Abstract
Oxidative stress based on mitochondrial dysfunction is assumed to be the principal molecular mechanism for the pathogenesis of many neurodegenerative disorders. However, the effects of oxidative stress on the neurodegeneration process in living patients remain to be elucidated. Molecular imaging with positron emission tomography (PET) can directly evaluate subtle biological changes, including the redox status. The present review focuses on recent advances in PET imaging for oxidative stress, in particular the use of the Cu-ATSM radioligand, in neurodegenerative disorders associated with mitochondrial dysfunction. Since reactive oxygen species are mostly generated by leakage of excess electrons from an over-reductive state due to mitochondrial respiratory chain impairment, PET with 62Cu-ATSM, the accumulation of which depends on an over-reductive state, is able to image oxidative stress. 62Cu-ATSM PET studies demonstrated enhanced oxidative stress in the disease-related brain regions of patients with mitochondrial disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Furthermore, the magnitude of oxidative stress increased with disease severity, indicating that oxidative stress based on mitochondrial dysfunction contributes to promoting neurodegeneration in these diseases. Oxidative stress imaging has improved our insights into the pathological mechanisms of neurodegenerative disorders, and is a promising tool for monitoring further antioxidant therapies.
1. Introduction
In neurodegenerative disorders such as Parkinson’s disease, amyotrophic lateral sclerosis (ALS), and Alzheimer’s disease, increasing evidence from pathological and biochemical studies suggests that oxidative stress and mitochondrial dysfunction are the principal molecular mechanisms in the neurodegenerative process [1,2]. Since impaired mitochondria are a major source of reactive oxygen species (ROS) [3,4], oxidative stress is closely linked to mitochondrial dysfunction and has been assumed to play a crucial role in the pathogenesis of neurodegenerative disorders associated with mitochondrial dysfunction [5]. Indeed, many studies demonstrated an increase in oxidized molecules, reduced antioxidant capacity, and impaired mitochondrial metabolism in various neurodegenerative disorders [2,6,7]. In addition, both oxidative stress and mitochondrial dysfunction induce aggregation of misfolded proteins, such as amyloid-β, tau, and α-synuclein, which is the primary pathogenesis of neurodegenerative disorders [8,9].
While direct and non-invasive evaluation of the cerebral redox status is difficult to perform in living patients, molecular imaging technology with positron emission tomography (PET) has enabled visualization of oxidative stress in the brain [10,11,12,13]. PET imaging allows us to evaluate minuscule changes in biological phenomena and processes, such as receptor distribution and metabolic activity, at the molecular level by the administration of specific radioligands [14]. Our recent studies with PET imaging demonstrated increased oxidative stress in the disease-related brain regions of patients with neurodegenerative disorders associated with mitochondrial dysfunction, including mitochondrial disease, Parkinson’s disease, and ALS [10,11,12,13].
In this review, we focus on the recent achievements and future potential prospects of PET imaging for oxidative stress in neurodegenerative disorders.
2. Oxidative Stress and Mitochondrial Dysfunction
2.1. Oxidative Stress Caused by Reactive Oxygen Species
Oxidative stress is classically defined as an imbalanced redox state in which the oxidation effect caused by increased ROS production exceeds the defense capacity of the antioxidant mechanism [5]. Enhanced oxidative stress due to excess ROS generation leads to oxidative damage to the cellular components, such as proteins, lipids, and DNA. Additionally, ROS change the expression of nuclear factor kappa B (NF-κB), a transcription factor responsible for inducing inflammation and apoptosis [15]. These ROS-induced pathological mechanisms provoke tissue and organ dysfunction, especially neuronal degeneration in the brain [6,7]. ROS such as superoxide (O2−), hydroxyl radical (OH), and hydrogen peroxide (H2O2), are derived from molecular oxygen by the reduction. In particular, superoxide and hydroxyl radical are classified as free radicals, which show high chemical reactivity due to their unpaired electrons [3]. ROS are endogenously produced in the mitochondria, peroxisomes, and endoplasmic reticulum of cells [16]. Among these organelles, mitochondria, which consume more than 90% of intravital oxygen during oxidative phosphorylation (i.e., the aerobic metabolism), are regarded as the principal endogenous source of ROS [17,18]. However, under a healthy condition with normal mitochondrial function, the amount of ROS leakage is so small that it can be eliminated by the endogenous biological antioxidants, such as superoxide dismutase (SOD) and glutathione (GSH) [5,19].
2.2. Mitochondria as a Major Source of Reactive Oxygen Species
The mitochondrion is an organelle that produces adenosine triphosphate (ATP) as energy essential for life activities using the intrinsic respiratory chains. In the mitochondrial respiratory chains (a.k.a. electron transport chains), which consist of five complexes (i.e., complex I-V), electrons obtained as the reduced form of nicotinamide adenine dinucleotide (NADH) from the metabolism of glucose (i.e., glycolysis), free fatty acids (i.e., β-oxidation), and the tricarboxylic acid cycle are transported to synthesize ATP [3,20,21]. Most of the transferred electrons are ultimately captured by oxygen in the four-electron reduction whereby electrons and oxygen are detoxified to harmless and stable water molecules [22]. However, in respiratory chain impairment due to mitochondrial dysfunction, deteriorated electron transport provokes excessive accumulation of electrons relative to the amount of oxygen, resulting in an over-reductive state [23,24]. Because ROS are produced by the reduction of molecular oxygen, redundant electrons that leak from the impaired respiratory chains in an over-reductive state readily react with oxygen, which generates ROS [4,25]. A total of nine sites have been identified as the sources of mitochondrial ROS; complex I produces superoxide solely in the matrix, while complex III generates superoxide in both the matrix and the intermembrane space [26]. As explained above, mitochondrial respiratory chain impairment provokes an over-reductive state, and this state under the normoxic condition results in oxidative stress, which suggests that the evaluation of an over-reductive state using molecular imaging would be a promising marker for oxidative stress.
2.3. Oxidative Stress Based on Mitochondrial Dysfunction in Neurodegenerative Disorders
As mentioned above, mitochondrial respiratory chain impairment causes oxidative stress due to an over-reductive state, in addition to an ATP production deficit [4]. Since mitochondria are distributed throughout the body, mitochondrial dysfunction may cause failures of various organs. In particular, the brain consumes 20% of intravital oxygen and has a relatively fragile antioxidant capacity [27,28], which underlies the vulnerability of the neurons and glial cells to oxidative stress due to mitochondrial dysfunction [18,29]. Besides reduced respiratory capacity of mitochondria, there are other possible causes of oxidative stress in the brain, e.g., neuroinflammation, protein aggregation, and decreased antioxidant defenses [7,30]. Aging is also a major factor in promoting these pathological mechanisms, especially decreased mitochondrial function and antioxidant potential, leading to the enhancement of cerebral oxidative stress in elderly people [17]. These factors explain why the prevalence of neurodegenerative disorders increases with advancing age, and many pathological and biochemical studies have demonstrated enhanced oxidative stress in various neurodegenerative disorders [1,2]. Interestingly, basic studies showed that aggregated misfolded proteins induce mitochondrial dysfunction and ROS generation [31,32]. Conversely, ROS may facilitate neurotoxic protein aggregation, such as amyloid-β (in AD), α-synuclein (in PD) and SOD1 (in ALS), as well as mitochondrial impairment, producing a vicious cycle among oxidative stress, mitochondrial dysfunction and protein aggregation [8,9]. These findings may indicate that the increase in ROS production precedes the appearance of plaque deposits and that mitochondrial dysfunction can be an early event that precedes protein aggregation in neurodegenerative disorders. [1,33,34].
3. Oxidative Stress Imaging with 62Cu-ATSM PET
3.1. Accumulation Mechanism of 62Cu-ATSM
We developed an imaging technique for oxidative stress due to an over-reductive state using PET with 62Cu-diacetyl-bis(N4-methylthiosemicarbazone) (62Cu-ATSM), and succeeded in demonstrating increased oxidative stress in the brains of living patients with various neurodegenerative disorders [10,11,12,13]. 62Cu-ATSM, a PET radioligand, is a chelate complex that contains a radioactive divalent copper (62Cu2+) [35] (Figure 1). 62Cu-ATSM has high membrane permeability owing to its high lipophilicity and low molecular weight, allowing it to cross the blood-brain barrier [35]. The 62Cu2+ in this radioligand has two aspects. One is radioactivity (62Cu); the 62Cu emits positrons by β+ decay. The accumulation site of 62Cu-ATSM can be visualized by a PET scanner that captures the annihilation radiation pairs of the positrons. The other is divalence (Cu2+); due to reduction of the divalent form (Cu2+) to the monovalent form (Cu1+) in sites with excess electrons (i.e., an over-reductive state), the reduced Cu1+ dissociates from the ATSM complex and is irreversibly retained in the sites [35,36,37,38,39].
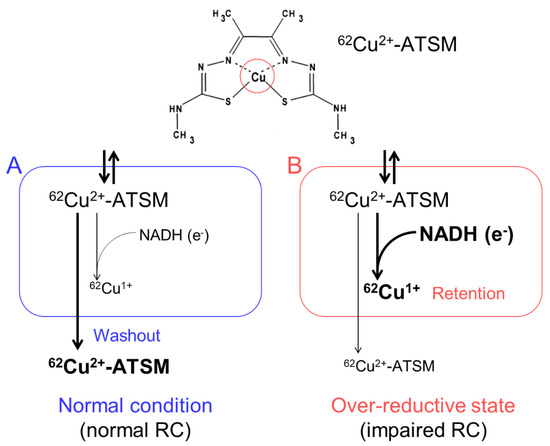
Figure 1.
The accumulation mechanism of 62Cu-ATSM: 62Cu-ATSM contains a radioactive divalent copper (62Cu2+) in the complex (circle in the upper panel). This radioligand is distributed by the blood flow after intravenous administration and readily penetrates cells. The distributed 62Cu-ATSM is rapidly washed out from the cells under the normal condition with intact mitochondria (A). In contrast, in sites with excess electrons supplied as the reduced form of nicotinamide adenine dinucleotide (NADH) (i.e., an over-reductive state) due to mitochondrial dysfunction, 62Cu-ATSM is retained in cells by a reduction of Cu2+ to Cu1+ (B). RC, mitochondrial respiratory chain.
