Sulforaphane Inhibits MGO-AGE-Mediated Neuroinflammation by Suppressing NF-?B, MAPK, and AGE–RAGE Signaling Pathways in Microglial Cells



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of AGEs
2.3. AGEs Competitive ELISA Assay
2.4. AGEs Formation Inhibition Assay
2.5. AGE Breakdown Activity Assay
2.6. Nitrite Production and Cell Viability Assays
2.7. LDH Production Assay
2.8. ROS Production Assay
2.9. NF-κB Assay (Nuclear and Cytosolic Extraction)
2.10. Western Blot Analysis
2.11. ELISA
2.12. Statistical Analysis
3. Results
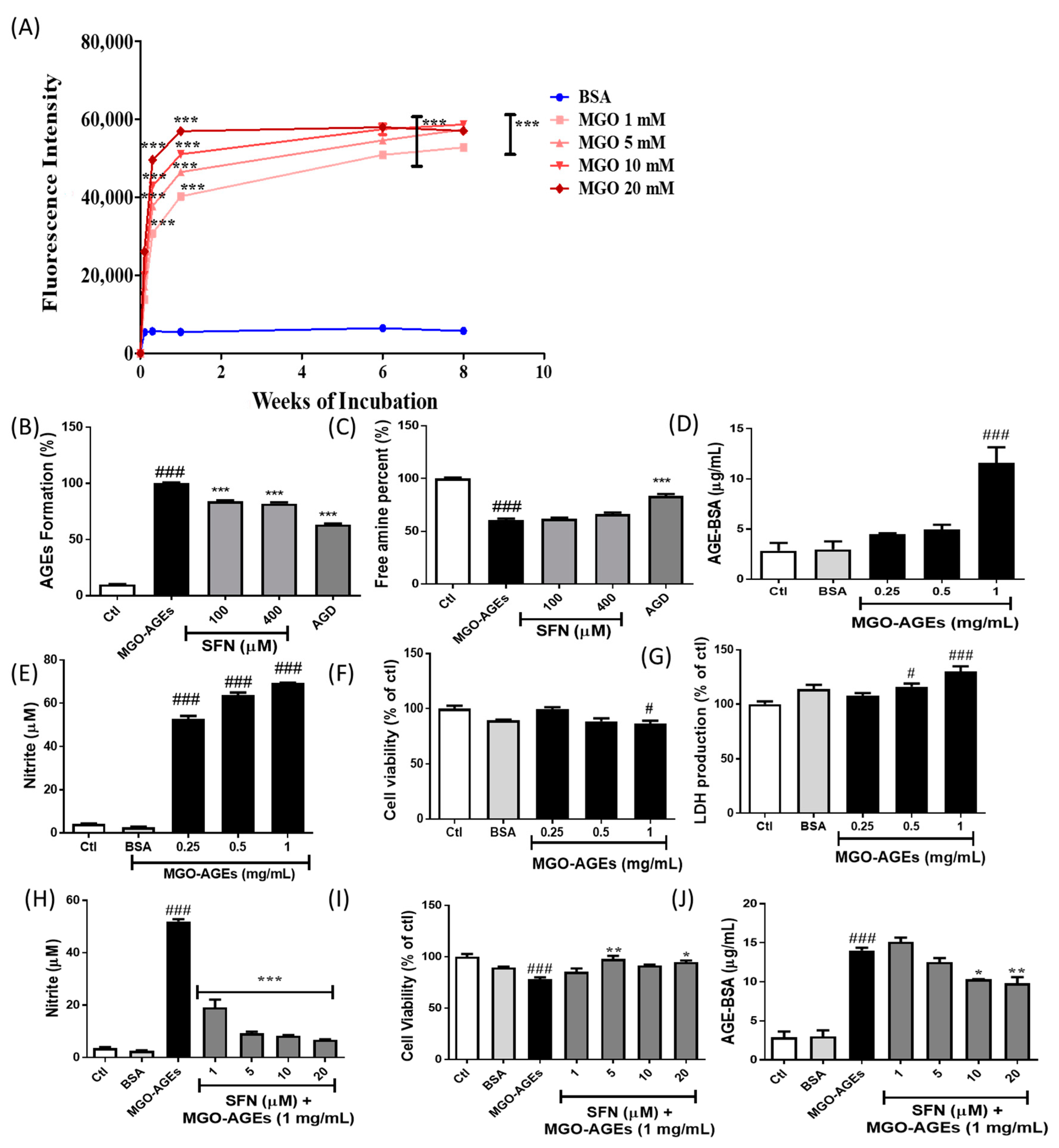
3.1. SFN Pretreatment Lowered MGO-AGEs Formation and Attenuated the Production of Neuroinflammatory Mediators Induced by MGO-AGE in BV2 Microglial Cells
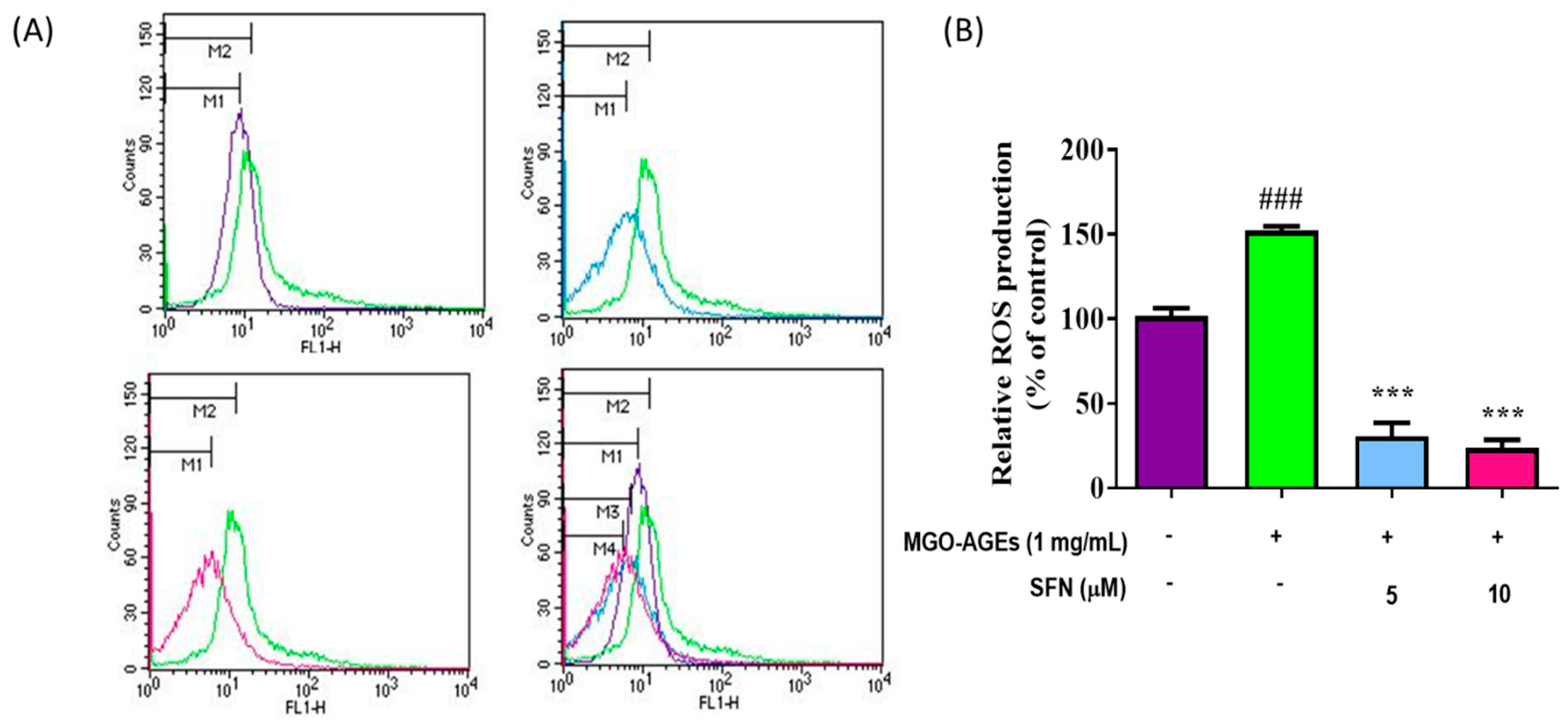
3.2. SFN Pretreatment Lowered Reactive Oxygen Species (ROS) Production Induced by MGO-AGEs in Microglial Cells
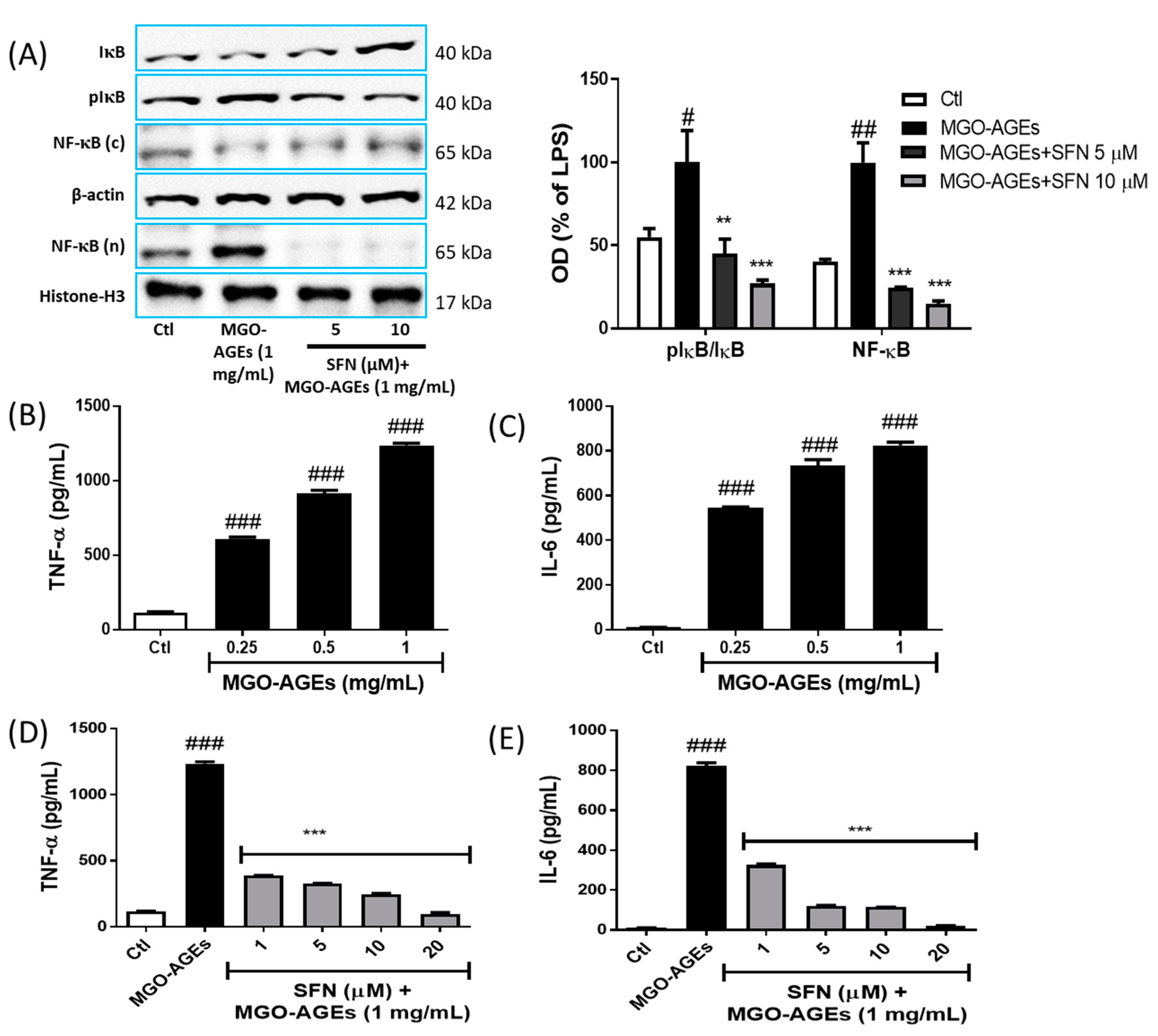
3.3. SFN Pretreatment Attenuated the Expression of Neuroinflammatory Proteins and MAPK Effector Signaling
3.4. SFN Pretreatment Reduced NF-κB Translocation and Proinflammatory Cytokine Production in MGO-AGEs-Activated Microglial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luevano-Contreras, C.; Chapman-Novakofski, K. Dietary advanced glycation end products and aging. Nutrients 2010, 2, 1247–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M. Serum Levels of Toxic AGEs (TAGE) May Be a Promising Novel Biomarker for the Onset/Progression of Lifestyle-Related Diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, M.; Kalantari, H.; Rezaei, M. Glyoxal toxicity in isolated rat liver mitochondria. Hum. Exp. Toxicol. 2018, 37, 532–539. [Google Scholar] [CrossRef]
- Lee, C.; Park, C. Bacterial Responses to Glyoxal and Methylglyoxal: Reactive Electrophilic Species. Int. J. Mol. Sci. 2017, 18, 169. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gkogkolou, P.; Bohm, M. Advanced glycation end products: Key players in skin aging? Dermatoendocrinology 2012, 4, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Basta, G.; Schmidt, A.M.; De Caterina, R. Advanced glycation end products and vascular inflammation: Implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res. 2004, 63, 582–592. [Google Scholar] [CrossRef]
