Microscopy and Spectroscopy Techniques for Characterization of Polymeric Membranes


Abstract
1. Introduction
2. Morphology Analysis
2.1. Scanning Electron Microscopy
2.1.1. Sample Preparation

2.1.2. Data Interpretation



2.1.3. Limitations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Dense surface | Polyetherimide | Cut samples in liquid nitrogen. Gold coat samples. | No pores thus a dense membrane. | [15] |
| Pore size measurements | Polyethersulfone | Cut samples in liquid nitrogen. Used an imaging software to determine the pore size. | Average pore size of 30 nm. | [25] |
| Membrane thickness | poly(1-trimethylsilyl-1-propyne) (PTMSP) | Immersed samples in isopropanol. Cut samples in liquid nitrogen. Coated samples with gold. | Average PTMSP thickness of 1.5 μm. | [17] |
| Particle size measurements | Copolymer of polysulfone (PS) and polyacrylic acid (PAA) | Freeze-dried samples. Sputter-coated samples with platinum. | Particle size ranging from 1 to 7 μm. | [21] |
2.2. Transmission Electron Microscopy
2.2.1. Sample Preparation
2.2.2. Data Interpretation

2.2.3. Limitations
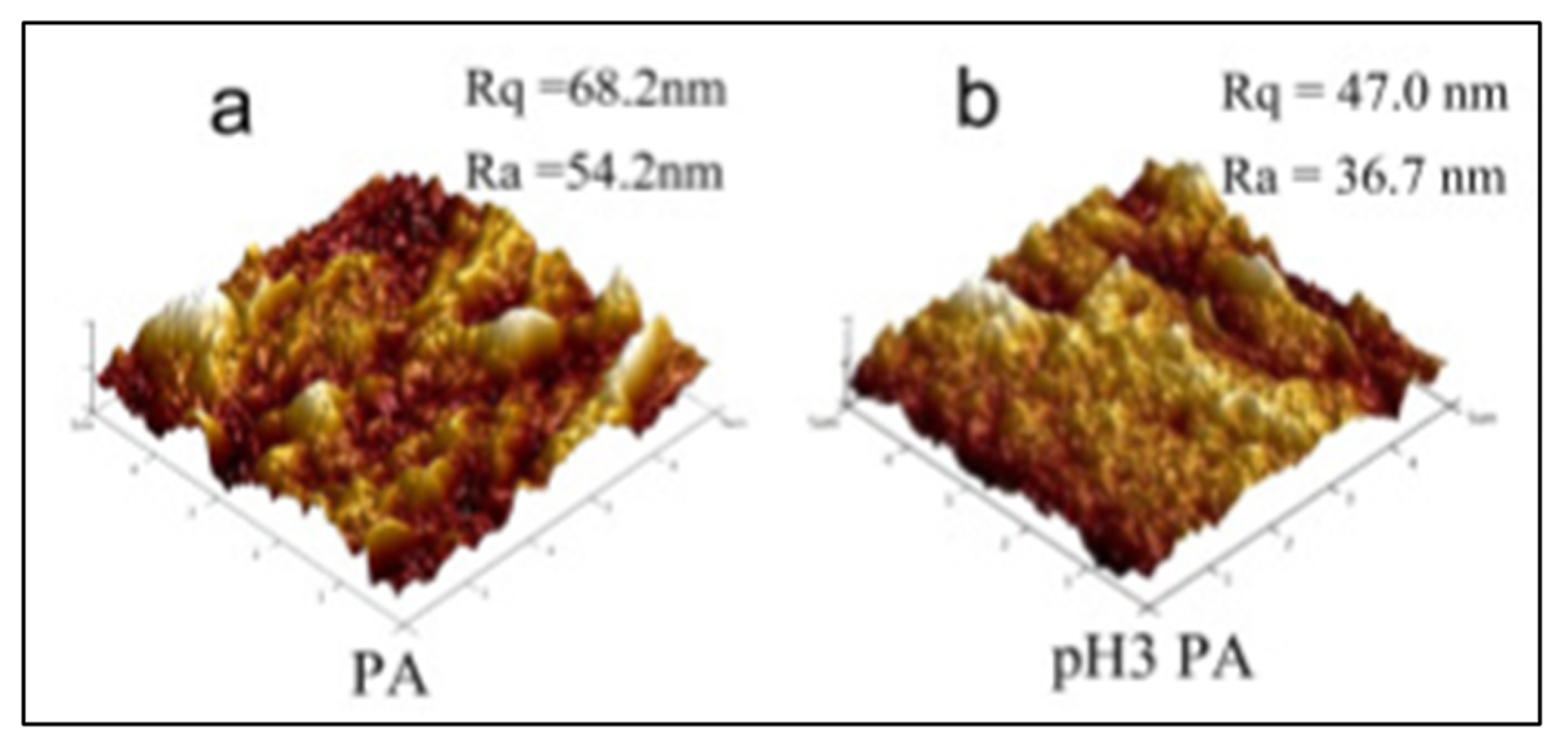
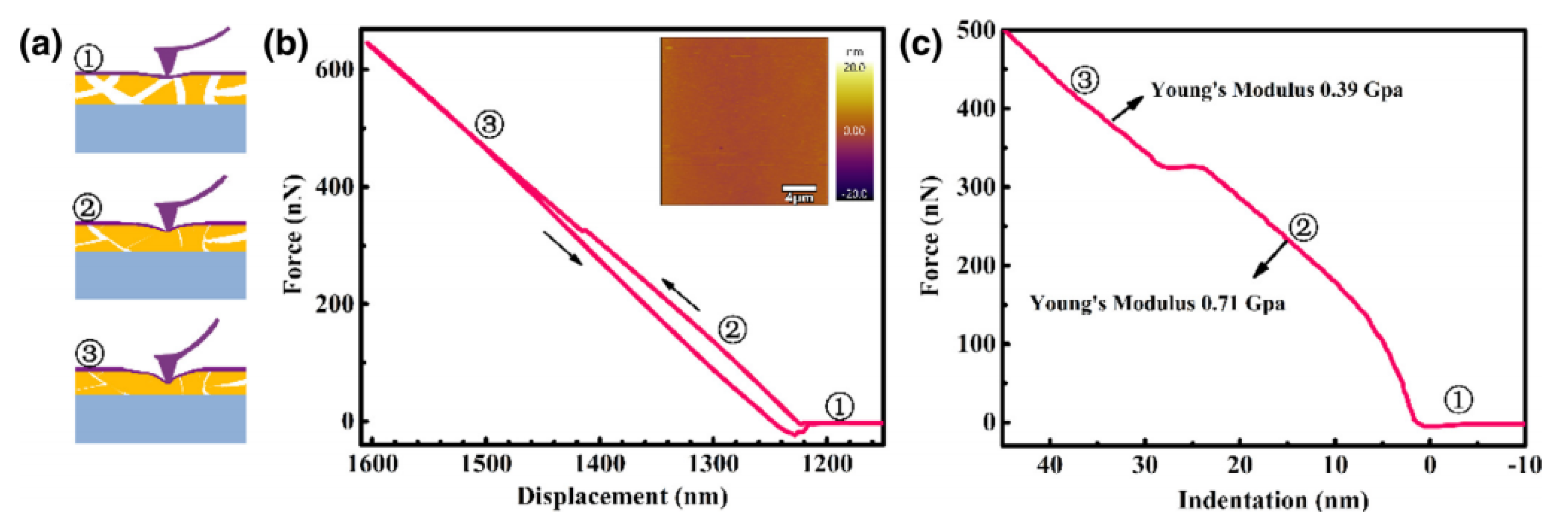
2.3. Atomic Force Microscopy
2.3.1. Sample Preparation
2.3.2. Data Interpretation


2.3.3. Limitations


| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Roughness measurements | Polyvinylidene fluoride (PVDF) /polyvinylalcohol (PA) | Samples in contact mode with silicon nitride probe. Scan area of 5 μm by 5 μm. Five areas were analyzed. | Roughness decreased from 54 to 37 nm indicating a fouling. | [46] |
| Pore size distribution | Polyvinylidene fluoride (PVDF) | Dried samples at 40 °C overnight. Semi-contact mode. Image processing by NT-MDT software. | Average pore size of 0.3 μm. | [50] |
| Membrane stiffness | Polymer of intrinsic microporosity (PIM-1) | Contact-mode analysis. Probe radius of 8 nm. Derjaguin–Muller–Toporov (DMT) method to calculate Young’s modulus. | Elasticity decrease with membrane depth. | [51] |
3. Crystal Structure Analysis
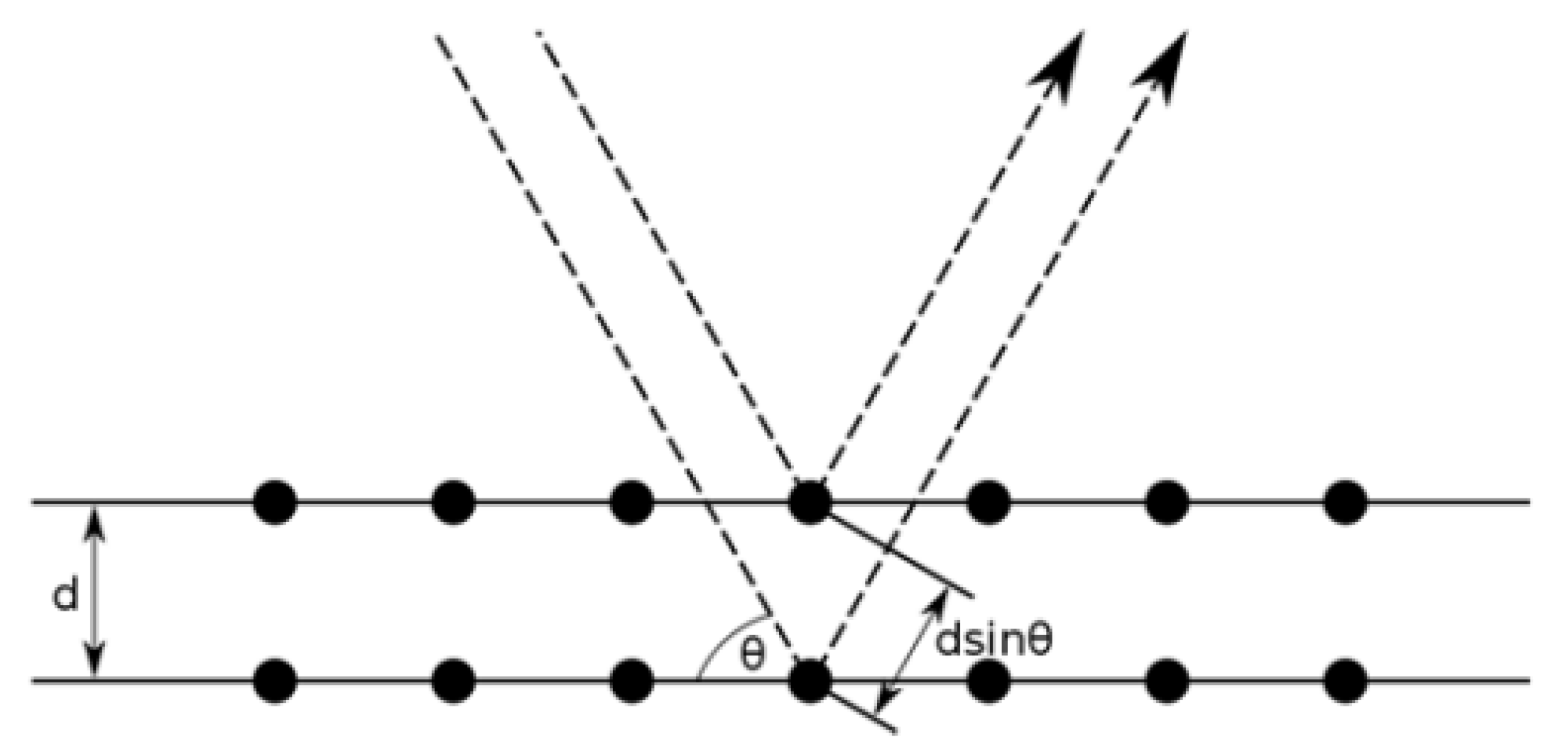
3.1. X-ray Diffraction
3.1.1. Sample Preparation
3.1.2. Data Interpretation
3.1.3. Limitations
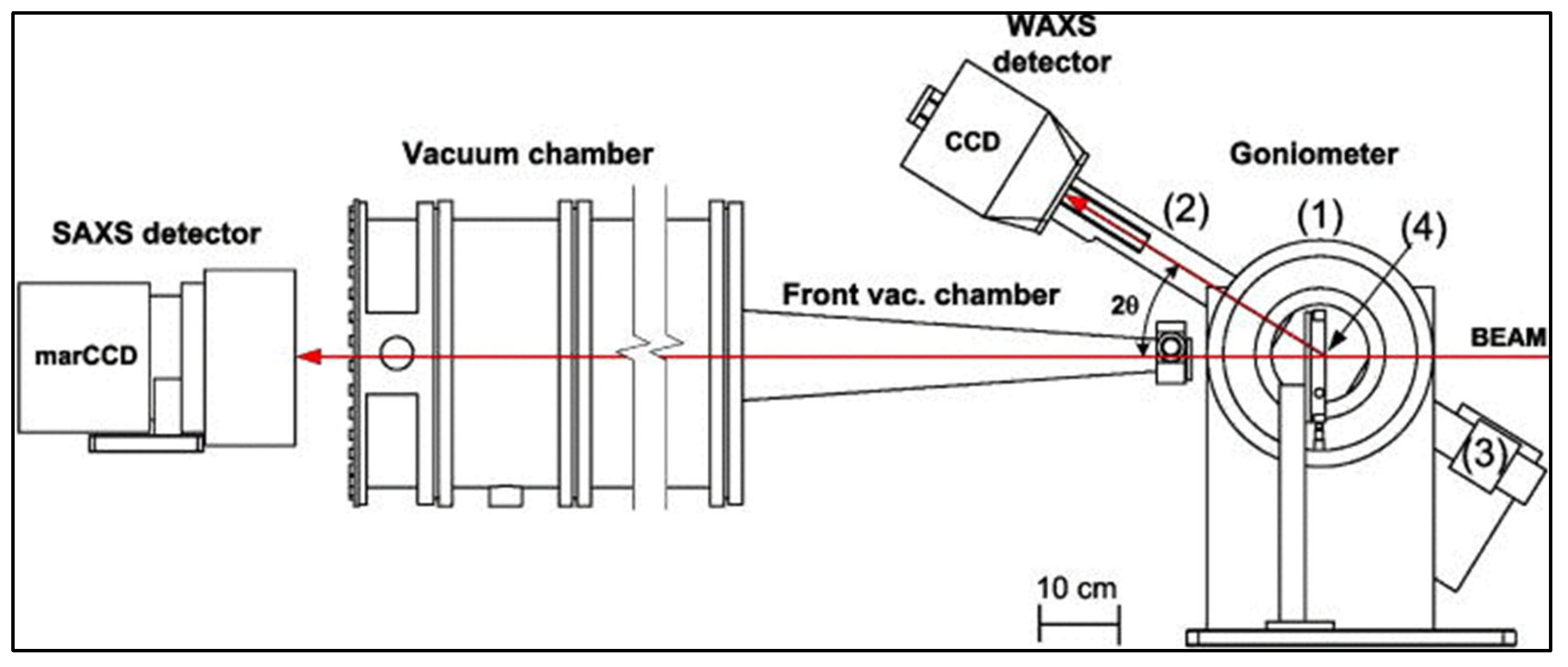
3.2. X-ray Scattering
3.2.1. Sample Preparation
3.2.2. Data Interpretation

3.2.3. Limitations
4. Functional Groups Analysis
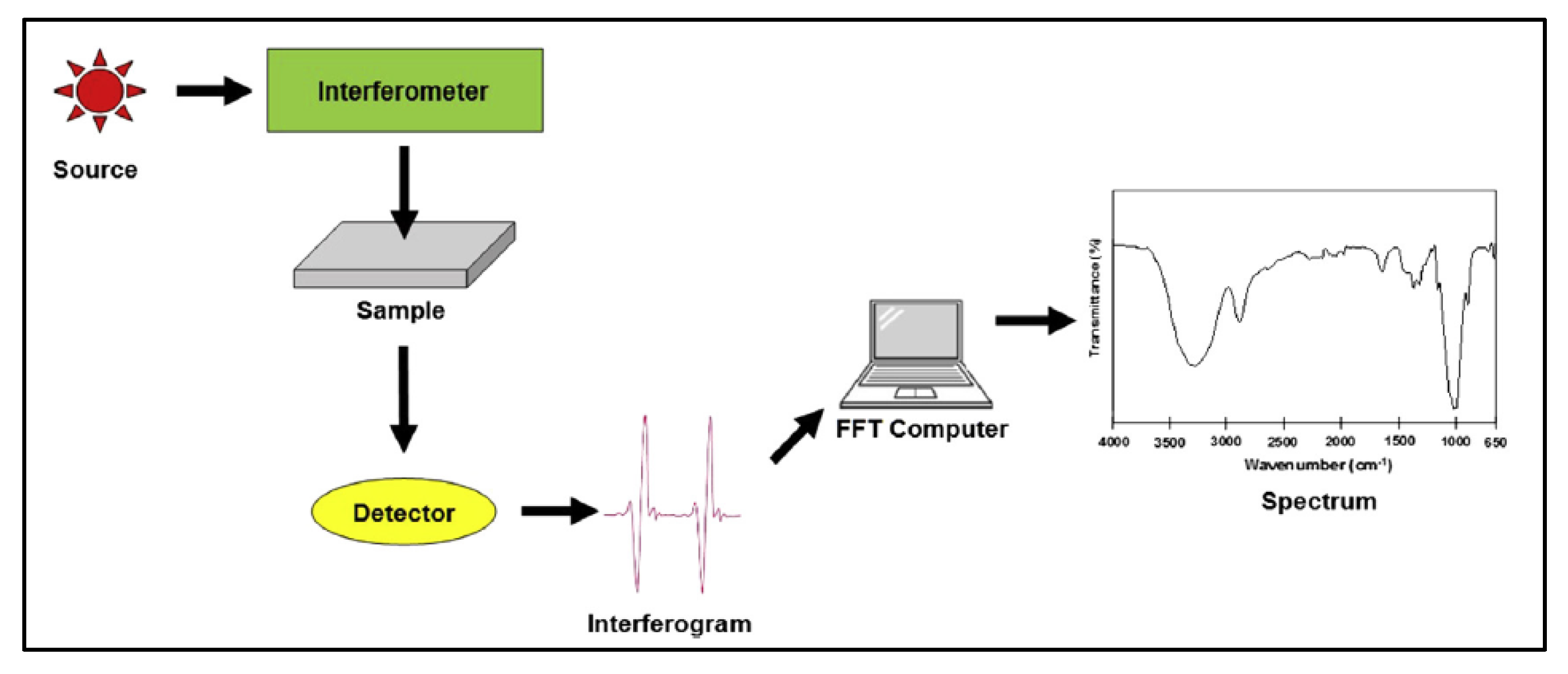
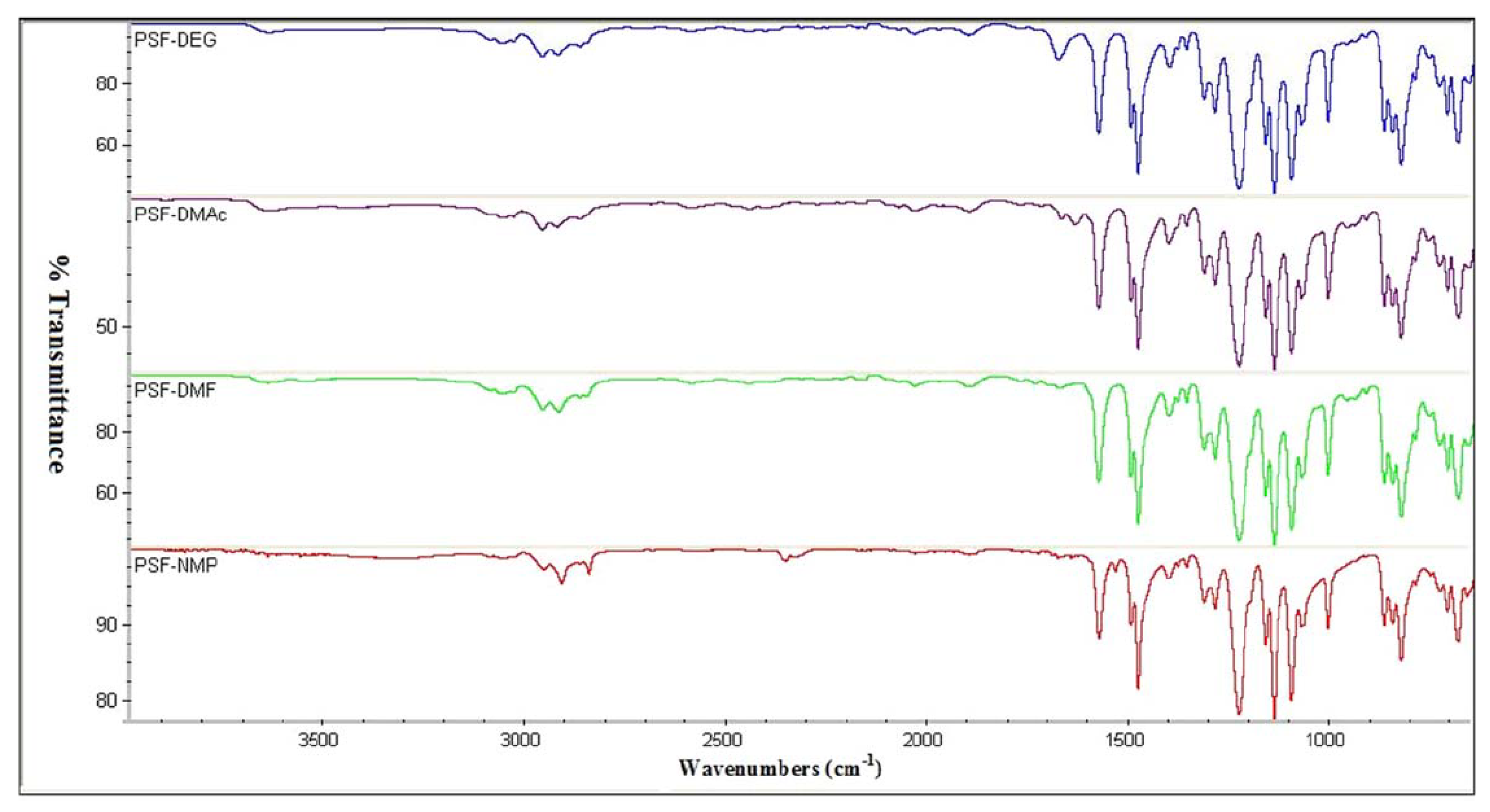
4.1. Fourier-Transform Infrared Spectroscopy
4.1.1. Sample Preparation
4.1.2. Data Interpretation
4.1.3. Limitations
| Wavelength (cm−1) | Functional Group | Chemical Class |
|---|---|---|
| 3200–3500 | OH– | Alcohols |
| 2500–3300 | OH– | Carboxylic acids |
| 2800–3000 | N–H | Amine salts |
| 3267–3333 | C–H | alkynes |
| 3000–3100 | C–H | alkenes |
| 2840–3000 | C–H | Alkanes |
| 2349 | O=C=O | carbon dioxide |
| 1380–1415 | S=O | sulfates |
| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Polymer-solvent compatibility | Polysulfone | Not reported. | Functional groups of only aryl ethers, aryl sulfones and methyl were detected indicating no interaction with the solvent (solvent is compatible). | [93] |
| Polymer-filler interaction | Polyetherimide with metal-organic framework filler (MIL-53) | FTIR Spectrum recorded at 4000–500 cm−1. | Peaks of (C-N), (Si-O), (CO2–), and (Al-O) indicated that MIL-53 was successfully incorporated in the polymer matrix. | [94] |
4.2. Raman Spectroscopy
4.2.1. Sample Preparation

4.2.2. Data Interpretation
