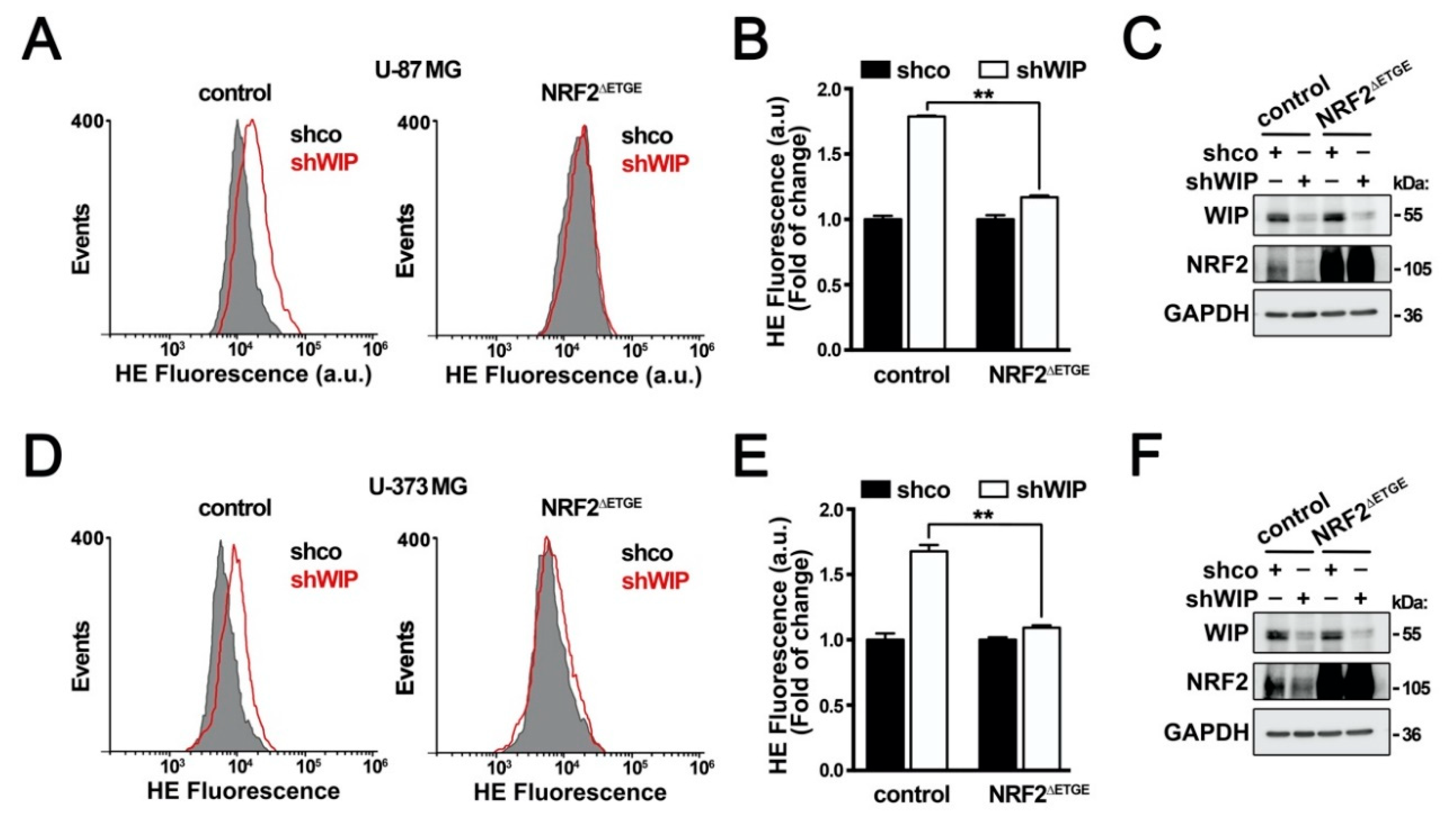
3.3. The Control of the Cellular Redox State by WIP Is NRF2-Dependent
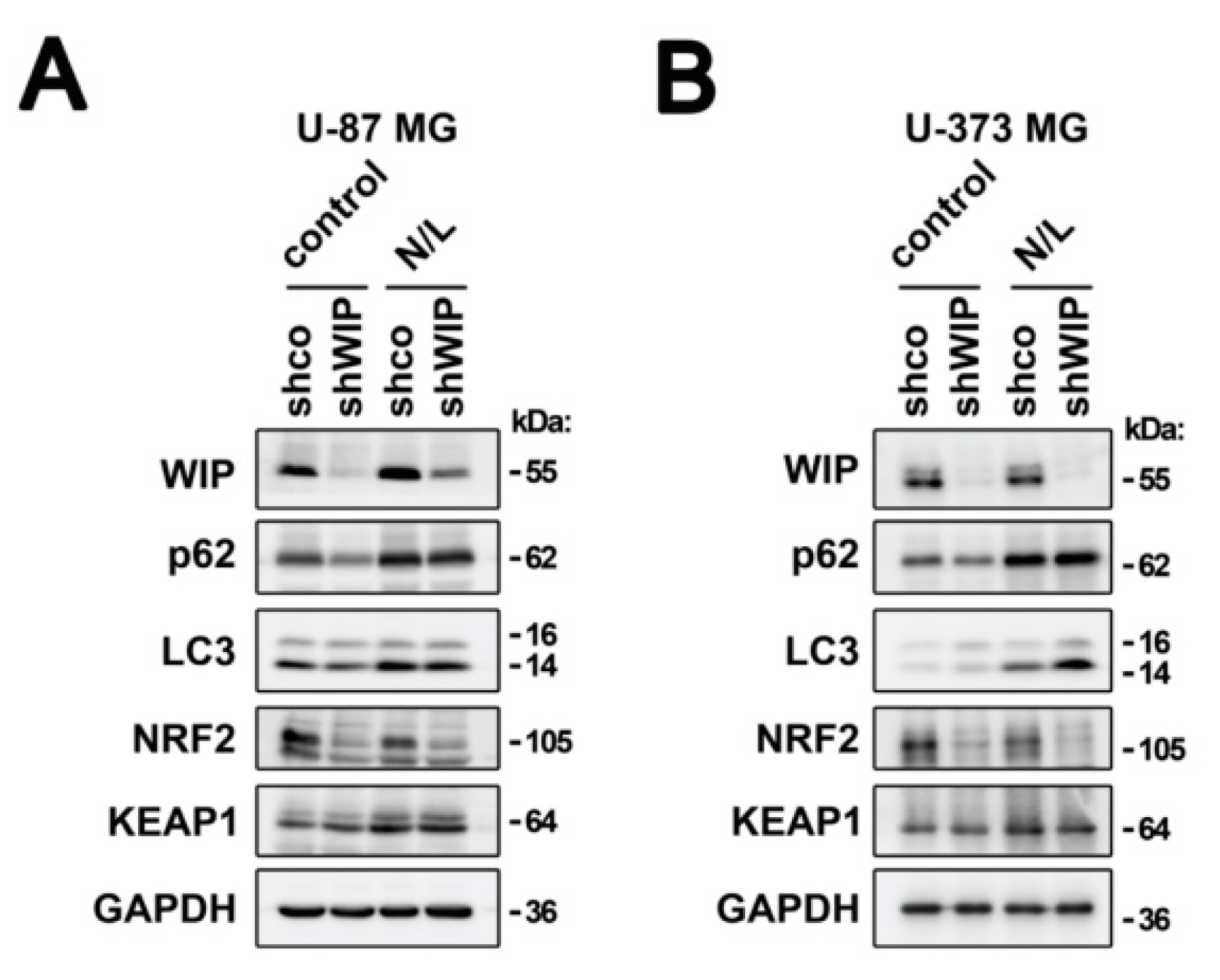
3.4. WIP Does Not Regulate KEAP1 Degradation
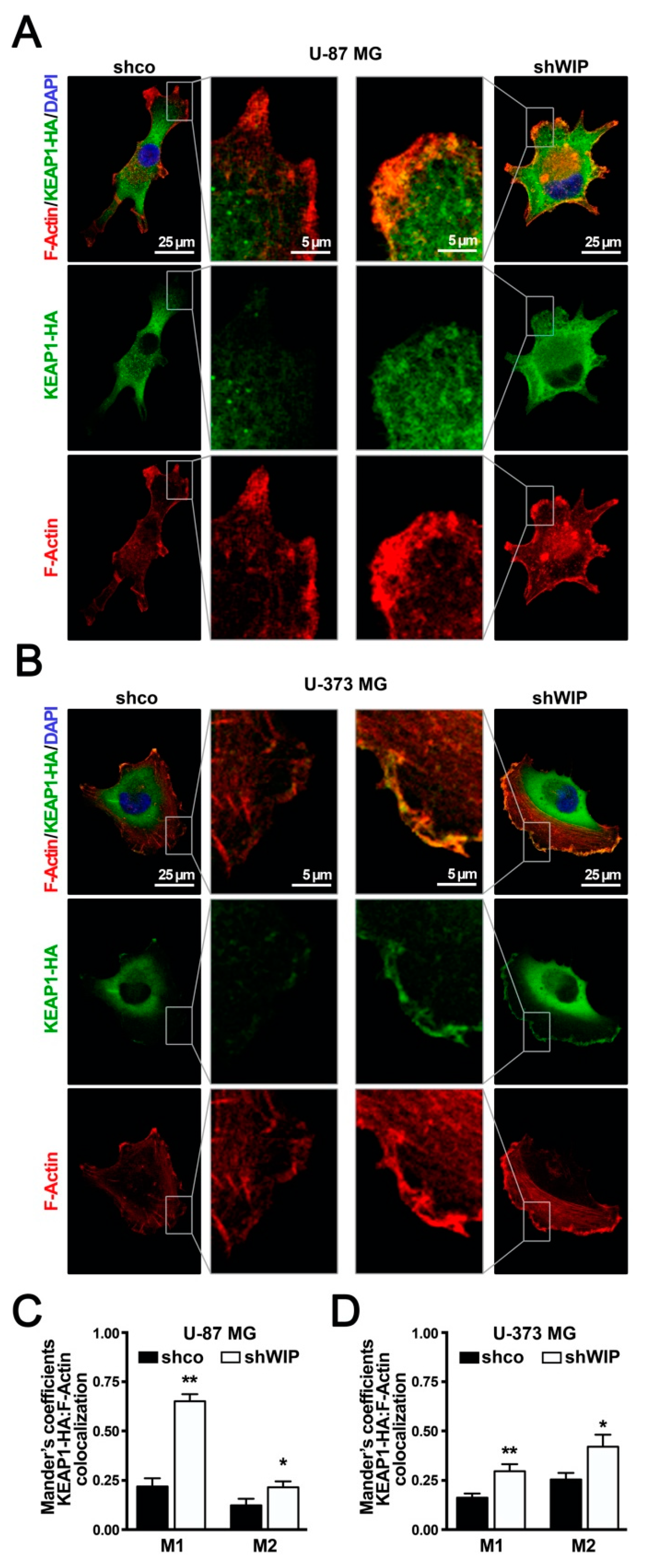
3.5. WIP-Depleted Cells Exhibit Increased Co-Localization between KEAP1 and F-Actin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Seetharaman, S.; Etienne-Manneville, S. Cytoskeletal crosstalk in cell migration. Trends Cell Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J. Mechanical tumor microenvironment and transduction: Cytoskeleton mediates cancer cell invasion and metastasis. Int. J. Biol. Sci. 2020, 16, 2014–2028. [Google Scholar] [CrossRef] [PubMed]
- Lehtimäki, J.; Hakala, M.; Lappalainen, P. Actin filament structures in migrating cells. Handb. Exp. Pharmacol. 2017, 235, 123–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, E.; Jones, G.E.; Machesky, L.M.; Antón, I.M. WIP: WASP-interacting proteins at invadopodia and podosomes. Eur. J. Cell Biol. 2012, 91, 869–877. [Google Scholar] [CrossRef]
- Antón, I.M.; Jones, G.E.; Wandosell, F.; Geha, R.; Ramesh, N. WASP-interacting protein (WIP): Working in polymerisation and much more. Trends Cell Biol. 2007, 17, 555–562. [Google Scholar] [CrossRef]
- Gargini, R.; Escoll, M.; García, E.; García-Escudero, R.; Wandosell, F.; Antón, I.M. WIP drives tumor progression through YAP/TAZ-dependent autonomous cell growth. Cell Rep. 2016, 17, 1962–1977. [Google Scholar] [CrossRef] [Green Version]
- Chou, H.-C.; Antón, I.M.; Holt, M.R.; Curcio, C.; Lanzardo, S.; Worth, A.; Burns, S.; Thrasher, A.J.; Jones, G.E.; Calle, Y. WIP regulates the stability and localization of WASP to podosomes in migrating dendritic cells. Curr. Biol. 2006, 16, 2337–2344. [Google Scholar] [CrossRef] [Green Version]