- Papagrigoraki, A.; Maurelli, M.; Del Giglio, M.; Gisondi, P.; Girolomoni, G. Advanced Glycation End Products in the Pathogenesis of Psoriasis. Int. J. Mol. Sci. 2017, 18, 2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, K.E.; Prasad, C.; Vijayagopal, P.; Juma, S.; Imrhan, V. Advanced Glycation End Products, Inflammation, and Chronic Metabolic Diseases: Links in a Chain? Crit. Rev. Food Sci. Nutr. 2016, 56, 989–998. [Google Scholar] [CrossRef]
- Wetzels, S.; Wouters, K.; Schalkwijk, C.G.; Vanmierlo, T.; Hendriks, J.J. Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N. Y. Acad. Sci. 2011, 1243, 88–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Sun, Z.; Jin, M.; Tu, Y.; Wang, S.; Yang, X.; Chen, Q.; Zhang, X.; Han, Y.; Pi, R. Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-kappaB pathway. J. Neuroimmunol. 2017, 305, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Talalay, P. Chemical structures of inducers of nicotinamide quinone oxidoreductase 1 (NQO1). Methods Enzymol. 2004, 382, 423–448. [Google Scholar] [CrossRef]
- Eren, E.; Tufekci, K.U.; Isci, K.B.; Tastan, B.; Genc, K.; Genc, S. Sulforaphane Inhibits Lipopolysaccharide-Induced Inflammation, Cytotoxicity, Oxidative Stress, and miR-155 Expression and Switches to Mox Phenotype through Activating Extracellular Signal-Regulated Kinase 1/2-Nuclear Factor Erythroid 2-Related Factor 2/Antioxidant Response Element Pathway in Murine Microglial Cells. Front. Immunol. 2018, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Axelsson, A.S.; Tubbs, E.; Mecham, B.; Chacko, S.; Nenonen, H.A.; Tang, Y.; Fahey, J.W.; Derry, J.M.J.; Wollheim, C.B.; Wierup, N.; et al. Sulforaphane reduces hepatic glucose production and improves glucose control in patients with type 2 diabetes. Sci. Transl. Med. 2017, 9, eaah4477. [Google Scholar] [CrossRef] [Green Version]
- Sotokawauchi, A.; Ishibashi, Y.; Matsui, T.; Yamagishi, S.I. Aqueous Extract of Glucoraphanin-Rich Broccoli Sprouts Inhibits Formation of Advanced Glycation End Products and Attenuates Inflammatory Reactions in Endothelial Cells. Evid.-Based Complement. Altern. Med. 2018, 2018, 9823141. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Nakamura, N.; Ojima, A.; Nishino, Y.; Yamagishi, S.I. Sulforaphane reduces advanced glycation end products (AGEs)-induced inflammation in endothelial cells and rat aorta. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 797–807. [Google Scholar] [CrossRef]