4.2.3. Limitation


| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Crystal structure | Polyvinylidene fluoride (PVDF) | Membrane layers were superimposed in a solid sampler. Spectrum recorded at 2 cm−1 resolution. | Additional peak at 795 cm−1 indicated formation of trans-Gauche sequence structure. | [101] |
| Membrane degradation | Perfluorinated sulfonic-acid (PFSA) | Samples mounted vertically. He-Ne laser and Peltier-cooled charge coupled device (CCD) to detect Raman spectrum. Resolution of < 2 cm−1. | Decrease in intensity of C–O–C, C–S and S–O bonds indicated membrane degradation. | [111] |
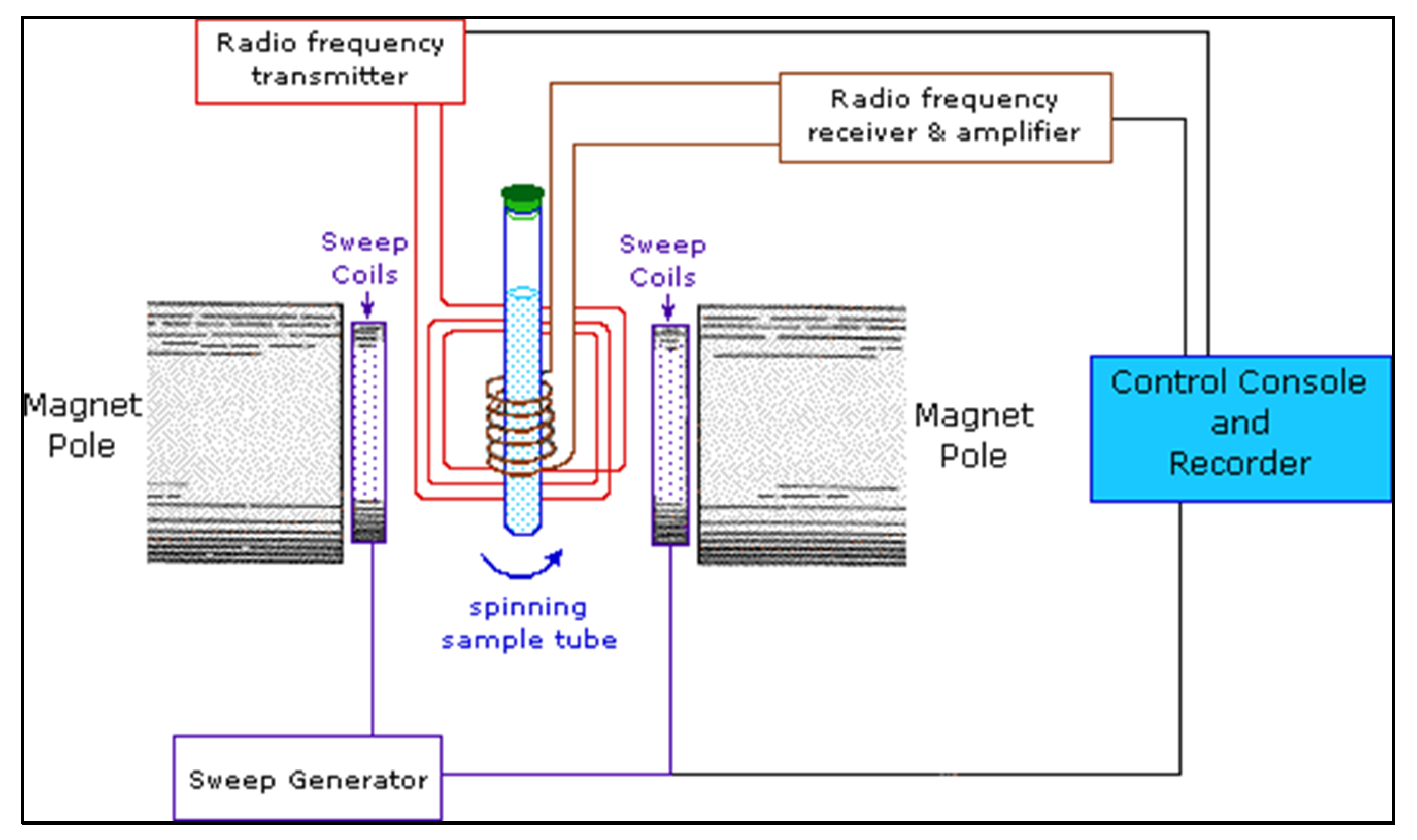
4.3. Nuclear Magnetic Resonance Spectroscopy
4.3.1. Sample Preparation
4.3.2. Data Interpretation




4.3.3. Limitations
5. Elemental Composition Analysis
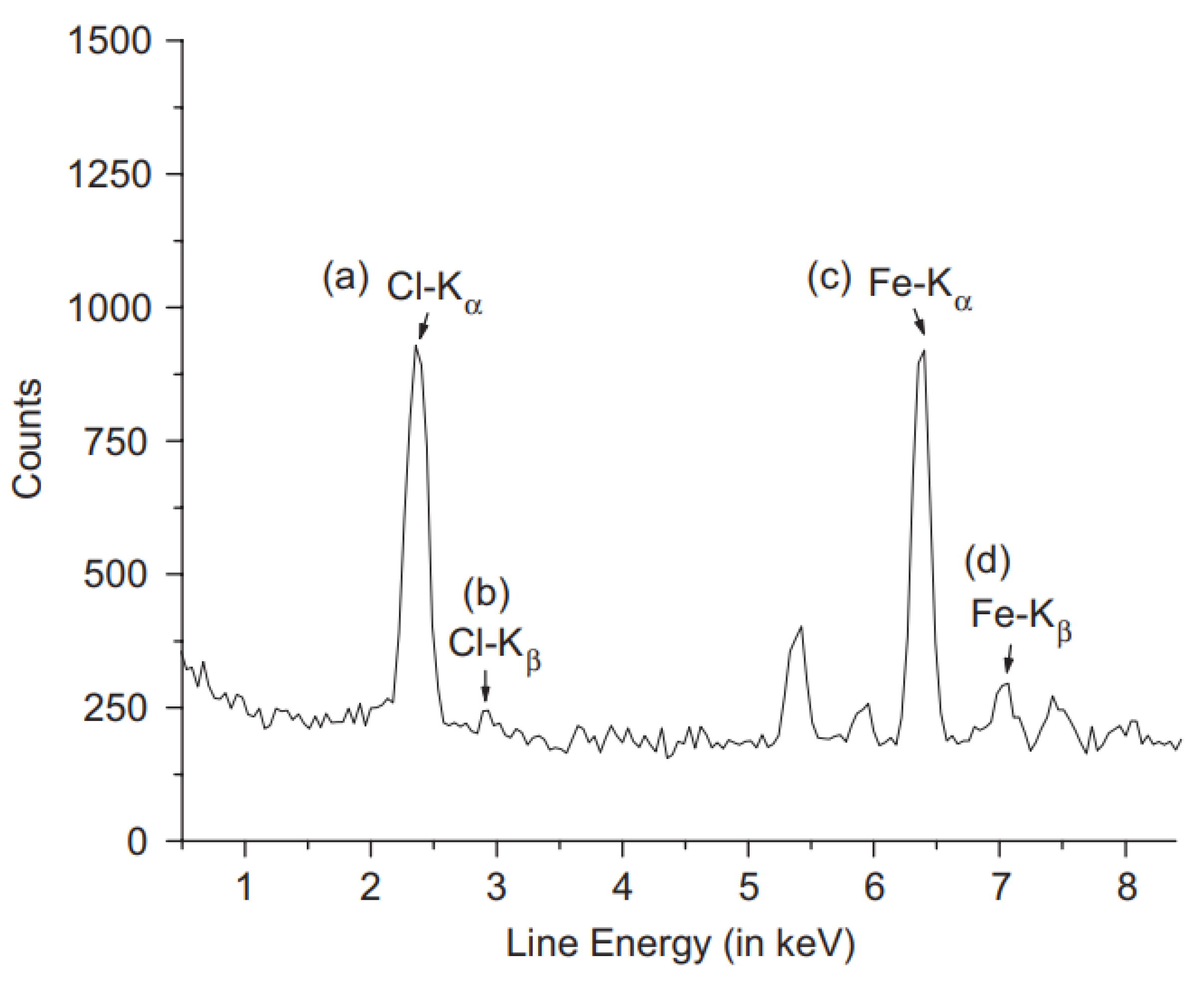
5.1. Energy-Dispersion X-Ray Spectroscopy
5.1.1. Sample Preparation
5.1.2. Data Interpretation

5.1.3. Limitations
5.2. X-Ray Fluorescence
5.2.1. Sample Preparation
5.2.2. Data Interpretation

5.2.3. Limitations
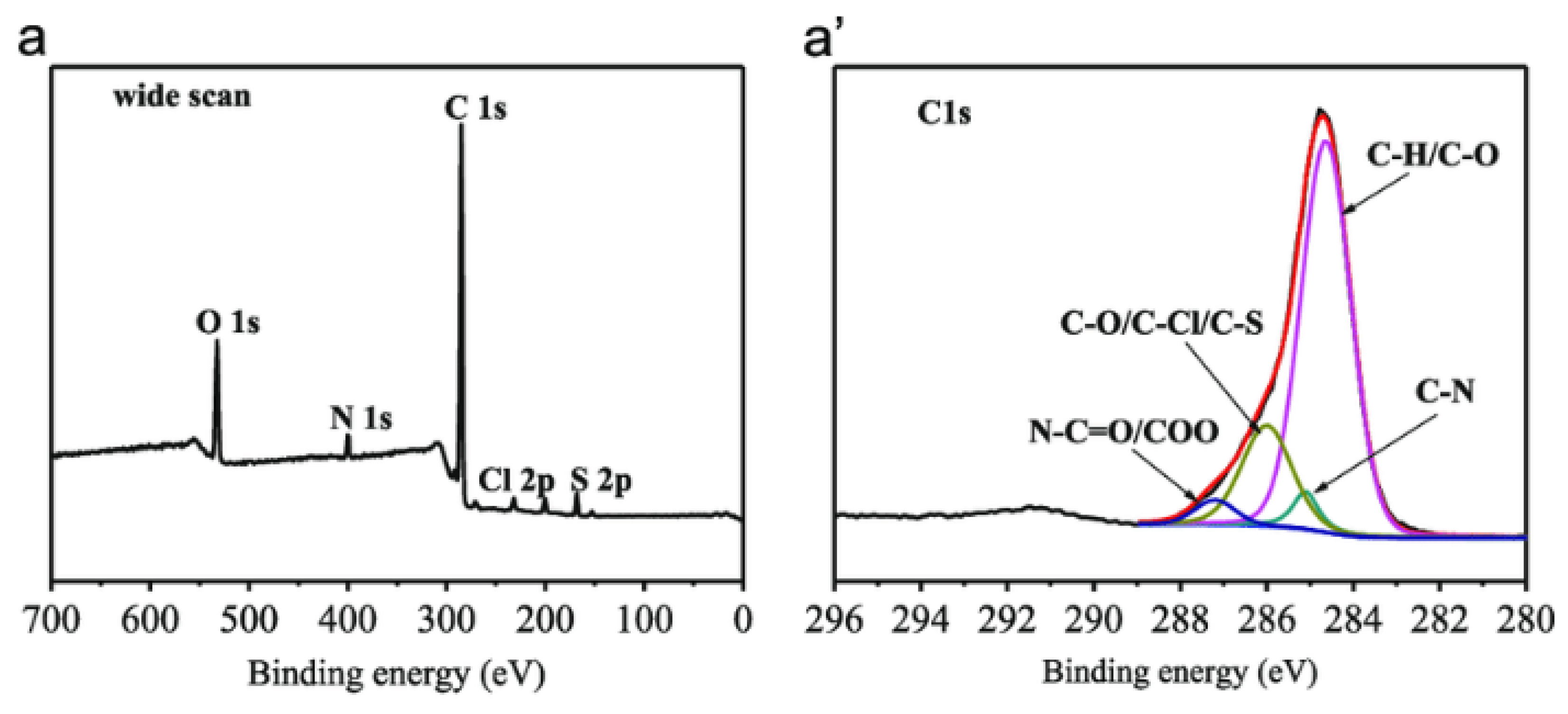
5.3. X-ray Photoelectron Spectroscopy
5.3.1. Sample Preparation
5.3.2. Data Interpretation

5.3.3. Limitations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Freeman, B. Basis of permeability/selectivity tradeoff relations in polymeric gas separation membranes. Macromolecules 1999, 32, 375–380. [Google Scholar] [CrossRef]
- Goh, P.; Ismail, A.; Sanip, S.; Ng, B.; Aziz, M. Recent advances of inorganic fillers in mixed matrix membrane for gas separation. Sep. Purif. Technol. 2011, 81, 243–264. [Google Scholar] [CrossRef]
- Czanderna, A.; Madey, T.; Powell, C. Beam Effects, Surface Topography, and Depth Profiling in Surface Analysis; Springer: New York, NY, USA, 1998. [Google Scholar]
- Ziel, R.; Haus, A.; Tulke, A. Quantification of the pore size distribution (porosity profiles) in microfiltration membranes by SEM, TEM and computer image analysis. J. Membr. Sci. 2008, 323, 241–246. [Google Scholar] [CrossRef]
- Mohamad, M.; Fong, Y. Preparation of defect—Free Polysulfone membrane: Optimization of fabrication method. J. Sci. Res. Dev. 2016, 3, 126–131. [Google Scholar]
- Ghorbanpour, M.; Wani, S. Advances in Phytonanotechnology; Academic Press: Cambridge, MA, USA, 2019; pp. 45–121. [Google Scholar]
- Hawkes, P.; Reimer, L. Scanning Electron Microscopy: Physics of Image Formation and Microanalysis; Springer: Berlin, Germany, 2013. [Google Scholar]
- Fails, A.; Magee, C. Anatomy and Physiology of Farm Animals; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Klapetek, P. Quantitative Data Processing in Scanning Probe Microscopy: SPM Applications for Nanometrology; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Aharinejad, S.; Lametschwandtner, A. Microvascular Corrosion Casting in Scanning Electron Microscopy: Techniques and Applications; Springer: Berlin, Germany, 2012. [Google Scholar]
- Sawyer, L.; Grubb, D.; Meyers, G. Polymer Microscopy; Springer: New York, NY, USA, 2008. [Google Scholar]
- Morozov, O.; Bulgakov, B.; Ivanchenko, A.; Shachneva, S.; Nechausov, S.; Bermeshev, M.; Kepman, A. Data on synthesis and characterization of sulfonated poly(phenylnorbornene) and polymer electrolyte membranes based on it. Data Brief 2019, 27, 104626. [Google Scholar] [CrossRef]
- Fleck, R.; Humbel, B. Biological Field Emission Scanning Electron Microscopy; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Choudhary, O.; Choudhary, P. Scanning electron microscope: Advantages and disadvantages in imaging components. Int. J. Curr. Microbiol. Appl. Sci. 2017, 6, 1877–1882. [Google Scholar] [CrossRef]