Several radioactive copper isotopes besides 62Cu, such as 60Cu, 61Cu, 64Cu, and 67Cu, can be used for labeling this ligand. These isotopes have different half-lives and decay modes, and thus the ligand can be adapted to the purpose and setting [40]. The distribution of Cu-ATSM is not affected by the type of the labeled copper isotopes, except for the background activity which depends on the time required for ligand washout [38,41,42].
Cu-ATSM was originally developed to visualize hypoxic regions [35]. Since an over-reductive state is one in which cells and tissues have excessive levels of electrons relative to oxygen, both hypoxia (i.e., relatively decreased oxygen) and impaired respiratory chains (i.e., relatively increased electrons) induce this state [35,43,44]. Indeed, many studies have shown the utility of Cu-ATSM to image tumor hypoxia or ischemic myocardium [36,37,38,39,43,44,45,46,47,48]. While Cu-ATSM can be available as a hypoxia imaging ligand, several basic studies indicated that accumulation of Cu-ATSM also occurs in normoxic tissue according to the presence of an intracellular over-reductive state with high NADH concentrations [24,35,49,50,51] (see also Section 4.2). Since neuronal degeneration is basically a non-hypoxic process, the cerebral accumulation of Cu-ATSM in patients with neurodegenerative disorders is assumed to depend on an over-reductive state caused mainly by reduced respiratory capacity of mitochondria, which results in oxidative stress [13].
3.2. PET Procedure with 62Cu-ATSM
62Cu-ATSM is obtained by simple mixing of generator eluate (62Cu-glycine) and ATSM solution (0.5 mM in dimethyl sulfoxide) [11,35,47,52]. The radiochemical purity of 62Cu-ATSM is confirmed by high-performance liquid chromatography (HPLC). 62Cu is eluted from a 62Zn/62Cu positron generator; the half-lives of 62Cu and the parent 62Zn are approximately 10 min and 9 h, respectively. Owing to the shorter half-life of 62Cu and the longer half-life of the parent 62Zn, 62Cu-ATSM can be eluted from a generator system every hour, permitting repeated PET scans during a whole day in clinical use [35,36].
PET scans with 62Cu-ATSM for the whole brain are performed for dynamic frames of 20 min or longer after intravenous bolus injection of 444-740 MBq 62Cu-ATSM [10,11,12,52]. The obtained PET data are usually reconstructed into static images in two phases: Early (up to 3 min) and delayed (10-20 min after injection). They are then converted into semiquantitative images with a unit of standardized uptake value (SUV), which is the tissue radioactivity concentration normalized by the injection dose and body weight (tissue concentration of radioactivity/[injection dose/body weight]) [13].
After intravenous injection, 62Cu-ATSM is distributed throughout the body including the brain by the blood flow in the early phase within 3 min. This radioligand readily penetrates cells but is rapidly washed out under normal conditions [10,35,36,52]. Contrastingly, in sites with an over-reductive state, this radioligand is retained in the delayed phase, approximately 7 min after injection [10,44]. Based on the aforementioned distribution property of 62Cu-ATSM, the accumulation of 62Cu-ATSM in early-phase images (obtained up to 3 min after injection) represents the blood flow distribution in the brain, whereas its accumulation in delayed-phase images (obtained 10–20 min after injection) reflects an over-reductive state [10,11,12,52]. Thus, in neurodegenerative disorders, delayed-phase images of 62Cu-ATSM PET indicate increased oxidative stress due to an over-reductive state caused mainly by mitochondrial dysfunction.
4. Oxidative Stress Imaging in Mitochondrial Disease
4.1. Oxidative Stress in Mitochondrial Diseases
Mitochondria incorporate mitochondrial DNA (mtDNA) as its own genome, which partially encodes the enzyme complexes of the respiratory chain [53]. Thus, mutations in mtDNA may induce mitochondrial dysfunction with impaired respiratory chains, which causes inherited mitochondrial diseases [54]. Among the mitochondrial diseases with mtDNA mutations, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome (OMIM 540000) is one of the most common phenotypes and is mainly caused by the A-to-G transition mutation at nucleotide position 3243 in mtDNA (m.3243A>G) [55]. Various characteristic symptoms, such as stroke-like episodes (SEs), myopathy, cardiomyopathy, diabetes, and hearing loss, are frequently observed in patients with MELAS syndrome [56].
Since mitochondrial dysfunction results in increased ROS production, enhancement of oxidative stress is assumed in mitochondrial diseases including MELAS [57], and increased oxidative stress in the blood of patients with MELAS has been observed [58]. Recent in vitro studies also showed respiratory defects and increased ROS generation in human cell lines (cybrids) carrying mutated mitochondria derived from a patient with MELAS harboring m.3243A>G [4,59], which suggests that oxidative stress is closely associated with the pathogenesis of mitochondrial diseases.
4.2. In Vitro Studies with 64Cu-ATSM in MELAS Model Cells
To demonstrate the feasibility of 62Cu-ATSM as a PET radioligand for imaging oxidative stress due to an over-reductive state based on mitochondrial dysfunction, in vitro and in vivo studies were performed on MELAS cybrids [10,24]. Since cybrids harboring m.3243A>G closely replicate the pathophysiological conditions of MELAS [4,59], the in vitro accumulation of 64Cu-ATSM, having a longer half-life (13 h) than 62Cu, was evaluated in MELAS cybrids with an intracellular over-reductive state caused by mitochondrial respiratory defects [24,51]. These in vitro studies showed 1.5-fold increased retention of 64Cu-ATSM in the MELAS cybrids compared with wild-type cybrids having normal mitochondria under normoxia. 64Cu-ATSM retention significantly increased with the intracellular levels of NADH, the biological reductant as an electron donor, in the cybrids, which suggests that Cu-ATSM accumulation reflects an intracellular over-reductive state caused by mitochondrial dysfunction [24]. Since ROS generation is enhanced in the MELAS cybrids with an over-reductive state [4,59], increased Cu-ATSM accumulation indicates enhanced oxidative stress due to an over-reductive state based on mitochondrial dysfunction.
4.3. PET Imaging for Oxidative Stress in Patients with Stroke-Like Episodes of MELAS
Alongside the above in vitro studies, PET imaging with 62Cu-ATSM was performed in a patient with MELAS carrying m.3243A>G who had brain lesions caused by SEs, to evaluate the clinical utility of 62Cu-ATSM PET for detecting cerebral oxidative stress based on mitochondrial dysfunction in vivo [10].
SEs occur repeatedly and determine the prognosis of patients with MELAS [56,60]. Several pathophysiological hypotheses have been proposed for SEs [61]: Mitochondrial angiopathy (endothelial dysfunction in cerebral blood vessels) [62,63], cytopathy (neuronal dysfunction due to mitochondrial metabolic failure) [64], and neuronal hyperexcitability [65]. The pathological findings, such as an increased number of abnormal mitochondria in the endothelial cells and disruption of the endothelial tight junctions in SE lesions [62,66], support these hypotheses. In addition, recent studies using magnetic resonance (MR) imaging with 1H-MR spectroscopy, apparent diffusion coefficient (ADC) maps, MR angiography, and arterial spin labeling (ASL) methods showed hyperperfusion and vasogenic edema with vasodilatation and lactic acid fermentation in acute lesions of SEs [67,68,69,70,71], which also suggests the involvement of mitochondrial angiopathy and cytopathy in the pathogenesis of SEs. Moreover, PET imaging with 15O tracers demonstrated a significant reduction in both the oxygen extraction fraction (OEF) and cerebral metabolic rate of oxygen (CMRO2) in acute SE lesions [72], which also indicates impaired mitochondrial oxygen consumption (i.e., mitochondrial cytopathy). Besides these pathophysiological hypotheses, oxidative stress caused by mitochondrial dysfunction has been assumed to play a crucial role in the pathogenesis of SEs [57]. A postmortem study showing an increase in the level of 8-hydroxy-2′-deoxyguanosine (8-OHdG; DNA oxides) in SE lesions supports this assumption [73].
In the study described above, the patient with MELAS underwent double brain PET imaging with 62Cu-ATSM and 18F-fluorodeoxyglucose (18F-FDG) to evaluate oxidative stress and the glucose metabolism, respectively [10]. At the time of the PET scans, the patient had three SE lesions: An acute lesion that occurred one day before the scans, a subacute lesion that developed one month before, and a chronic lesion that appeared half a year before. 18F-FDG PET demonstrated increased uptake in the acute lesion, but the subacute and chronic lesions showed decreased uptake. In contrast, 62Cu-ATSM PET revealed significant accumulation in the subacute lesion (1.8-fold SUV increase compared with that in the acute lesion) (Figure 2). The acute lesion showed slightly increased uptake of 62Cu-ATSM, and decreased uptake was observed in the chronic lesion. These findings indicate that enhanced glycolysis is followed by increased oxidative stress due to an over-reductive state, which eventually leads to neuronal cell death in SEs [10]. Combined with the results of imaging studies, we proposed the pathophysiological process of SEs as Figure 2 [10,74]. In addition to elucidating the pathogenesis of SEs, this PET study demonstrates the feasibility of 62Cu-ATSM PET for the evaluation of cerebral oxidative stress based on mitochondrial dysfunction in living patients.
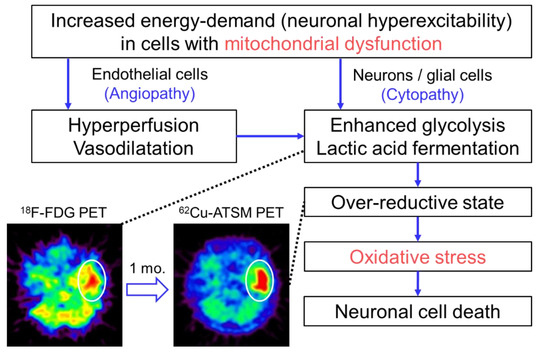
Figure 2.
The pathophysiological process of stroke-like episodes (SEs) in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) hypothesized based on imaging studies. Brain positron emission tomography (PET) with 18F-FDG exhibited increased uptake in the acute SE lesion (circle) in a patient with MELAS who had an SE attack immediately before the PET scan. Interestingly, the lesion showed increased accumulation of 62Cu-ATSM on PET imaging (circle) performed one month after the SE attack (i.e., subacute phase). These imaging findings suggest that enhanced glycolysis is followed by increased oxidative stress due to an over-reductive state based on mitochondrial dysfunction in the pathophysiological process of SEs.