- Banon-Rodriguez, I.; Saez de Guinoa, J.; Bernardini, A.; Ragazzini, C.; Fernandez, E.; Carrasco, Y.R.; Jones, G.E.; Wandosell, F.; Anton, I.M. WIP regulates persistence of cell migration and ruffle formation in both mesenchymal and amoeboid modes of motility. PLoS ONE 2013, 8, e70364. [Google Scholar] [CrossRef] [Green Version]
- Antón, I.M.; de la Fuente, M.A.; Sims, T.N.; Freeman, S.; Ramesh, N.; Hartwig, J.H.; Dustin, M.L.; Geha, R.S. WIP deficiency reveals a differential role for WIP and the actin cytoskeleton in T and B cell activation. Immunity 2002, 16, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Escoll, M.; Gargini, R.; Cuadrado, A.; Anton, I.M.; Wandosell, F. Mutant p53 oncogenic functions in cancer stem cells are regulated by WIP through YAP/TAZ. Oncogene 2017, 36, 3515–3527. [Google Scholar] [CrossRef]
- Pan, Y.; Lu, F.; Xiong, P.; Pan, M.; Zhang, Z.; Lin, X.; Pan, M.; Huang, H. WIPF1 antagonizes the tumor suppressive effect of miR-141/200c and is associated with poor survival in patients with PDAC. J. Exp. Clin. Cancer Res. 2018, 37, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Hua, Y.; Qiu, H.; Hao, J.; Zou, K.; Li, Z.; Hu, S.; Guo, P.; Chen, M.; Sui, S.; et al. PD-L1 promotes tumor growth and progression by activating WIP and β-catenin signaling pathways and predicts poor prognosis in lung cancer. Cell Death Dis. 2020, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Staub, E.; Groene, J.; Heinze, M.; Mennerich, D.; Roepcke, S.; Klaman, I.; Hinzmann, B.; Castanos-Velez, E.; Pilarsky, C.; Mann, B.; et al. An expression module of WIPF1-coexpressed genes identifies patients with favorable prognosis in three tumor types. J. Mol. Med. 2009, 87, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, A.C.; Massagué, J. Molecular basis of metastasis. N. Engl. J. Med. 2008, 359, 2814–2823. [Google Scholar] [CrossRef] [Green Version]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive oxygen and nitrogen species-induced