- Maeda, S.; Matsui, T.; Ojima, A.; Takeuchi, M.; Yamagishi, S. Sulforaphane inhibits advanced glycation end product-induced pericyte damage by reducing expression of receptor for advanced glycation end products. Nutr. Res. 2014, 34, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Cho, K.; Park, Y.U.; Choi, H.J.; Kim, S.Y. Sulforaphane-Enriched Broccoli Sprouts Pretreated by Pulsed Electric Fields Reduces Neuroinflammation and Ameliorates Scopolamine-Induced Amnesia in Mouse Brain through Its Antioxidant Ability via Nrf2-HO-1 Activation. Oxidative Med. Cell. Longev. 2019, 2019, 3549274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-kappaB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Do, M.H.; Lee, J.H.; Jeong, M.; Lim, O.K.; Kim, S.Y. Inhibitory Effect of Arachis hypogaea (Peanut) and Its Phenolics against Methylglyoxal-Derived Advanced Glycation End Product Toxicity. Nutrients 2017, 9, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiho, T.; Kato, M.; Usui, S.; Hirano, K. Effect of buformin and metformin on formation of advanced glycation end products by methylglyoxal. Clin. Chim. Acta 2005, 358, 139–145. [Google Scholar] [CrossRef]
- Furlani, R.E.; Richardson, M.A.; Podell, B.K.; Ackart, D.F.; Haugen, J.D.; Melander, R.J.; Basaraba, R.J.; Melander, C. Second generation 2-aminoimidazole based advanced glycation end product inhibitors and breakers. Bioorg. Med. Chem. Lett. 2015, 25, 4820–4823. [Google Scholar] [CrossRef] [Green Version]
- Subedi, L.; Baek, S.H.; Kim, S.Y. Genetically Engineered Resveratrol-Enriched Rice Inhibits Neuroinflammation in Lipopolysaccharide-Activated BV2 Microglia Via Downregulating Mitogen-Activated Protein Kinase-Nuclear Factor Kappa B Signaling Pathway. Oxidative Med. Cell. Longev. 2018, 2018, 8092713. [Google Scholar] [CrossRef] [Green Version]
- Gaire, B.P.; Kwon, O.W.; Park, S.H.; Chun, K.H.; Kim, S.Y.; Shin, D.Y.; Choi, J.W. Neuroprotective effect of 6-paradol in focal cerebral ischemia involves the attenuation of neuroinflammatory responses in activated microglia. PLoS ONE 2015, 10, e0120203. [Google Scholar] [CrossRef]