5. Oxidative Stress Imaging in Parkinson’s Disease
5.1. Oxidative Stress and Mitochondrial Dysfunction in Parkinson’s Disease
Parkinson’s disease is the most common motor neurodegenerative disorder, with a prevalence of more than 1% in elderly individuals [75]. The pathological findings are characterized by the degeneration of dopaminergic neurons in the nigrostriatal system with the appearance of Lewy bodies composed of α-synuclein aggregation in the remaining neurons [76]. While various pathogenetic molecular mechanisms as well as genetic and environmental factors are believed to be associated with the cause and pathogenesis, a number of pathobiochemical and genetic studies have indicated that oxidative stress and mitochondrial dysfunction play a major role in the nigrostriatal neurodegeneration in Parkinson’s disease (Figure 3) [77,78].
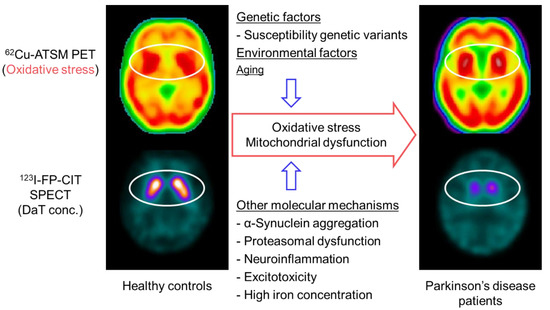
Figure 3.
Brain positron emission tomography (PET) imaging with 62Cu-ATSM for oxidative stress and putative molecular mechanisms in patients with Parkinson’s disease. Single-photon emission computed tomography (SPECT) with 123I-FP-CIT, which reflects the density of dopamine transporters (DaT) in the striatal dopaminergic nerve terminals, showed decreased uptake in the striatum of patients with Parkinson’s disease compared with that of healthy controls (circles in the lower images). In contrast, 62Cu-ATSM PET revealed increased striatal accumulation in patients with Parkinson’s disease compared with healthy controls, which suggests enhanced oxidative stress in the striatal dopaminergic neurons in Parkinson’s disease (circles in the upper images).
Since the endogenous dopamine metabolism with a high iron concentration in the nigrostriatal neurons inevitably induces auto-oxidation of dopamine to generate dopamine quinones and ROS [79,80], the involvement of oxidative stress in the pathogenesis of Parkinson’s disease has been assumed. In fact, multiple postmortem studies showed increases in the levels of 8-OHdG, protein carbonyls, 4-hydroxy-2-nonenal (4-HNE) histidine, or lipid peroxidation products (i.e., oxides of DNA, proteins, and lipids, respectively) in the nigrostriatal system of patients with Parkinson’s disease [81,82,83]. Changes in the levels of antioxidant molecules, such as manganese SOD and GSH, were demonstrated in other pathological studies [84,85]. Interestingly, a recent study using a mouse model with α-synuclein overexpression showed that oxidative stress induced by paraquat exposure resulted in the aggregation and propagation of α-synuclein, and marked neurodegeneration [86]. These findings suggest that oxidative stress contributes to the pathological processes of Parkinson’s disease.
The involvement of mitochondrial respiratory dysfunction in the pathogenesis of Parkinson’s disease has been strongly suggested by postmortem and in vivo model studies [77,87], such as activity deficiency of the mitochondrial respiratory chain complex I in the substantia nigra of patients and an animal model induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), an inhibitor of complex I [88,89,90]. In addition, genetic studies have revealed that many causal genes of monogenic Parkinson’s disease, such as Parkin (PARK2) [91], PINKl (PARK6) [92], DJ-1 (PARK7) [93], Omi/HtrA2 (PARK13) [94], and CHCHD2 (PARK22) [95,96], are associated with mitochondrial functions and quality control [77,97]. Especially, PINK1 recruits Parkin to induce autophagic degradation of the impaired mitochondria with low membrane potential (i.e., mitophagy), which suggests that these genetic products are instrumental for mitochondrial quality control [98,99].
Similar to Parkinson’s disease, a recent study revealed COQ2 gene mutations in patients with familial multiple-system atrophy, which presents with Parkinsonian symptoms due to nigrostriatal degeneration [100]. COQ2 is essential for the biosynthesis of coenzyme Q10 [101], a key component in mitochondrial respiratory chains, which also indicates the involvement of mitochondrial dysfunction in the dopaminergic neurodegeneration. Thus, these pathological and genetic studies strongly suggest that mitochondrial respiratory chain impairment leading to oxidative stress is one of the principal pathogenetic mechanisms in Parkinson’s and its related diseases. However, the effects of oxidative stress due to mitochondrial failure on the neurodegeneration process in living patients remain unknown.
5.2. PET Imaging for Oxidative Stress in Patients with Parkinson’s Disease
Based on the oxidative stress hypothesis, a PET study with 62Cu-ATSM was performed to investigate cerebral oxidative stress in patients with sporadic Parkinson’s disease [11]. This PET study found a significant increase in 62Cu-ATSM uptake in the bilateral striata in the patient group as compared to that in healthy subjects (6% increase in the striatum-to-cerebellum SUV ratio) (Figure 3). The uptake in the striatum was positively correlated with the clinical severity in patients, as estimated by the Unified Parkinson’s Disease Rating Scale (UPDRS) score. These findings strongly indicate the involvement of oxidative stress based on mitochondrial dysfunction in the neurodegeneration process in Parkinson’s disease [11,13]. However, the density of the remaining nigrostriatal dopaminergic neurons decreases markedly with the progression of Parkinson’s disease [102], which may offset the striatal uptake of 62Cu-ATSM in patients with advanced Parkinson’s disease.
To resolve this problem, single-photon emission computed tomography (SPECT) with 123I-N-ω-fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl)nortropane (123I-FP-CIT) was performed along with 62Cu-ATSM PET in patients with Parkinson’s disease in another study (Figure 3) [103]. 123I-FP-CIT accumulates in the dopamine transporters that are highly expressed in the striatal dopaminergic nerve terminals. The striatal 123I-FP-CIT binding thereby approximately reflects the density of the remaining striatal pre-synaptic dopaminergic neurons [104]. Thus, the 62Cu-ATSM uptake levels corrected by 123I-FP-CIT binding levels for the striatum may precisely indicate oxidative stress in the remaining striatal neurons. The corrected 62Cu-ATSM uptake significantly increased with disease severity, as estimated by the UPDRS scores in patients with Parkinson’s disease, when compared with the non-corrected 62Cu-ATSM uptake [103]. These results suggest that oxidative stress is enhanced in the remaining dopaminergic neurons, which may facilitate the dopaminergic neurodegenerative process even in advanced Parkinson’s disease.
According to the above substantial evidence implicating oxidative stress based on mitochondrial dysfunction in the pathogenesis of Parkinson’s disease, many pharmaceutical molecules having effects of oxidative stress reduction and/or mitochondrial function improvement have been developed to modify the disease course [78]. While only a few agents, such as inosine (a precursor of urate) and the reduced form of CoQ10, have shown possible therapeutic efficiency in patients with Parkinson’s disease [105,106,107], the results of 62Cu-ATSM PET studies suggest that optimized antioxidative agents would be effective even in the advanced disease stage. Interestingly, Hung et al. showed the therapeutic effects of non-radioactive Cu-ATSM in multiple animal models of Parkinson’s disease [108]. Rodent models treated with oral administration of Cu-ATSM exhibited restoration of motor function and dopamine biosynthesis and the prevention of nigral dopaminergic cell death. Besides its utility as an imaging ligand for oxidative stress, these findings suggest the therapeutic potential of Cu-ATSM as an antioxidant in Parkinson’s disease.
5.3. PET Imaging for Neuroinflammation and Mitochondrial Activity in Parkinson’s Disease
Neuroinflammation mediated by the activated microglia is another principal molecular mechanism of the neurodegenerative process [109]. Oxidative stress and mitochondrial dysfunction are induced by neuroinflammation [110,111]; conversely, ROS induces inflammation via the activation of NF-κB [15], which suggests an inextricable relationship between these pathological factors [7]. Several PET studies with radioligands for 18-kDa translocator protein (TSPO), which is highly expressed in activated microglia in the brain [112], such as 11C-(R)-PK11195 and 11C-DPA713, revealed increased cortical uptake in patients with Parkinson’s disease, especially in the early stage of the disease [113,114,115]. While recent PET studies with other TSPO radioligands, 11C-PBR28 and 18F-FEPPA, showed conflicting results [116,117], the PET studies that yielded positive results indicated that neuroinflammation due to microglia activation occurs in the early phase of neurodegeneration in Parkinson’s disease [113,114,115]. These findings of neuroinflammation in the early stage contrast with the 62Cu-ATSM PET studies showing that oxidative stress increases with disease progression [11,13,103]. The relationship between oxidative stress and neuroinflammation in the pathophysiological process of Parkinson’s disease requires further clarification [118]. Interestingly, Metzger et al. showed increased uptake of both 11C-PBR28 and 61Cu-ATSM in the myocardium one week after systemic administration of 6-hydroxydopamine (6-OHDA), which causes cardiac catecholaminergic neuronal degeneration due to induction of oxidative stress and inflammation, in non-human primates [119]. While it was a cardiac investigation, this study suggested that oxidative stress and neuroinflammation arise concurrently, at least in the acute intoxication model.
Recently, a promising PET radioligand, 18F-BCPP-EF, for imaging of the activity of mitochondrial complex I has been developed [120]. A preclinical PET study with 18F-BCPP-EF showed decreased uptake in the cortex and basal ganglia in non-human primates treated with MPTP as an animal model of Parkinson’s disease [121]. While clinical studies with 18F-BCPP-EF PET are still ongoing [122], a direct comparison between mitochondrial activity and oxidative stress is anticipated in patients with Parkinson’s disease using these PET imaging techniques.