- Alqaheem, Y.; Alomair, A.; Alhendi, A.; Alkandari, S.; Tanoli, N.; Alnajdi, N.; Quesada-Peréz, A. Preparation of polyetherimide membrane from non-toxic solvents for the separation of hydrogen from methane. Chem. Cent. J. 2018, 12, 80. [Google Scholar] [CrossRef]
- Ren, Y.; Zhu, J.; Cong, S.; Wang, J.; Van der Bruggen, B.; Liu, J.; Zhang, Y. High flux thin film nanocomposite membranes based on porous organic polymers for nanofiltration. J. Membr. Sci. 2019, 585, 19–28. [Google Scholar] [CrossRef]
- Bazhenov, S.; Borisov, I.; Bakhtin, D.; Rybakova, A.; Khotimskiy, V.; Molchanov, S.; Volkov, V. High-permeance crosslinked PTMSP thin-film composite membranes as supports for CO2 selective layer formation. Green Energy Environ. 2016, 1, 235–245. [Google Scholar] [CrossRef]
- Gaur, G. Quantum Dot Integrated Silicon Photonic Devices for Optical Sensor Applications. Ph.D. Thesis, Vanderbilt University, Nashville, TN, USA, 2015. [Google Scholar]
- Almijbilee, M.; Wu, X.; Zhou, A.; Zheng, X.; Cao, X.; Li, W. Polyetheramide organic solvent nanofiltration membrane prepared via an interfacial assembly and polymerization procedure. Sep. Purif. Technol. 2020, 234, 116033. [Google Scholar] [CrossRef]
- Diab, M.; El-Sonbati, A.; El-Bindary, A.; Abd El-Ghany, H. Thermal stability and degradation of poly (N-phenylpropionamide) homopolymer and copolymer of N-phenylpropionamide with methyl methacrylate. Arabian J. Chem. 2017, 10, S3732–S3739. [Google Scholar] [CrossRef]
- Alvarez, J.; Saudino, G.; Musteata, V.; Madhavan, P.; Genovese, A.; Behzad, A.; Sougrat, R.; Boi, C.; Peinemann, K.-V.; Nunes, S. 3D analysis of ordered porous polymeric particles using complementary electron microscopy methods. Sci. Rep. 2019, 9, 13987. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Ams, M.; Bustamante, H.; Vorreiter, L.; Withford, M.; Clark, S. A practical methodology to assess corrosion in concrete sewer pipes. MATEC Web Conf. 2018, 199, 6010. [Google Scholar] [CrossRef][Green Version]
- Kaufmann, E. Characterization of Materials; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Brunette, D.; Tengvall, P.; Textor, M.; Thomsen, P. Titanium in Medicine: Material Science, Surface Science, Engineering, Biological Responses, and Medical Applications; Springer: New York, NY, USA, 2001. [Google Scholar]
- Kellenberger, C.; Pfleiderer, F.; Raso, R.; Burri, C.; Schumacher, C.; Grass, R.; Stark, W. Limestone nanoparticles as nanopore templates in polymer membranes: Narrow pore size distribution and use as self-wetting dialysis membranes. RSC Adv. 2014, 4, 61420–61426. [Google Scholar] [CrossRef]
- Bhowmick, A. Current Topics in Elastomers Research; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Sharma, S.; Verma, D.; Khan, L.; Kumar, S.; Khan, S. Handbook of Materials Characterization; Springer International Publishing: New York, NY, USA, 2018. [Google Scholar]
- Li, Z. Industrial Applications of Electron Microscopy; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Freger, V.; Gilron, J.; Belfer, S. TFC polyamide membranes modified by grafting of hydrophilic polymers: An FT-IR/AFM/TEM study. J. Membr. Sci. 2002, 209, 283–292. [Google Scholar] [CrossRef]
- Knez, S.; Stražišar, J.; Golob, J.; Horvat, A. Agglomeration of zeolite in the fluidized bed. Acta Chimica Slovenica 2001, 48, 487–504. [Google Scholar]