protein modifications: Implication in carcinogenesis and anticancer therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Gogvadze, V.; Norberg, E.; Venkatesh, R.; Orrenius, S.; Zhivotovsky, B. Reactive oxygen species generated in different compartments induce cell death, survival, or senescence. Free Radic. Biol. Med. 2013, 57, 176–187. [Google Scholar] [CrossRef]
- Vurusaner, B.; Poli, G.; Basaga, H. Tumor suppressor genes and ROS: Complex networks of interactions. Free Radic. Biol. Med. 2012, 52, 7–18. [Google Scholar] [CrossRef]
- Moon, D.-O.; Kim, M.-O.; Choi, Y.H.; Hyun, J.W.; Chang, W.Y.; Kim, G.-Y. Butein induces G(2)/M phase arrest and apoptosis in human hepatoma cancer cells through ROS generation. Cancer Lett. 2010, 288, 204–213. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pani, G.; Galeotti, T.; Chiarugi, P. Metastasis: Cancer cell’s escape from oxidative stress. Cancer Metastasis Rev. 2010, 29, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef]
- Robledinos-Antón, N.; Rojo, A.I.; Ferreiro, E.; Núñez, Á.; Krause, K.-H.; Jaquet, V.; Cuadrado, A. Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol. 2017, 13, 393–401. [Google Scholar] [CrossRef]
- MANDERS, E.M.M.; VERBEEK, F.J.; ATEN, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/β-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.-G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velichkova, M.; Hasson, T. Keap1 in adhesion complexes. Cell Motil. Cytoskelet. 2003, 56, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of NRF2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Kanamori, M.; Higa, T.; Sonoda, Y.; Murakami, S.; Dodo, M.; Kitamura, H.; Taguchi, K.; Shibata, T.; Watanabe, M.; Suzuki, H.; et al. Activation of the NRF2 pathway and its impact on the prognosis of anaplastic glioma patients. Neuro. Oncol. 