6. Oxidative Stress Imaging in Amyotrophic Lateral Sclerosis (ALS)
6.1. Oxidative Stress and Mitochondrial Dysfunction in ALS
ALS is an intractable neurodegenerative disorder characterized by progressive degeneration of both upper and lower motor neurons [123]. Various pathophysiological mechanisms have been assumed to be involved in the pathogenesis of ALS, including glutamate-induced excitotoxicity, cytoplasmic protein aggregates, autophagy, disrupted axonal transport system, inflammation caused by activated microglia, RNA processing defects, and endoplasmic reticulum stress (Figure 4) [123,124]. In addition to these factors, a number of investigations have indicated that oxidative stress based on mitochondrial dysfunction plays a principal role in the motor neuron degeneration in ALS [125,126]. Multiple postmortem studies have demonstrated the accumulation of cellular component oxides (i.e., DNA, proteins, and lipids) indicated by increases in the levels of 8-OHdG, protein carbonyls, or 4-HNE histidine in the motor cortex and/or spinal cord in patients with ALS [127,128,129,130]. Similarly, biochemical studies showed increased concentrations of 8-OHdG or 4-HNE in patients’ blood, urine, or cerebrospinal fluid [131,132,133]. Genetic factors, especially SOD1 and TDP-43 gene mutations frequently found in familial ALS, also indicate the involvement of oxidative stress in the pathogenesis [134]. Linked with oxidative stress, postmortem investigations showed mitochondrial alterations and mitochondrial respiratory chain impairment in the spinal cord or muscles in patients with ALS [135,136]. Mitochondrial abnormalities were also observed in transgenic mice and cell culture models carrying SOD1 gene mutations [137]. These biopathological and genetic studies provide collateral evidence, but cannot directly evaluate regional changes of the redox status in living patients with ALS.
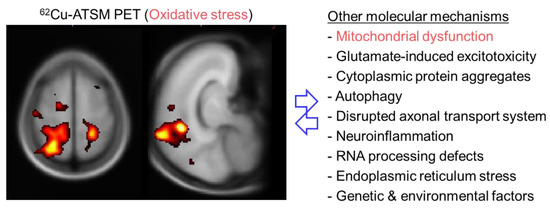
Figure 4.
Brain positron emission tomography (PET) imaging with 62Cu-ATSM for oxidative stress and putative molecular mechanisms in patients with amyotrophic lateral sclerosis (ALS). The left panel shows the T-map generated by statistical parametric mapping analysis, displaying the regions in which 62Cu-ATSM accumulation was higher in patients with ALS than in healthy controls. A significantly greater accumulation of 62Cu-ATSM in patients with ALS than in controls was observed in the bilateral cortices around the central sulcus, including the motor cortex, and the right superior parietal lobule. These results demonstrate increased oxidative stress, primarily in the motor cortex, in patients with ALS.
6.2. PET Imaging for Oxidative Stress in Patients with ALS
To evaluate cerebral oxidative stress and its relationship with the clinical features, a PET study with 62Cu-ATSM was performed in patients with sporadic ALS [12]. This study demonstrated a significantly greater accumulation of 62Cu-ATSM in patients with ALS than in healthy controls, mainly in the bilateral cortices around the central sulcus, including the motor cortex and motor-related parietal areas (9% SUV increase after global normalization in the bilateral cortices around the central sulcus) (Figure 4). Furthermore, 62Cu-ATSM accumulation in these regions correlated positively with the clinical severity in patients, as estimated by the revised ALS Functional Rating Scale (ALSFRS-R) score [12]. These findings indicated that increased oxidative stress in motor and motor-related cortices strongly correlated with the disease severity in patients with ALS, which is consistent with biochemical studies showing that serum 4-HNE or urine 8-OHdG levels increased with clinical severity in patients with ALS [131,133]. Thus, the results of 62Cu-ATSM PET imaging successfully indicated that oxidative stress based on mitochondrial dysfunction is associated with motor neuron degeneration in ALS.
Focusing on the involvement of oxidative stress in the pathogenesis of ALS, some therapeutic agents showing antioxidant effects have been developed [138]. The therapeutic efficacy of edaravone, a free radical scavenger, has been demonstrated in patients with ALS [139,140]. In addition, similar to Parkinson’s disease, several therapeutic studies yielded positive outcomes of non-radioactive Cu-ATSM in SOD1-mutant mouse models of ALS, including improved locomotive function and overall survival [141,142]. Cu-ATSM has the potential to detoxify mutant SOD1 by supplying Cu, as well as antioxidant activity inhibiting lipid peroxidation and iron accumulation (i.e., ferroptosis), which may result in neuroprotective effects [143,144,145]. Based on these basic studies, a Phase 2 clinical trial using Cu-ATSM in patients with ALS commenced in Australia in 2019 (NCT04082832). Thus, in addition to its imaging utility, Cu-ATSM is also a potential therapeutic agent for ALS [146].
6.3. PET Imaging for Other Factors Associated with Oxidative Stress in ALS
Besides oxidative stress, many pathological molecular factors have been investigated using PET imaging in patients with ALS [147,148]. Interestingly, several PET studies to image neuroinflammation using TSPO radioligands, such as 11C-(R)-PK11195, 18F-DPA714, and 11C-PBR28, showed increased uptake mainly in the primary motor cortex in ALS patients [149,150,151,152]. Taken together with the 62Cu-ATSM PET results, this indicates that oxidative stress and neuroinflammation may occur concurrently in the brain and may contribute together to neurodegeneration in ALS.
In contrast to neuroinflammation, 18F-FDG PET studies showed a decreased glucose metabolism in the primary motor cortex in patients with ALS [153,154,155]. Based on the aforementioned PET study in MELAS showing increased 18F-FDG uptake followed by elevated 62Cu-ATSM accumulation in SE lesions [10], a period of enhanced oxidative stress may be inconsistent with that of hypermetabolism in the cerebral cortex. Unlike the upper motor neurons of the motor cortex, several studies showed increased 18F-FDG uptake in the brainstem and spinal cord, which include lower motor neurons, in patients with ALS [153,155,156,157]. The uptake value correlated positively with disease progression, which suggests a difference in metabolic mechanism between these regions and the cerebral cortex [155].
7. Application of 62Cu-ATSM PET to Cerebrovascular Diseases
7.1. Pathophysiological Changes in Brain Misery Perfusion
Chronic steno-occlusive changes in the major cerebral arteries are known to be a risk factor for ischemic cerebral infarction [158]. Especially, the region with “misery perfusion” that exhibits decreased cerebral blood flow (CBF) relative to normal oxygen consumption shows a high risk of infarction [159]. In misery perfusion, the OEF is increased to sustain the aerobic energy metabolism in mitochondria (i.e., CMRO2) (Figure 5); however, chronic ischemic changes gradually induce mitochondrial dysfunction [160,161]. Thus, enhanced OEF with mild mitochondrial respiratory chain impairment may induce an over-reductive state in chronic misery perfusion, which would ultimately lead to oxidative stress and neuronal damage [158,161]. To precisely detect misery perfusion carrying a high risk of exacerbation, the assessment of hemodynamic parameters including OEF and CBF is indispensable [159].
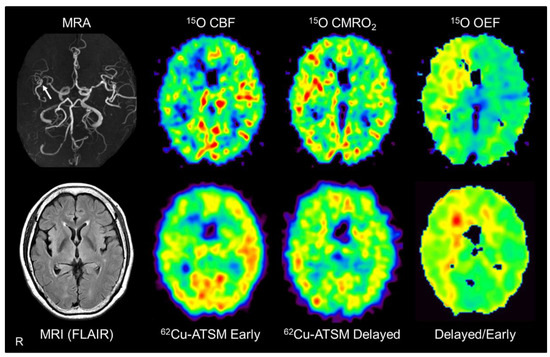
Figure 5.
Brain positron emission tomography (PET) imaging with 62Cu-ATSM and 15O tracers (15O-water, 15O2, and C15O) in a representative patient with major cerebral arterial occlusive disease. Brain magnetic resonance imaging (MRI) showed severe stenosis in the right middle cerebral artery (arrow) but no apparent infarction. PET images with 15O tracers (upper images) showed decreased cerebral blood flow (CBF) with increased cerebral metabolic rate of oxygen (CMRO2) and oxygen extraction fraction (OEF) in the right cerebral hemisphere, which indicates misery perfusion in this area. Alongside 15O-gas and water PET, early, delayed, and delayed-to-early ratio images from 62Cu-ATSM PET (lower images) correspond well with the CBF, CMRO2, and OEF images, respectively. MRA: MR angiography; FLAIR: Fluid attenuation inversion recovery.
7.2. PET Imaging in Patients with Misery Perfusion
Based on the above assumption, PET scans with 62Cu-ATSM and 15O tracers (15O-water, 15O2, and C15O) performed in patients with unilateral steno-occlusive lesions in the major cerebral arteries were compared [52]. This double PET study showed that the delayed-to-early ratio of 62Cu-ATSM correlated significantly with the OEF value derived from PET with 15O tracers in these patients (Figure 5). In addition, the early-phase accumulation of 62Cu-ATSM correlated well with the CBF value obtained from 15O-gas and water PET, as expected (see Section 3). While 15O-gas and water studies can provide accurate CBF and OEF values [160,161], PET imaging with 15O tracers has some disadvantages, such as the need for invasive arterial blood sampling and cumbersome handling of the tracers. The results of the 62Cu-ATSM PET study indicate the feasibility of a simple PET scan with a single injection of 62Cu-ATSM for detecting chronic misery perfusion with increased OEF and reduced blood flow [52].
8. Recent Progress and Perspective of Oxidative Stress Imaging
8.1. PET Imaging for Oxidative Stress in Patients with Alzheimer’s Disease
Alzheimer’s disease is the most common neurodegenerative disorder causing progressive dementia. The pathological findings of extracellular deposits of amyloid-β plaques and intraneuronal neurofibrillary tangles containing aggregated tau protein are observed as diagnostic markers in the brains of patients with Alzheimer’s disease [162,163]. PET imaging with radioligands for amyloid-β or tau protein, such as 11C-PiB and 18F-MK-6240, respectively, is already being used to diagnose Alzheimer’s disease in living patients [164,165,166], replacing pathological diagnosis.