- Akhtar, F.; Kumar, M.; Villalobos, L.; Shevate, R.; Vovusha, H.; Schwingenschlogl, U.; Peinemann, K.-V. Polybenzimidazole-based mixed membranes with exceptional high water vapor permeability and selectivity. J. Mater.l Chem. A 2017, 5, 21807–21819. [Google Scholar] [CrossRef]
- Hilal, N.; Ismail, A.; Matsuura, T.; Oatley-Radcliffe, D. Membrane Characterization; Elsevier Science: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Erinosho, M.; Akinlabi, E.; Johnson, O. Characterization of surface roughness of laser deposited titanium alloy and copper using AFM. Appl. Surf. Sci. 2018, 435, 393–397. [Google Scholar] [CrossRef]
- Sharpe, W. Springer Handbook of Experimental Solid Mechanics; Springer: New York, NY, USA, 2008. [Google Scholar]
- Kargarzadeh, H.; Ahmad, I.; Thomas, S.; Dufresne, A. Handbook of Nanocellulose and Cellulose Nanocomposites; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
- Giordano, N. College Physics: Reasoning and Relationships; Cengage Learning: Boston, MA, USA, 2009. [Google Scholar]
- Grant, C.; Twigg, P.; Bell, G.; Lu, J. AFM relative stiffness measurement of the plasticising effect of a non-ionic surfactant on plant leaf wax. J. Colloid Interface Sci. 2008, 321, 360–364. [Google Scholar] [CrossRef]
- Cahn, R.; Lifshitz, E. Concise Encyclopedia of Materials Characterization; Elsevier Science: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Cohen, S.; Lightbody, M. Atomic Force Microscopy/Scanning Tunneling Microscopy 3; Springer US: New York, NY, USA, 2007. [Google Scholar]
- Mohammad, A.; Hilal, N.; Lim, Y.; Amin, I.; Raslan, R. Atomic force microscopy as a tool for asymmetric polymeric membrane characterization. Sains Malays. 2011, 40, 237–244. [Google Scholar]
- Shamohammadi, M.; Hormozi, E.; Moradinezhad, M.; Moradi, M.; Skini, M.; Rakhshan, V. Surface topography of plain nickel-titanium (NiTi), as-received aesthetic (coated) NiTi, and aesthetic NiTi archwires sterilized by autoclaving or glutaraldehyde immersion: A profilometry/SEM/AFM study. Int. Orthod. 2019, 17, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Khulbe, K.; Feng, C.; Matsuura, T. Synthetic Polymeric Membranes: Characterization by Atomic Force Microscopy; Springer: Berlin, Germany, 2007. [Google Scholar]
- ElHadidy, A.; Peldszus, S.; Van Dyke, M. Development of a pore construction data analysis technique for investigating pore size distribution of ultrafiltration membranes by atomic force microscopy. J. Membr. Sci. 2013, 429, 373–383. [Google Scholar] [CrossRef]
- Ochoa, N.; Prádanos, P.; Palacio, L.; Pagliero, C.; Marchese, J.; Hernández, A. Pore size distributions based on AFM imaging and retention of multidisperse polymer solutes: Characterisation of polyethersulfone UF membranes with dopes containing different PVP. J. Membr. Sci. 2001, 187, 227–237. [Google Scholar] [CrossRef]
- Arita, M.; Sakaguchi, N. Electron Microscopy: Novel Microscopy Trends; IntechOpen: London, UK, 2018. [Google Scholar]
- Meng, X.; Tang, W.; Wang, L.; Wang, X.; Huang, D.; Chen, H.; Zhang, N. Mechanism analysis of membrane fouling behavior by humic acid using atomic force microscopy: Effect of solution pH and hydrophilicity of PVDF ultrafiltration membrane interface. J. Membr. Sci. 2015, 487, 180–188. [Google Scholar] [CrossRef]
- Najafi, M.; Sadeghi, M.; Bolverdi, A.; Pourafshari Chenar, M.; Pakizeh, M. Gas permeation properties of cellulose acetate/silica nanocomposite membrane. Adv. Polym. Technol. 2018, 37, 2043–2052. [Google Scholar] [CrossRef]