- Subedi, L.; Gaire, B.P.; Do, M.H.; Lee, T.H.; Kim, S.Y. Anti-neuroinflammatory and neuroprotective effects of the Lindera neesiana fruit in vitro. Phytomedicine 2016, 23, 872–881. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Cho, K.S.; Jeon, S.J.; Kwon, O.W.; Jang, D.S.; Kim, S.Y.; Ryu, J.H.; Choi, J.W. Eupatilin exerts neuroprotective effects in mice with transient focal cerebral ischemia by reducing microglial activation. PLoS ONE 2017, 12, e0171479. [Google Scholar] [CrossRef] [Green Version]
- Subedi, L.; Teli, M.K.; Lee, J.H.; Gaire, B.P.; Kim, M.H.; Kim, S.Y. A Stilbenoid Isorhapontigenin as a Potential Anti-Cancer Agent against Breast Cancer through Inhibiting Sphingosine Kinases/Tubulin Stabilization. Cancers 2019, 11, 1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, L.; Lee, T.H.; Wahedi, H.M.; Baek, S.H.; Kim, S.Y. Resveratrol-Enriched Rice Attenuates UVB-ROS-Induced Skin Aging via Downregulation of Inflammatory Cascades. Oxidative Med. Cell. Longev. 2017, 2017, 8379539. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Venkatesan, R.; Kim, S.Y. Neuroprotective and Anti-Inflammatory Activities of Allyl Isothiocyanate through Attenuation of JNK/NF-kappaB/TNF-alpha Signaling. Int. J. Mol. Sci. 2017, 18, 1423. [Google Scholar] [CrossRef] [Green Version]
- Younessi, P.; Yoonessi, A. Advanced glycation end-products and their receptor-mediated roles: Inflammation and oxidative stress. Iran. J. Med. Sci. 2011, 36, 154–166. [Google Scholar]
- Hobbs, S.; Reynoso, M.; Geddis, A.V.; Mitrophanov, A.Y.; Matheny, R.W., Jr. LPS-stimulated NF-kappaB p65 dynamic response marks the initiation of TNF expression and transition to IL-10 expression in RAW 264.7 macrophages. Physiol. Rep. 2018, 6, e13914. [Google Scholar] [CrossRef] [Green Version]
- Aragno, M.; Mastrocola, R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 2017, 9, 385. [Google Scholar] [CrossRef] [Green Version]