While various pathophysiological mechanisms including genetic and environmental factors have been proposed for the pathogenesis of Alzheimer’s disease [163], a number of investigations using postmortem specimens or serum samples have emphasized the involvement of oxidative stress and mitochondrial dysfunction, similar to other neurodegenerative disorders [6,167,168]. In support of this hypothesis, a recent PET study with 18F-BCPP-EF, a radioligand to image mitochondrial complex I activity, demonstrated decreased uptake in the parahippocampus in patients with early-stage Alzheimer’s disease [169], which supports the involvement of mitochondrial dysfunction.
We are engaged in PET imaging for oxidative stress using 64Cu-ATSM, instead of 62Cu-ATSM, combined with PET imaging with 11C-PiB and 18F-MK-6240 for amyloid-β and tau protein, respectively, in patients with Alzheimer’s disease. 64Cu-ATSM has a longer radioactive half-life (~13 h) than 62Cu-ATSM (~10 min), and is thus expected to more precisely evaluate cerebral oxidative stress [170]. By combining these PET imaging protocols, investigation of the relationships among protein accumulation, disease progression, and oxidative stress would shed light on the molecular mechanisms of Alzheimer’s disease.
8.2. Development of Imaging Techniques for Oxidative Stress
Besides 62Cu-/64Cu-ATSM, several PET radioligands for imaging of the redox status have been developed, such as 18F-FASu for the cystine/glutamate transporter [171], 18F-ROStrace for superoxide [172], and 18F-FDHM for ROS [173]. These new radioligands are promising but are still in the preclinical stage with animals. In addition to PET, a new MR imaging method using hyperpolarized 13C-MR spectroscopy permits measurement of the reduction rate of 1-13C-dehydroascorbic acid in tumor cells, which reflects the capacity of tumor cells to resist oxidative stress [174,175]. Several MR contrast agents, such as paramagnetic nitroxide radicals (e.g., mito-TEMPO, 3-carbamoyl-PROXYL) and paramagnetic chemical exchange saturation transfer agents, have also been developed to evaluate the redox status in vivo [176,177,178]. As new MR techniques for imaging oxygen metabolism advance, the OEF and oxygen metabolism, as well as oxidative stress, will also be elucidated by MR imaging [179]. However, PET has the advantage of higher detection sensitivity at the nanomolar level [14], which suggests that the optimal approach will be multimodal imaging combining PET and MR [165,180]. Imaging techniques for oxidative stress will further develop in support of clinical research aimed at clarifying the pathophysiological mechanisms of various neurodegenerative disorders.
The use of PET imaging is not limited to elucidating the pathogenesis; it can also directly evaluate the effects of therapeutic agents on the pathological targets. In addition to the aforementioned studies in Parkinson’s disease and ALS, several clinical trials testing new therapeutic molecules having antioxidant effects, such as vatiquinone (EPI-743) and elamipretide (MTP-131), are ongoing on patients with mitochondrial diseases [181,182]. Oxidative stress imaging is thus also able to assess the therapeutic efficacy of these agents against oxidative stress in living patients.
9. Conclusions
This review provides an overview of the availability and future potential of recent imaging techniques for oxidative stress, especially 62Cu-/64Cu-ATSM PET, in neurodegenerative disorders. Clinical 62Cu-ATSM PET studies showed increased uptake in brain regions of pathologically responsible sites of neurodegeneration, i.e., the SE lesions of mitochondrial disease (MELAS), the striatum of Parkinson’s disease, and the motor and motor-related cortices of ALS [10,11,12,13]. These PET studies delineated enhancement of oxidative stress in the disease-related brain regions, suggesting that oxidative stress based on mitochondrial dysfunction is closely associated with the neurodegenerative process in these diseases. Similarly, future studies with oxidative stress and neuroinflammation imaging would also focus on the cardinal brain pathophysiology of dementia and other neurodegenerative disorders, such as amyloid and tau deposition in Alzheimer’s disease. PET imaging for oxidative stress improves our insight into the pathogenesis of neurodegenerative disorders, and is a promising tool for monitoring further antioxidant and mitochondrial therapies.
Author Contributions
Conceptualization, M.I. and H.O.; resources, M.I. and H.O.; writing—original draft preparation, M.I.; writing—review and editing, M.I., H.O. and M.Y.; supervision, H.O. and Y.N.; project administration, M.I. and M.Y.; funding acquisition, M.I., M.Y. and H.O. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by Grants-in-Aid for Scientific Research (B) (18H02763 to H.O.) and (C) (20K07900 to M.I.) from the Japan Society for the Promotion of Science and a Grant for Strategic Issue Promotion Research from Fukui Prefectural University (to M.Y.).
Acknowledgments
The authors thank staff of the Biomedical Imaging Research Center and doctors in the Departments of Neurology and Psychiatry, Faculty of Medical Sciences, University of Fukui for technical and clinical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, S.; Jaimes, E.A. Mitochondria and reactive oxygen species: Physiology and pathophysiology. Int. J. Mol. Sci. 2013, 14, 6306–6344. [Google Scholar] [CrossRef] [PubMed]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. [Google Scholar] [CrossRef]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef]
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive oxygen species: Stuck in the middle of neurodegeneration. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S357–S367. [Google Scholar] [CrossRef]
- Tabner, B.J.; El-Agnaf, O.M.; German, M.J.; Fullwood, N.J.; Allsop, D. Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem. Soc. Trans. 2005, 33, 1082–1086. [Google Scholar] [CrossRef]
- Levy, E.; El Banna, N.; Baille, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.E.; Beringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Arakawa, K.; Kudo, T.; Kimura, H.; Fujibayashi, Y.; Kuriyama, M.; Yoneda, M. PET imaging of redox and energy states in stroke-like episodes of MELAS. Mitochondrion 2009, 9, 144–148. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Kudo, T.; Kuriyama, M.; Fujibayashi, Y.; Yoneda, M. Evaluation of striatal oxidative stress in patients with Parkinson’s disease using [62Cu]ATSM PET. Nucl. Med. Biol. 2011, 38, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Matsunaga, A.; Yamamura, O.; Mori, T.; Hamano, T.; Kiyono, Y.; Nakamoto, Y.; Yoneda, M. Increased oxidative stress is related to disease severity in the ALS motor cortex: A PET study. Neurology 2015, 84, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Okazawa, H.; Ikawa, M.; Tsujikawa, T.; Kiyono, Y.; Yoneda, M. Brain imaging for oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Q. J. Nucl. Med. Mol. Imaging 2014, 58, 387–397. [Google Scholar] [PubMed]
- Phelps, M.E. PET: The merging of biology and imaging into molecular imaging. J. Nucl. Med. 2000, 41, 661–681. [Google Scholar]
- Bubici, C.; Papa, S.; Dean, K.; Franzoso, G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: Molecular basis and biological significance. Oncogene 2006, 25, 6731–6748. [Google Scholar] [CrossRef]
- Muller, F. The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging. J. Am. Aging Assoc. 2000, 23, 227–253. [Google Scholar] [CrossRef]
- Lenaz, G.; Bovina, C.; D’Aurelio, M.; Fato, R.; Formiggini, G.; Genova, M.L.; Giuliano, G.; Merlo Pich, M.; Paolucci, U.; Parenti Castelli, G.; et al. Role of mitochondria in oxidative stress and aging. Ann. N. Y. Acad. Sci. 2002, 959, 199–213. [Google Scholar] [CrossRef]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef]
- Orrenius, S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab. Rev. 2007, 39, 443–455. [Google Scholar] [CrossRef]
- Ikawa, M.; Kawai, Y.; Arakawa, K.; Tsuchida, T.; Miyamori, I.; Kuriyama, M.; Tanaka, M.; Yoneda, M. Evaluation of respiratory chain failure in mitochondrial cardiomyopathy by assessments of 99mTc-MIBI washout and 123I-BMIPP/99mTc-MIBI mismatch. Mitochondrion 2007, 7, 164–170. [Google Scholar] [CrossRef]
- Ikawa, M.; Yoneda, M.; Tanaka, M. Energy states in mitochondrial cardiomyopathy. In vivo functional imaging and L-arginine therapy. Circ. J. 2010, 74, 2560–2561. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Kami, K.; Fujita, Y.; Igarashi, S.; Koike, S.; Sugawara, S.; Ikeda, S.; Sato, N.; Ito, M.; Tanaka, M.; Tomita, M.; et al. Metabolomic profiling rationalized pyruvate efficacy in cybrid cells harboring MELAS mitochondrial DNA mutations. Mitochondrion 2012, 12, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, Y.; Yoneda, M.; Ikawa, M.; Furukawa, T.; Kiyono, Y.; Mori, T.; Yoshii, H.; Oyama, N.; Okazawa, H.; Saga, T.; et al. Radiolabeled Cu-ATSM as a novel indicator of overreduced intracellular state due to mitochondrial dysfunction: Studies with mitochondrial DNA-less ρ0 cells and cybrids carrying MELAS mitochondrial DNA mutation. Nucl. Med. Biol. 2012, 39, 177–185. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc. Soc. Exp. Biol. Med. 1999, 222, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Valera-Alberni, M.; Canto, C. Mitochondrial stress management: A dynamic journey. Cell Stress 2018, 2, 253–274. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants (Basel) 2020, 9, 743. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Dolgacheva, L.P. Interaction of misfolded proteins and mitochondria in neurodegenerative disorders. Biochem. Soc. Trans. 2017, 45, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42 (Suppl. 3), S125–S152. [Google Scholar] [CrossRef]