- Ross, R. Microelectronics Failure Analysis Desk Reference, 6th ed.; ASM International: Geauga County, OH, USA, 2011. [Google Scholar]
- Mittal, V.; Matsko, N. Analytical Imaging Techniques for Soft Matter Characterization; Springer: Berlin, Germany, 2012. [Google Scholar]
- Mistry, R.; Saxena, M.; Ray, P.; Singh, P. Octadecyl-silica—PVDF membrane of superior MD desalination performance. J. Appl. Polym. Sci. 2018, 135, 46043. [Google Scholar] [CrossRef]
- Yang, Q.; Li, W.; Dong, C.; Ma, Y.; Yin, Y.; Wu, Q.; Xu, Z.; Ma, W.; Fan, C.; Sun, K. PIM-1 as an artificial solid electrolyte interphase for stable lithium metal anode in high-performance batteries. J. Energy Chem. 2020, 42, 83–90. [Google Scholar] [CrossRef]
- Chowdhury, G.; Kruczek, B.; Matsuura, T. Polyphenylene Oxide and Modified Polyphenylene Oxide Membranes: Gas, Vapor and Liquid Separation; Springer: New York, NY, USA, 2013. [Google Scholar]
- Li, B.; Wang, B.; Liu, Z.; Qing, G. Synthesis of nanoporous PVDF membranes by controllable crystallization for selective proton permeation. J. Membr. Sci. 2016, 517, 111–120. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, B.; Zhao, L.; Dutta, P.; Winston Ho, W. New Pebax®/zeolite Y composite membranes for CO2 capture from flue gas. J. Membr. Sci. 2015, 495, 415–423. [Google Scholar] [CrossRef]
- Powell, C.; Qiao, G. Polymeric CO2/N2 gas separation membranes for the capture of carbon dioxide from power plant flue gases. J. Membr. Sci. 2006, 279, 1–49. [Google Scholar] [CrossRef]
- Minelli, M.; Sarti, G. Elementary prediction of gas permeability in glassy polymers. J. Membr. Sci. 2017, 521, 73–83. [Google Scholar] [CrossRef]
- Saberi, M.; Rouhi, P.; Teimoori, M. Estimation of dual mode sorption parameters for CO2 in the glassy polymers using group contribution approach. J. Membr. Sci. 2020, 595, 117481. [Google Scholar] [CrossRef]
- Swallowe, G. Mechanical Properties and Testing of Polymers: An a–z Reference; Springer: Dordrecht The Netherlands, 2013. [Google Scholar]
- Ismail, A.; Khulbe, K.; Matsuura, T. Gas Separation Membranes: Polymeric and Inorganic; Springer International Publishing: New York, NY, USA, 2015. [Google Scholar]
- Weller, M. Inorganic Materials Chemistry; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Suryanarayana, C.; Norton, M. X-ray Diffraction: A Practical Approach; Springer: New York, NY, USA, 2013. [Google Scholar]
- Thomas, S.; Rouxel, D.; Ponnamma, D. Spectroscopy of Polymer Nanocomposites; William Andrew Publishing: Oxford, UK, 2016. [Google Scholar]
- Zakhariev, Z. Polycrystalline Materials: Theoretical and Practical Aspects; IntechOpen: London, UK, 2012. [Google Scholar]
- Shahien, M.; Yamada, M.; Yasui, T.; Fukumoto, M. Reactive atmospheric plasma spraying of AlN coatings: Influence of aluminum feedstock particle size. J. Therm. Spray Technol. 2011, 20, 580–589. [Google Scholar] [CrossRef]
- Shen, Q.; Cong, S.; He, R.; Wang, Z.; Jin, Y.; Li, H.; Cao, X.; Wang, J.; Van der Bruggen, B.; Zhang, Y. SIFSIX-3-Zn/PIM-1 mixed matrix membranes with enhanced permeability for propylene/propane separation. J. Membr. Sci. 2019, 588, 117201. [Google Scholar] [CrossRef]
- Velu, S.; Arthanareeswaran, G.; Lade, H. Removal of organic and inorganic substances from industry wastewaters using modified aluminosilicate-based polyethersulfone ultrafiltration membranes. Environ. Prog. Sustain. Energy 2017, 36, 1612–1620. [Google Scholar] [CrossRef]