2015, 17, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Escoll, M.; Lastra, D.; Pajares, M.; Robledinos-Antón, N.; Rojo, A.I.; Fernández-Ginés, R.; Mendiola, M.; Martínez-Marín, V.; Esteban, I.; López-Larrubia, P.; et al. Transcription factor NRF2 uses the Hippo pathway effector TAZ to induce tumorigenesis in glioblastomas. Redox Biol. 2020, 30, 101425. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Tang, Z.; Chen, D.; Moughon, D.; Ding, X.; Chen, S.; Zhu, M.; Zhong, Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 2010, 6, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.P.; Lobato-Márquez, D.; Pramanik, N.; Sirianni, A.; Daza-Cajigal, V.; Rivers, E.; Cavazza, A.; Bouma, G.; Moulding, D.; Hultenby, K.; et al. Wiskott-Aldrich syndrome protein regulates autophagy and inflammasome activity in innate immune cells. Nat. Commun. 2017, 8, 1576. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Araysi, L.M.; Fathallah-Shaykh, H.M. c-Src and neural Wiskott-Aldrich syndrome protein (N-WASP) promote low oxygen-induced accelerated brain invasion by gliomas. PLoS ONE 2013, 8, e75436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziv-Av, A.; Giladi, N.D.; Lee, H.K.; Cazacu, S.; Finniss, S.; Xiang, C.; Pauker, M.H.; Barda-Saad, M.; Poisson, L.; Brodie, C. RTVP-1 regulates glioma cell migration and invasion via interaction with N-WASP and hnRNPK. Oncotarget 2015, 6, 19826–19840. [Google Scholar] [CrossRef]
- Antón, I.M.; Jones, G.E. WIP: A multifunctional protein involved in actin cytoskeleton regulation. Eur. J. Cell Biol. 2006, 85, 295–304. [Google Scholar] [CrossRef]
- Martinez-Quiles, N.; Rohatgi, R.; Antón, I.M.; Medina, M.; Saville, S.P.; Miki, H.; Yamaguchi, H.; Takenawa, T.; Hartwig, J.H.; Geha, R.S.; et al. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nat. Cell Biol. 2001, 3, 484–491. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- van der Kammen, R.; Song, J.