- Fujibayashi, Y.; Taniuchi, H.; Yonekura, Y.; Ohtani, H.; Konishi, J.; Yokoyama, A. Copper-62-ATSM: A new hypoxia imaging agent with high membrane permeability and low redox potential. J. Nucl. Med. 1997, 38, 1155–1160. [Google Scholar] [PubMed]
- Fujibayashi, Y.; Cutler, C.S.; Anderson, C.J.; McCarthy, D.W.; Jones, L.A.; Sharp, T.; Yonekura, Y.; Welch, M.J. Comparative studies of Cu-64-ATSM and C-11-acetate in an acute myocardial infarction model: Ex vivo imaging of hypoxia in rats. Nucl. Med. Biol. 1999, 26, 117–121. [Google Scholar] [CrossRef]
- Lewis, J.S.; McCarthy, D.W.; McCarthy, T.J.; Fujibayashi, Y.; Welch, M.J. Evaluation of 64Cu-ATSM in vitro and in vivo in a hypoxic tumor model. J. Nucl. Med. 1999, 40, 177–183. [Google Scholar]
- Lewis, J.S.; Sharp, T.L.; Laforest, R.; Fujibayashi, Y.; Welch, M.J. Tumor uptake of copper-diacetyl-bis(N4-methylthiosemicarbazone): Effect of changes in tissue oxygenation. J. Nucl. Med. 2001, 42, 655–661. [Google Scholar]
- Lewis, J.S.; Herrero, P.; Sharp, T.L.; Engelbach, J.A.; Fujibayashi, Y.; Laforest, R.; Kovacs, A.; Gropler, R.J.; Welch, M.J. Delineation of hypoxia in canine myocardium using PET and copper(II)-diacetyl-bis(N(4)-methylthiosemicarbazone). J. Nucl. Med. 2002, 43, 1557–1569. [Google Scholar] [PubMed]
- Blower, P.J.; Lewis, J.S.; Zweit, J. Copper radionuclides and radiopharmaceuticals in nuclear medicine. Nucl. Med. Biol. 1996, 23, 957–980. [Google Scholar] [CrossRef]
- Lewis, J.S.; Laforest, R.; Dehdashti, F.; Grigsby, P.W.; Welch, M.J.; Siegel, B.A. An imaging comparison of 64Cu-ATSM and 60Cu-ATSM in cancer of the uterine cervix. J. Nucl. Med. 2008, 49, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Mori, T.; Tsujikawa, T.; Ogai, K.; Sugama, J.; Kiyono, Y.; Kawai, K.; Okazawa, H. Comparison of image quality with 62Cu and 64Cu-labeled radiotracers in positron emission tomography whole-body phantom imaging. Hell. J. Nucl. Med. 2015, 18, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Fujibayashi, Y.; Yonekura, Y.; Welch, M.J.; Waki, A.; Tsuchida, T.; Sadato, N.; Sugimoto, K.; Itoh, H. Evaluation of 62Cu labeled diacetyl-bis(N4-methylthiosemicarbazone) as a hypoxic tissue tracer in patients with lung cancer. Ann. Nucl. Med. 2000, 14, 323–328. [Google Scholar] [CrossRef]
- Obata, A.; Yoshimi, E.; Waki, A.; Lewis, J.S.; Oyama, N.; Welch, M.J.; Saji, H.; Yonekura, Y.; Fujibayashi, Y. Retention mechanism of hypoxia selective nuclear imaging/radiotherapeutic agent cu-diacetyl-bis(N4-methylthiosemicarbazone) (Cu-ATSM) in tumor cells. Ann. Nucl. Med. 2001, 15, 499–504. [Google Scholar] [CrossRef]
- Dehdashti, F.; Grigsby, P.W.; Lewis, J.S.; Laforest, R.; Siegel, B.A.; Welch, M.J. Assessing tumor hypoxia in cervical cancer by PET with 60Cu-labeled diacetyl-bis(N4-methylthiosemicarbazone). J. Nucl. Med. 2008, 49, 201–205. [Google Scholar] [CrossRef]
- Dietz, D.W.; Dehdashti, F.; Grigsby, P.W.; Malyapa, R.S.; Myerson, R.J.; Picus, J.; Ritter, J.; Lewis, J.S.; Welch, M.J.; Siegel, B.A. Tumor hypoxia detected by positron emission tomography with 60Cu-ATSM as a predictor of response and survival in patients undergoing Neoadjuvant chemoradiotherapy for rectal carcinoma: A pilot study. Dis. Colon Rectum 2008, 51, 1641–1648. [Google Scholar] [CrossRef]
- Lohith, T.G.; Kudo, T.; Demura, Y.; Umeda, Y.; Kiyono, Y.; Fujibayashi, Y.; Okazawa, H. Pathophysiologic correlation between 62Cu-ATSM and 18F-FDG in lung cancer. J. Nucl. Med. 2009, 50, 1948–1953. [Google Scholar] [CrossRef]
- Liu, T.; Karlsen, M.; Karlberg, A.M.; Redalen, K.R. Hypoxia imaging and theranostic potential of [(64)Cu][Cu(ATSM)] and ionic Cu(II) salts: A review of current evidence and discussion of the retention mechanisms. EJNMMI Res. 2020, 10, 33. [Google Scholar] [CrossRef]
- Takahashi, N.; Fujibayashi, Y.; Yonekura, Y.; Welch, M.J.; Waki, A.; Tsuchida, T.; Sadato, N.; Sugimoto, K.; Nakano, A.; Lee, J.D.; et al. Copper-62 ATSM as a hypoxic tissue tracer in myocardial ischemia. Ann. Nucl. Med. 2001, 15, 293–296. [Google Scholar] [CrossRef]
- Holland, J.P.; Barnard, P.J.; Collison, D.; Dilworth, J.R.; Edge, R.; Green, J.C.; McInnes, E.J. Spectroelectrochemical and computational studies on the mechanism of hypoxia selectivity of copper radiopharmaceuticals. Chemistry 2008, 14, 5890–5907. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Liddell, J.R.; Lim, S.; Paterson, B.M.; Cater, M.A.; Savva, M.S.; Mot, A.I.; James, J.L.; Trounce, I.A.; White, A.R.; et al. An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis(4-methylthiosemicarbazonato)copper II. Proc. Natl. Acad. Sci. USA 2012, 109, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, M.; Kiyono, Y.; Arai, Y.; Kudo, T.; Mori, T.; Maruyama, R.; Kikuta, K.; Okazawa, H. Feasibility of 62Cu-ATSM PET for evaluation of brain ischaemia and misery perfusion in patients with cerebrovascular disease. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1075–1082. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S. Mitochondrial encephalomyopathies--fifty years on: The Robert Wartenberg Lecture. Neurology 2013, 81, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, S.G.; Phillips, P.C.; DiMauro, S.; De Vivo, D.C.; Rowland, L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984, 16, 481–488. [Google Scholar] [CrossRef]
- Hayashi, G.; Cortopassi, G. Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 2015, 88, 10–17. [Google Scholar] [CrossRef]
- Ikawa, M.; Arakawa, K.; Hamano, T.; Nagata, M.; Nakamoto, Y.; Kuriyama, M.; Koga, Y.; Yoneda, M. Evaluation of systemic redox states in patients carrying the MELAS A3243G mutation in mitochondrial DNA. Eur. Neurol. 2012, 67, 232–237. [Google Scholar] [CrossRef]
- Zhang, J.; Yoneda, M.; Naruse, K.; Borgeld, H.J.; Gong, J.S.; Obata, S.; Tanaka, M.; Yagi, K. Peroxide production and apoptosis in cultured cells carrying mtDNA mutation causing encephalomyopathy. Biochem. Mol. Biol. Int. 1998, 46, 71–79. [Google Scholar] [CrossRef]
- Yatsuga, S.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T.; Kakuma, T.; Koga, Y.; Taro Matsuoka for, M.S.G.i.J. MELAS: A nationwide prospective cohort study of 96 patients in Japan. Biochim. Biophys. Acta 2012, 1820, 619–624. [Google Scholar] [CrossRef]
- Koga, Y.; Povalko, N.; Nishioka, J.; Katayama, K.; Yatsuga, S.; Matsuishi, T. Molecular pathology of MELAS and L-arginine effects. Biochim. Biophys. Acta 2012, 1820, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Ohama, E.; Ohara, S.; Ikuta, F.; Tanaka, K.; Nishizawa, M.; Miyatake, T. Mitochondrial angiopathy in cerebral blood vessels of mitochondrial encephalomyopathy. Acta Neuropathol. 1987, 74, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Akita, Y.; Junko, N.; Yatsuga, S.; Povalko, N.; Fukiyama, R.; Ishii, M.; Matsuishi, T. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology 2006, 66, 1766–1769. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, J.; Jackson, S.; Schafer, J.; Reichmann, H. Mitochondrial cytopathies. J. Neurol. 2003, 250, 267–277. [Google Scholar] [CrossRef]
- Iizuka, T.; Sakai, F.; Suzuki, N.; Hata, T.; Tsukahara, S.; Fukuda, M.; Takiyama, Y. Neuronal hyperexcitability in stroke-like episodes of MELAS syndrome. Neurology 2002, 59, 816–824. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Takahashi, R.; Nakayama, T.; Shishikura, K.; Suzuki, H.; Hirayama, Y.; Osawa, M.; Oda, H. Disruption of endothelial tight junctions in a patient with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS). Neuropediatrics 2010, 41, 72–74. [Google Scholar] [CrossRef]
- Yoneda, M.; Maeda, M.; Kimura, H.; Fujii, A.; Katayama, K.; Kuriyama, M. Vasogenic edema on MELAS: A serial study with diffusion-weighted MR imaging. Neurology 1999, 53, 2182–2184. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Yoneda, M.; Shimizu, Y.; Uematsu, H.; Toyooka, M.; Ikawa, M.; Kudo, T.; Okazawa, H.; Kuriyama, M.; Kimura, H. Pathophysiologic evaluation of MELAS strokes by serially quantified MRS and CASL perfusion images. Brain Dev. 2010, 32, 143–149. [Google Scholar] [CrossRef]
- Li, R.; Xiao, H.F.; Lyu, J.H.; JJ Wang, D.; Ma, L.; Lou, X. Differential diagnosis of mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) and ischemic stroke using 3D pseudocontinuous arterial spin labeling. J. Magn. Reson. Imaging 2017, 45, 199–206. [Google Scholar] [CrossRef]