- Sawada, H.; Takahashi, Y.; Miyata, S.; Kanehashi, S.; Sato, S.; Nagai, K. Gas transport properties and crystalline structures of poly(lactic acid) membranes. Trans. Mater. Res. Soc. Japan 2010, 35, 241–246. [Google Scholar] [CrossRef]
- Karimi, S.; Firouzfar, E.; Khoshchehreh, M. Assessment of gas separation properties and CO2 plasticization of polysulfone/polyethylene glycol membranes. J. Pet. Sci. Eng. 2019, 173, 13–19. [Google Scholar] [CrossRef]
- Lasseuguette, E.; Malpass-Evans, R.; Carta, M.; McKeown, N.; Ferrari, M.-C. Temperature and pressure dependence of gas permeation in a microporous Tröger’s base polymer. Membranes 2018, 8, 132. [Google Scholar] [CrossRef]
- Ali, A. Failure Analysis and Prevention; IntechOpen: London, UK, 2017. [Google Scholar]
- Fendler, J. Nanoparticles and Nanostructured Films: Preparation, Characterization, and Applications; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Mishra, S. Fibre Structure; Woodhead Publishing: Cambridge, UK, 2016. [Google Scholar]
- Paradies, H. Particle size distribution and determination of characteristic properties of colloidal bismuth—silica compounds by small-angle x-ray scattering and inelastic light scattering. Colloids Surf. A Physicochem. Eng. Asp. 1993, 74, 57–69. [Google Scholar] [CrossRef]
- Radlinski, A.; Mastalerz, M.; Hinde, A.; Hainbuchner, M.; Rauch, H.; Baron, M.; Lin, J.; Fan, L.; Thiyagarajan, P. Application of SAXS and SANS in evaluation of porosity, pore size distribution and surface area of coal. Int. J. Coal Geol. 2004, 59, 245–271. [Google Scholar] [CrossRef]
- Agbabiaka, A.; Wiltfong, M.; Park, C. Small angle X-Ray scattering technique for the particle size distribution of nonporous nanoparticles. J. Nanopartic. 2013, 2013, 11. [Google Scholar] [CrossRef]
- Schneider, K.; Schöne, A.; Jun, T.-S.; Korsunsky, A. Investigation of changes in crystalline and amorphous structure during deformation of nano-reinforced semi-crystalline polymers by space-resolved synchrotron SAXS and WAXS. Procedia Eng. 2009, 1, 159–162. [Google Scholar] [CrossRef]
- Niu, Q.; Pan, J.; Jin, Y.; Wang, H.; Li, M.; Ji, Z.; Wang, K.; Wang, Z. Fractal study of adsorption-pores in pulverized coals with various metamorphism degrees using N2 adsorption, X-ray scattering and image analysis methods. J. Pet. Sci. Eng. 2019, 176, 584–593. [Google Scholar] [CrossRef]
- Baran, M.; Braten, M.; Sahu, S.; Baskin, A.; Meckler, S.; Li, L.; Maserati, L.; Carrington, M.; Chiang, Y.-M.; Prendergast, D.; et al. Design rules for membranes from polymers of intrinsic microporosity for crossover-free aqueous electrochemical devices. Joule 2019, 3, 2968–2985. [Google Scholar] [CrossRef]
- Rueda, D.; García-Gutiérrez, M.; Nogales, A.; Capitán, M.; Ezquerra, T.; Labrador, A.; Fraga, E.; Beltrán, D.; Juanhuix, J.; Herranz, J.; et al. Versatile wide angle diffraction setup for simultaneous wide and small angle x-ray scattering measurements with synchrotron radiation. Rev. Sci. Instru. 2006, 77, 033904. [Google Scholar] [CrossRef]
- De Prado, S.A.L.; Ponce, M.; Funari, S.; Schulte, K.; Garamus, V.; Willumeit, R.; Nunes, S. SAXS/WAXS characterization of proton-conducting polymer membranes containing phosphomolybdic acid. J. Non-Cryst. Solids 2005, 351, 2194–2199. [Google Scholar] [CrossRef]
- Mrozowich, T.; McLennan, S.; Overduin, M.; Patel, T. Structural studies of macromolecules in solution using small angle x-ray scattering. JoVE 2018, 141, e58538. [Google Scholar] [CrossRef]
- Kikhney, A.; Svergun, D. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef]