- Maessen, D.E.; Hanssen, N.M.; Scheijen, J.L.; van der Kallen, C.J.; van Greevenbroek, M.M.; Stehouwer, C.D.; Schalkwijk, C.G. Post-Glucose Load Plasma alpha-Dicarbonyl Concentrations Are Increased in Individuals With Impaired Glucose Metabolism and Type 2 Diabetes: The CODAM Study. Diabetes Care 2015, 38, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzels, S.; Wouters, K.; Miyata, T.; Scheijen, J.; Hendriks, J.J.A.; Schalkwijk, C.G.; Vanmierlo, T. Advanced Glycation Endproducts Are Increased in the Animal Model of Multiple Sclerosis but Cannot Be Reduced by Pyridoxamine Treatment or Glyoxalase 1 Overexpression. Int. J. Mol. Sci. 2018, 19, 1311. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Wake, H.; Morioka, Y.; Liu, K.; Teshigawara, K.; Shibuya, M.; Zhou, J.; Mori, S.; Takahashi, H.; Nishibori, M. Phagocytosis of Advanced Glycation End Products (AGEs) in Macrophages Induces Cell Apoptosis. Oxidative Med. Cell. Longev. 2017, 2017, 8419035. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Takino, J.; Furuno, S.; Shirai, H.; Kawakami, M.; Muramatsu, M.; Kobayashi, Y.; Yamagishi, S. Assessment of the concentrations of various advanced glycation end-products in beverages and foods that are commonly consumed in Japan. PLoS ONE 2015, 10, e0118652. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Puentes, F.; Baker, D.; Van Der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Z.; Zhang, J.H.; Tang, J.; Liu, X.; Tan, L.; Huang, Q.Y.; Feng, H. Receptor for advanced glycation end-product antagonist reduces blood-brain barrier damage after intracerebral hemorrhage. Stroke 2015, 46, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Vasdev, S.; Gill, V.; Singal, P. Role of advanced glycation end products in hypertension and atherosclerosis: Therapeutic implications. Cell Biochem. Biophys. 2007, 49, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Tuttle, K.R. Advanced glycation end products and nephrotoxicity of high-protein diets. Clin. J. Am. Soc. Nephrol. 2006, 1, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Ji, E.; Shin, D.; Jin, J.; Yeo, J.H.; Kim, S.Y. Equol, a Dietary Daidzein Gut Metabolite Attenuates Microglial Activation and Potentiates Neuroprotection In Vitro. Nutrients 2017, 9, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Sun, P.; Zhang, J.C.; Zhang, Q.; Yao, S.L. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, R.; Kastrisianaki, E.; Giambanco, I.; Donato, R. S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J. Biol. Chem. 2011, 286, 7214–7226. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia Polarization with M1/M2 Phenotype Changes in rd1 Mouse Model of Retinal Degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Sathiya Priya, C.; Vidhya, R.; Kalpana, K.; Anuradha, C.V. Indirubin-3′-monoxime prevents aberrant activation of GSK-3beta/NF-kappaB and alleviates high fat-high fructose induced Abeta-aggregation, gliosis and apoptosis in mice brain. Int. Immunopharmacol. 2019, 70, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Yang, C.; Huang, W.; Du, S.; Mai, H.; Xiao, J.; Lu, T. Sulforaphane attenuates microglia-mediated neuronal necroptosis through down-regulation of MAPK/NF-kappaB signaling pathways in LPS-activated BV-2 microglia. Pharmacol. Res. 2018, 133, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol.-Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137–4142. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Araki, E. Mechanism-based antioxidant therapies promise to prevent diabetic complications? J. Diabetes Investig. 2013, 4, 105–107. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subedi, L.; Lee, J.H.; Gaire, B.P.; Kim, S.Y. Sulforaphane Inhibits MGO-AGE-Mediated Neuroinflammation by Suppressing NF-?B, MAPK, and AGE–RAGE Signaling Pathways in Microglial Cells. Antioxidants 2020, 9, 792. https://doi.org/10.3390/antiox9090792
Subedi L, Lee JH, Gaire BP, Kim SY. Sulforaphane Inhibits MGO-AGE-Mediated Neuroinflammation by Suppressing NF-?B, MAPK, and AGE–RAGE Signaling Pathways in Microglial Cells. Antioxidants. 2020; 9(9):792. https://doi.org/10.3390/antiox9090792
Chicago/Turabian StyleSubedi, Lalita, Jae Hyuk Lee, Bhakta Prasad Gaire, and Sun Yeou Kim. 2020. "Sulforaphane Inhibits MGO-AGE-Mediated Neuroinflammation by Suppressing NF-?B, MAPK, and AGE–RAGE Signaling Pathways in Microglial Cells" Antioxidants 9, no. 9: 792. https://doi.org/10.3390/antiox9090792
APA StyleSubedi, L., Lee, J. H., Gaire, B. P., & Kim, S. Y. (2020). Sulforaphane Inhibits MGO-AGE-Mediated Neuroinflammation by Suppressing NF-?B, MAPK, and AGE–RAGE Signaling Pathways in Microglial Cells. Antioxidants, 9(9), 792. https://doi.org/10.3390/antiox9090792