- Ikawa, M.; Yoneda, M.; Muramatsu, T.; Matsunaga, A.; Tsujikawa, T.; Yamamoto, T.; Kosaka, N.; Kinoshita, K.; Yamamura, O.; Hamano, T.; et al. Detection of preclinically latent hyperperfusion due to stroke-like episodes by arterial spin-labeling perfusion MRI in MELAS patients. Mitochondrion 2013, 13, 676–680. [Google Scholar] [CrossRef]
- Ikawa, M.; Povalko, N.; Koga, Y. Arginine therapy in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Curr. Opin. Clin. Nutr. Metab. Care 2020, 23, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Tohgi, H.; Yonezawa, H.; Obara, S.; Nagane, Y. Cerebral blood flow and oxygen metabolism before and after a stroke-like episode in patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). J. Neurol. Sci. 1998, 158, 58–64. [Google Scholar] [CrossRef]
- Katayama, Y.; Maeda, K.; Iizuka, T.; Hayashi, M.; Hashizume, Y.; Sanada, M.; Kawai, H.; Kashiwagi, A. Accumulation of oxidative stress around the stroke-like lesions of MELAS patients. Mitochondrion 2009, 9, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Ikawa, M.; Arakawa, K.; Kudo, T.; Kimura, H.; Fujibayashi, Y.; Okazawa, H. In vivo functional brain imaging and a therapeutic trial of L-arginine in MELAS patients. Biochim. Biophys. Acta 2012, 1820, 615–618. [Google Scholar] [CrossRef]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Abeliovich, A. Parkinson’s disease: Mitochondrial damage control. Nature 2010, 463, 744–745. [Google Scholar] [CrossRef]
- Chang, K.H.; Chen, C.M. The Role of Oxidative Stress in Parkinson’s Disease. Antioxidants (Basel) 2020, 9, 597. [Google Scholar] [CrossRef]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef]
- Mochizuki, H.; Choong, C.J.; Baba, K. Parkinson’s disease and iron. J. Neural. Transm. (Vienna) 2020, 127, 181–187. [Google Scholar] [CrossRef]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Yoritaka, A.; Hattori, N.; Uchida, K.; Tanaka, M.; Stadtman, E.R.; Mizuno, Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Saggu, H.; Cooksey, J.; Dexter, D.; Wells, F.R.; Lees, A.; Jenner, P.; Marsden, C.D. A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J. Neurochem. 1989, 53, 692–697. [Google Scholar] [CrossRef]
- Sofic, E.; Lange, K.W.; Jellinger, K.; Riederer, P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci. Lett. 1992, 142, 128–130. [Google Scholar] [CrossRef]
- Musgrove, R.E.; Helwig, M.; Bae, E.J.; Aboutalebi, H.; Lee, S.J.; Ulusoy, A.; Di Monte, D.A. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular alpha-synuclein transfer. J. Clin. Investig. 2019, 129, 3738–3753. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Mizuno, Y.; Sone, N.; Saitoh, T. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J. Neurochem. 1987, 48, 1787–1793. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Muller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: A genome-wide linkage and sequencing study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 2017, 8, 15500. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Multiple-System Atrophy Research, C. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N. Engl. J. Med. 2013, 369, 233–244. [Google Scholar] [CrossRef]
- Forsgren, M.; Attersand, A.; Lake, S.; Grunler, J.; Swiezewska, E.; Dallner, G.; Climent, I. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem. J. 2004, 382, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Neishi, H.; Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Arishima, H.; Kikuta, K.I.; Yoneda, M. Precise evaluation of striatal oxidative stress corrected for severity of dopaminergic neuronal degeneration in patients with Parkinson’s disease: A study with 62Cu-ATSM PET and 123I-FP-CIT SPECT. Eur. Neurol. 2017, 78, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Stoessl, A.J.; Lehericy, S.; Strafella, A.P. Imaging insights into basal ganglia function, Parkinson’s disease, and dystonia. Lancet 2014, 384, 532–544. [Google Scholar] [CrossRef]
- Parkinson Study Group, S.-P.D.I.; Schwarzschild, M.A.; Ascherio, A.; Beal, M.F.; Cudkowicz, M.E.; Curhan, G.C.; Hare, J.M.; Hooper, D.C.; Kieburtz, K.D.; Macklin, E.A.; et al. Inosine to increase serum and cerebrospinal fluid urate in Parkinson disease: A randomized clinical trial. JAMA Neurol. 2014, 71, 141–150. [Google Scholar] [CrossRef]
- Yoritaka, A.; Kawajiri, S.; Yamamoto, Y.; Nakahara, T.; Ando, M.; Hashimoto, K.; Nagase, M.; Saito, Y.; Hattori, N. Randomized, double-blind, placebo-controlled pilot trial of reduced coenzyme Q10 for Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 911–916. [Google Scholar] [CrossRef]
- Crotty, G.F.; Ascherio, A.; Schwarzschild, M.A. Targeting urate to reduce oxidative stress in Parkinson disease. Exp. Neurol. 2017, 298, 210–224. [Google Scholar] [CrossRef]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S.; et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Hassanzadeh, K.; Rahimmi, A. Oxidative stress and neuroinflammation in the story of Parkinson’s disease: Could targeting these pathways write a good ending? J. Cell Physiol. 2018, 234, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapere, J.J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Terada, T.; Yokokura, M.; Yoshikawa, E.; Futatsubashi, M.; Kono, S.; Konishi, T.; Miyajima, H.; Hashizume, T.; Ouchi, Y. Extrastriatal spreading of microglial activation in Parkinson’s disease: A positron emission tomography study. Ann. Nucl. Med. 2016, 30, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Koshimori, Y.; Ko, J.H.; Mizrahi, R.; Rusjan, P.; Mabrouk, R.; Jacobs, M.F.; Christopher, L.; Hamani, C.; Lang, A.E.; Wilson, A.A.; et al. Imaging Striatal Microglial Activation in Patients with Parkinson’s Disease. PLoS ONE 2015, 10, e0138721. [Google Scholar] [CrossRef]
- Varnas, K.; Cselenyi, Z.; Jucaite, A.; Halldin, C.; Svenningsson, P.; Farde, L.; Varrone, A. PET imaging of [11C]PBR28 in Parkinson’s disease patients does not indicate increased binding to TSPO despite reduced dopamine transporter binding. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 367–375. [Google Scholar] [CrossRef]
- Belloli, S.; Morari, M.; Murtaj, V.; Valtorta, S.; Moresco, R.M.; Gilardi, M.C. Translation Imaging in Parkinson’s Disease: Focus on Neuroinflammation. Front. Aging Neurosci. 2020, 12, 152. [Google Scholar] [CrossRef]
- Metzger, J.M.; Moore, C.F.; Boettcher, C.A.; Brunner, K.G.; Fleddermann, R.A.; Matsoff, H.N.; Resnikoff, H.A.; Bondarenko, V.; Kamp, T.J.; Hacker, T.A.; et al. In vivo imaging of inflammation and oxidative stress in a nonhuman primate model of cardiac sympathetic neurodegeneration. NPJ Parkinsons Dis. 2018, 4, 22. [Google Scholar] [CrossRef]
- Tsukada, H.; Ohba, H.; Kanazawa, M.; Kakiuchi, T.; Harada, N. Evaluation of 18F-BCPP-EF for mitochondrial complex 1 imaging in the brain of conscious monkeys using PET. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 755–763. [Google Scholar] [CrossRef]
- Tsukada, H.; Kanazawa, M.; Ohba, H.; Nishiyama, S.; Harada, N.; Kakiuchi, T. PET Imaging of Mitochondrial Complex I with 18F-BCPP-EF in the Brains of MPTP-Treated Monkeys. J. Nucl. Med. 2016, 57, 950–953. [Google Scholar] [CrossRef]
- Mansur, A.; Rabiner, E.A.; Comley, R.A.; Lewis, Y.; Middleton, L.T.; Huiban, M.; Passchier, J.; Tsukada, H.; Gunn, R.N.; Consortium, M.-M. Characterization of 3 PET tracers for quantification of mitochondrial and synaptic function in healthy human brain: 18F-BCPP-EF, 11C-SA-4503, and 11C-UCB-J. J. Nucl. Med. 2020, 61, 96–103. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Fitzmaurice, P.S.; Shaw, I.C.; Kleiner, H.E.; Miller, R.T.; Monks, T.J.; Lau, S.S.; Mitchell, J.D.; Lynch, P.G. Evidence for DNA damage in amyotrophic lateral sclerosis. Muscle Nerve 1996, 19, 797–798. [Google Scholar]
- Shaw, P.J.; Ince, P.G.; Falkous, G.; Mantle, D. Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann. Neurol. 1995, 38, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, G.; Xu, Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion 2005, 5, 77–87. [Google Scholar] [CrossRef]
- Murata, T.; Ohtsuka, C.; Terayama, Y. Increased mitochondrial oxidative damage in patients with sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 2008, 267, 66–69. [Google Scholar] [CrossRef]
- Muyderman, H.; Chen, T. Mitochondrial dysfunction in amyotrophic lateral sclerosis—A valid pharmacological target? Br. J. Pharmacol. 2014, 171, 2191–2205. [Google Scholar] [CrossRef]
- Carrera-Julia, S.; Moreno, M.L.; Barrios, C.; de la Rubia Orti, J.E.; Drehmer, E. Antioxidant Alternatives in the Treatment of Amyotrophic Lateral Sclerosis: A Comprehensive Review. Front. Physiol. 2020, 11, 63. [Google Scholar] [CrossRef]
- Writing, G.; Edaravone, A.L.S.S.G. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, S.Y.; Park, D.; Park, J.S. Effect of edaravone therapy in Korean amyotrophic lateral sclerosis (ALS) patients. Neurol. Sci. 2020, 41, 119–123. [Google Scholar] [CrossRef]
- Soon, C.P.; Donnelly, P.S.; Turner, B.J.; Hung, L.W.; Crouch, P.J.; Sherratt, N.A.; Tan, J.L.; Lim, N.K.; Lam, L.; Bica, L.; et al. Diacetylbis(N4-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 2011, 286, 44035–44044. [Google Scholar] [CrossRef] [PubMed]