- Tant, M.; Mauritz, K.; Wilkes, G. Ionomers: Synthesis, Structure, Properties and Applications; Springer: Dordrecht, The Netherlands, 1997. [Google Scholar]
- Xiao, T.; Yuan, H.; Ma, Q.; Guo, X.; Wu, Y. An approach for in situ qualitative and quantitative analysis of moisture adsorption in nanogram-scaled lignin by using micro-FTIR spectroscopy and partial least squares regression. Int. J. Biol. Macromol. 2019, 132, 1106–1111. [Google Scholar] [CrossRef]
- Wypych, G. Handbook of Solvents; Noyes Publications: Park Ridge, NJ, USA, 2001. [Google Scholar]
- Sun, D. Modern Techniques for food Authentication; Elsevier Science: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Bouis, P. Reagent Chemicals: Specifications and Procedures; American Chemical Society Specifications: Washington, DC, USA, 2006. [Google Scholar]
- Mishra, M. Encyclopedia of Polymer Applications; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Townshend, A. Encyclopedia of Analytical Science: Gast-Lip; Academic Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Dalton, D. Foundations of Organic Chemistry: Unity and Diversity of Structures, Pathways, and Reactions; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Bonilla, J.; Srivatsa, G. Handbook of Analysis of Oligonucleotides and Related Products; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Hansen, S.; Pedersen-Bjergaard, S.; Rasmussen, K. Introduction to Pharmaceutical Chemical Analysis; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Adewole, J.; Ahmad, A.; Ismail, S.; Leo, C.; Sultan, A. Comparative studies on the effects of casting solvent on physico-chemical and gas transport properties of dense polysulfone membrane used for CO2/CH4 separation. J. Appl. Polym. Sci. 2015, 132, 42205. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, L.; Jie, X.; Liu, D.; Cao, Y. Improved interfacial affinity and CO2 separation performance of asymmetric mixed matrix membranes by incorporating postmodified MIL-53(Al). ACS Appl. Mater. Interfaces 2016, 8, 22696–22704. [Google Scholar] [CrossRef] [PubMed]
- Grdadolnik, J. ATR-FTIR spectroscopy: Its advantages and limitations. Acta Chimica Slovenica 2002, 49, 631–642. [Google Scholar]
- Smith, B. Fundamentals of Fourier Transform Infrared Spectroscopy; Taylor & Francis: Abingdon-on-Thames, UK, 1995. [Google Scholar]
- Winter, A. Organic Chemistry I for Dummies; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar]
- Zhang, S. Raman Spectroscopy and its Application in Nanostructures; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Korb, C. Differential Absorption Lidars for Remote Sensing of Atmospheric Pressure and Temperature Profiles; National Aeronautics and Space Administration: Washington, DC, USA, 1995.
- Correia, J.; Detrich, H. Biophysical Tools for Biologists: In Vivo Techniques; Elsevier Science: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Boccaccio, T.; Bottino, A.; Capannelli, G.; Piaggio, P. Characterization of PVDF membranes by vibrational spectroscopy. J. Membr. Sci. 2002, 210, 315–329. [Google Scholar] [CrossRef]
- Andrews, D.; Nann, T.; Lipson, R. Comprehensive Nanoscience and Nanotechnology; Elsevier Science: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Amer, M. Raman Spectroscopy for Soft Matter Applications; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Katoh, T.; Imamura, G.; Obata, S.; Bhanuchandra, M.; Copley, G.; Yorimitsu, H.; Saiki, K. The influence of source molecule structure on the low temperature growth of nitrogen-doped graphene. Phys. Chem. Chem. Phys. 2015, 17, 14115–14121. [Google Scholar] [CrossRef] [PubMed]