-Y.; de Rink, I.; Janssen, H.; Madonna, S.; Scarponi, C.; Albanesi, C.; Brugman, W.; Innocenti, M. Knockout of the Arp2/3 complex in epidermis causes a psoriasis-like disease hallmarked by hyperactivation of transcription factor Nrf2. Development 2017, 144, 4588–4603. [Google Scholar] [CrossRef] [Green Version]
- Sokolik, C.G.; Qassem, N.; Chill, J.H. The disordered cellular multi-tasker WIP and its protein-protein interactions: A structural view. Biomolecules 2020, 10, 1084. [Google Scholar] [CrossRef]
- Haider, N.; Larose, L. Activation of the PDGFRα-Nrf2 pathway mediates impaired adipocyte differentiation in bone marrow mesenchymal stem cells lacking Nck1. Cell. Commun. Signal. 2020, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, M.; Kuleva, N.; Hoffmann, R. Identification of cysteine, methionine and tryptophan residues of actin oxidized in vivo during oxidative stress. J. Proteome Res. 2010, 9, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Diaz, B.; Shani, G.; Pass, I.; Anderson, D.; Quintavalle, M.; Courtneidge, S.A. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci. Signal. 2009, 2, ra53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escoll, M.; Lastra, D.; Robledinos-Antón, N.; Wandosell, F.; Antón, I.M.; Cuadrado, A. WIP Modulates Oxidative Stress through NRF2/KEAP1 in Glioblastoma Cells. Antioxidants 2020, 9, 773. https://doi.org/10.3390/antiox9090773
Escoll M, Lastra D, Robledinos-Antón N, Wandosell F, Antón IM, Cuadrado A. WIP Modulates Oxidative Stress through NRF2/KEAP1 in Glioblastoma Cells. Antioxidants. 2020; 9(9):773. https://doi.org/10.3390/antiox9090773
Chicago/Turabian StyleEscoll, Maribel, Diego Lastra, Natalia Robledinos-Antón, Francisco Wandosell, Inés María Antón, and Antonio Cuadrado. 2020. "WIP Modulates Oxidative Stress through NRF2/KEAP1 in Glioblastoma Cells" Antioxidants 9, no. 9: 773. https://doi.org/10.3390/antiox9090773