- McAllum, E.J.; Lim, N.K.; Hickey, J.L.; Paterson, B.M.; Donnelly, P.S.; Li, Q.X.; Liddell, J.R.; Barnham, K.J.; White, A.R.; Crouch, P.J. Therapeutic effects of CuII(atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013, 14, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Lim, N.K.; McAllum, E.J.; Donnelly, P.S.; Hare, D.J.; Doble, P.A.; Turner, B.J.; Price, K.A.; Lim, S.C.; Paterson, B.M.; et al. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 8021–8031. [Google Scholar] [CrossRef] [PubMed]
- Southon, A.; Szostak, K.; Acevedo, K.M.; Dent, K.A.; Volitakis, I.; Belaidi, A.A.; Barnham, K.J.; Crouch, P.J.; Ayton, S.; Donnelly, P.S.; et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br. J. Pharmacol. 2020, 177, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.R.; Trias, E.; Beilby, P.R.; Lopez, N.I.; Labut, E.M.; Bradford, C.S.; Roberts, B.R.; McAllum, E.J.; Crouch, P.J.; Rhoads, T.W.; et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis. 2016, 89, 1–9. [Google Scholar] [CrossRef]
- Hilton, J.B.; Mercer, S.W.; Lim, N.K.; Faux, N.G.; Buncic, G.; Beckman, J.S.; Roberts, B.R.; Donnelly, P.S.; White, A.R.; Crouch, P.J. CuII(atsm) improves the neurological phenotype and survival of SOD1G93A mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci. Rep. 2017, 7, 42292. [Google Scholar] [CrossRef]
- Willekens, S.M.; Van Weehaeghe, D.; Van Damme, P.; Van Laere, K. Positron emission tomography in amyotrophic lateral sclerosis: Towards targeting of molecular pathological hallmarks. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 533–547. [Google Scholar] [CrossRef]
- Chew, S.; Atassi, N. Positron Emission Tomography Molecular Imaging Biomarkers for Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 135. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.L.; et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef]
- Van Weehaeghe, D.; Babu, S.; De Vocht, J.; Zurcher, N.R.; Chew, S.; Tseng, C.J.; Loggia, M.L.; Koole, M.; Rezaei, A.; Schramm, G.; et al. Moving towards multicenter therapeutic trials in ALS: Feasibility of data pooling using different TSPO positron emission tomography (PET) radioligands. J. Nucl. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Alshikho, M.J.; Zurcher, N.R.; Loggia, M.L.; Cernasov, P.; Reynolds, B.; Pijanowski, O.; Chonde, D.B.; Izquierdo Garcia, D.; Mainero, C.; Catana, C.; et al. Integrated magnetic resonance imaging and [11 C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann. Neurol. 2018, 83, 1186–1197. [Google Scholar] [CrossRef]
- Pagani, M.; Chio, A.; Valentini, M.C.; Oberg, J.; Nobili, F.; Calvo, A.; Moglia, C.; Bertuzzo, D.; Morbelli, S.; De Carli, F.; et al. Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurology 2014, 83, 1067–1074. [Google Scholar] [CrossRef]
- Pagani, M.; Oberg, J.; De Carli, F.; Calvo, A.; Moglia, C.; Canosa, A.; Nobili, F.; Morbelli, S.; Fania, P.; Cistaro, A.; et al. Metabolic spatial connectivity in amyotrophic lateral sclerosis as revealed by independent component analysis. Hum. Brain Mapp. 2016, 37, 942–953. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Morbelli, S.; Cistaro, A.; Campi, C.; Caponnetto, C.; Bauckneht, M.; Bellini, A.; Buschiazzo, A.; Calamia, I.; Beltrametti, M.C.; et al. Interplay between spinal cord and cerebral cortex metabolism in amyotrophic lateral sclerosis. Brain 2018, 141, 2272–2279. [Google Scholar] [CrossRef]
- Marini, C.; Cistaro, A.; Campi, C.; Calvo, A.; Caponnetto, C.; Nobili, F.M.; Fania, P.; Beltrametti, M.C.; Moglia, C.; Novi, G.; et al. A PET/CT approach to spinal cord metabolism in amyotrophic lateral sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Hatakeyama, T.; Sato, K.; Fukui, Y.; Hishikawa, N.; Ohta, Y.; Nishiyama, Y.; Kawai, N.; Tamiya, T.; Abe, K. Flow-metabolism uncoupling in the cervical spinal cord of ALS patients. Neurol. Sci. 2017, 38, 659–665. [Google Scholar] [CrossRef]
- Powers, W.J. Cerebral hemodynamics in ischemic cerebrovascular disease. Ann. Neurol. 1991, 29, 231–240. [Google Scholar] [CrossRef]
- Grubb, R.L., Jr.; Derdeyn, C.P.; Fritsch, S.M.; Carpenter, D.A.; Yundt, K.D.; Videen, T.O.; Spitznagel, E.L.; Powers, W.J. Importance of hemodynamic factors in the prognosis of symptomatic carotid occlusion. JAMA 1998, 280, 1055–1060. [Google Scholar] [CrossRef]
- Yamauchi, H.; Fukuyama, H.; Nagahama, Y.; Nabatame, H.; Ueno, M.; Nishizawa, S.; Konishi, J.; Shio, H. Significance of increased oxygen extraction fraction in five-year prognosis of major cerebral arterial occlusive diseases. J. Nucl. Med. 1999, 40, 1992–1998. [Google Scholar]
- Okazawa, H.; Kudo, T. Clinical impact of hemodynamic parameter measurement for cerebrovascular disease using positron emission tomography and 15O-labeled tracers. Ann. Nucl. Med. 2009, 23, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergstrom, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 2004, 55, 306–319. [Google Scholar] [CrossRef]
- Okazawa, H.; Ikawa, M.; Jung, M.; Maruyama, R.; Tsujikawa, T.; Mori, T.; Rahman, M.G.M.; Makino, A.; Kiyono, Y.; Kosaka, H. Multimodal analysis using [11C]PiB-PET/MRI for functional evaluation of patients with Alzheimer’s disease. EJNMMI Res. 2020, 10, 30. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Therriault, J.; Benedet, A.L.; Savard, M.; Lussier, F.Z.; Chamoun, M.; Tissot, C.; Qureshi, M.N.I.; Kang, M.S.; Mathotaarachchi, S.; et al. 18F-MK-6240 PET for early and late detection of neurofibrillary tangles. Brain 2020. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Nunomura, A.; Tamaoki, T.; Motohashi, N.; Nakamura, M.; McKeel, D.W., Jr.; Tabaton, M.; Lee, H.G.; Smith, M.A.; Perry, G.; Zhu, X. The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J. Neuropathol. Exp. Neurol. 2012, 71, 233–241. [Google Scholar] [CrossRef]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020. [Google Scholar] [CrossRef]
- Crouch, P.J. Comment: Cu-ATSM to treat and image amyotrophic lateral sclerosis. Neurology 2015, 84, 2038. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.M.; Morton, C.A.; Johnson, B.F.; Yang, H.; Rishel, M.J.; Lee, B.D.; Miao, Q.; Pabba, C.; Yapp, D.T.; Schaffer, P. Functional imaging of oxidative stress with a novel PET imaging agent, 18F-5-fluoro-L-aminosuberic acid. J. Nucl. Med. 2014, 55, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Hsieh, C.J.; Li, S.; Lee, H.; Graham, T.J.; Xu, K.; Weng, C.C.; Doot, R.K.; Chu, W.; Chakraborty, S.K.; et al. Development of a Positron Emission Tomography Radiotracer for Imaging Elevated Levels of Superoxide in Neuroinflammation. ACS Chem. Neurosci. 2018, 9, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Egami, H.; Nakagawa, S.; Katsura, Y.; Kanazawa, M.; Nishiyama, S.; Sakai, T.; Arano, Y.; Tsukada, H.; Inoue, O.; Todoroki, K.; et al. 18F-Labeled dihydromethidine: Positron emission tomography radiotracer for imaging of reactive oxygen species in intact brain. Org. Biomol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Keshari, K.R.; Kurhanewicz, J.; Bok, R.; Larson, P.E.; Vigneron, D.B.; Wilson, D.M. Hyperpolarized 13C dehydroascorbate as an endogenous redox sensor for in vivo metabolic imaging. Proc. Natl. Acad. Sci. USA 2011, 108, 18606–18611. [Google Scholar] [CrossRef]
- Timm, K.N.; Hu, D.E.; Williams, M.; Wright, A.J.; Kettunen, M.I.; Kennedy, B.W.; Larkin, T.J.; Dzien, P.; Marco-Rius, I.; Bohndiek, S.E.; et al. Assessing Oxidative Stress in Tumors by Measuring the Rate of Hyperpolarized [1-13C]Dehydroascorbic Acid Reduction Using 13C Magnetic Resonance Spectroscopy. J. Biol. Chem. 2017, 292, 1737–1748. [Google Scholar] [CrossRef]
- Zhelev, Z.; Bakalova, R.; Aoki, I.; Lazarova, D.; Saga, T. Imaging of superoxide generation in the dopaminergic area of the brain in Parkinson’s disease, using mito-TEMPO. ACS Chem. Neurosci. 2013, 4, 1439–1445. [Google Scholar] [CrossRef]
- Kuroda, Y.; Togashi, H.; Uchida, T.; Haga, K.; Yamashita, A.; Sadahiro, M. Oxidative stress evaluation of skeletal muscle in ischemia-reperfusion injury using enhanced magnetic resonance imaging. Sci. Rep. 2020, 10, 10863. [Google Scholar] [CrossRef] [PubMed]
- Do, Q.N.; Ratnakar, J.S.; Kovacs, Z.; Sherry, A.D. Redox- and hypoxia-responsive MRI contrast agents. ChemMedChem 2014, 9, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.P.; An, H.; Moradi, F.; Rosenberg, J.; Ishii, Y.; Nariai, T.; Okazawa, H.; Zaharchuk, G. Quantification of brain oxygen extraction and metabolism with [15O]-gas PET: A technical review in the era of PET/MRI. Neuroimage 2020, 220, 117136. [Google Scholar] [CrossRef]
- Drzezga, A.; Barthel, H.; Minoshima, S.; Sabri, O. Potential Clinical Applications of PET/MR Imaging in Neurodegenerative Diseases. J. Nucl. Med. 2014, 55, 47S–55S. [Google Scholar] [CrossRef]
- Steele, H.E.; Horvath, R.; Lyon, J.J.; Chinnery, P.F. Monitoring clinical progression with mitochondrial disease biomarkers. Brain 2017, 140, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Muraresku, C.C.; McCormick, E.M.; Falk, M.J. Mitochondrial Disease: Advances in clinical diagnosis, management, therapeutic development, and preventative strategies. Curr. Genet. Med. Rep. 2018, 6, 62–72. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).