- Jacox, M. Vibrational and electronic energy levels of polyatomic transient molecules. Supplement B. J. Phys. Chem. Ref. Data 2003, 32, 1–441. [Google Scholar] [CrossRef]
- Grumezescu, A. Nanostructures for the Engineering of Cells, Tissues and Organs: From Design to Applications; Elsevier Science: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Asundi, A.K.; Rao, V.V. Measurement of Methanol Proportion in Methanol-Gasoline Mixtures—An Application of Fiber Optic Raman Spectroscopy; SAE International: Warrendale, PA, USA, 2000. [Google Scholar]
- Wild, D. The Immunoassay Handbook: Theory and Applications of Ligand Binding, ELISA and Related Techniques; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Pathak, Y.; Lokhande, J. Handbook of Metallonutraceuticals; Taylor & Francis: Abingdon, UK, 2014. [Google Scholar]
- Schreiner, M.; Strlič, M.; Salimbeni, R. Handbook of the Use of Lasers in Conservation and Conservation Science; COST Office: Brussels, Belgium, 2008. [Google Scholar]
- Holber, M.; Johansson, P.; Jacobsson, P. Raman spectroscopy of an aged low temperature polymer electrolyte fuel cell membrane. Fuel Cells 2011, 11, 459–464. [Google Scholar] [CrossRef]
- Sundarapandiyan, S.; Renitha, T.; Sridevi, J.; Saravanan, P.; Chandrasekaran, B.; Raju, G. Photocatalytic degradation of highly refractive phenolic polymer—mechanistic insights as revealed by electron spin resonance (ESR) and solid-state 13C NMR spectroscopy. Chem. Eng. J. 2017, 313, 1112–1121. [Google Scholar] [CrossRef]
- Sangroniz, A.; Gonzalez, A.; Martin, L.; Irusta, L.; Iriarte, M.; Etxeberria, A. Miscibility and degradation of polymer blends based on biodegradable poly(butylene adipate-co-terephthalate). Polym. Degrad. Stab. 2018, 151, 25–35. [Google Scholar] [CrossRef]
- Ebbing, D.; Gammon, S. General Chemistry, 10th ed.; Cengage Learning: Boston, MA, USA, 2012. [Google Scholar]
- Emwas, A.; Roy, R.; McKay, R.; Tenori, L.; Saccenti, E.; Gowda, G.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR spectroscopy for metabolomics research. Metabolites 2019, 9, 7. [Google Scholar] [CrossRef]
- Claridge, T.; Claridge, N. High-Resolution NMR Techniques in Organic Chemistry; Elsevier Science: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Pavia, D.; Kriz, G.; Lampman, G.; Engel, R. A Microscale Approach to Organic Laboratory Techniques; Cengage Learning: Boston, MA, USA, 2016. [Google Scholar]
- Kobayashi, Y.; Nakamitsu, Y.; Zheng, Y.; Takashima, Y.; Yamaguchi, H.; Harada, A. Preparation of cyclodextrin-based porous polymeric membrane by bulk polymerization of ethyl acrylate in the presence of cyclodextrin. Polymer 2019, 177, 208–213. [Google Scholar] [CrossRef]
- Mueller, R.; Zhang, S.; Zhang, C.; Lively, R.; Vasenkov, S. Relationship between long-range diffusion and diffusion in the ZIF-8 and polymer phases of a mixed-matrix membrane by high field NMR diffusometry. J. Membr. Sci. 2015, 477, 123–130. [Google Scholar] [CrossRef]
- Becker, E. High Resolution NMR: Theory and Chemical Applications; Elsevier Science: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Nechifor, A.; Panait, V.; Naftanaila, L.; Batalu, D.; Voicu, Ş. Symmetrically polysulfone membranes obtained by solvent evaporation using carbon nanotubes as additives. Synthesis, characterization and applications. Dig. J. Nanomater. Biostruc. 2013, 8, 875–884. [Google Scholar]
- Reusch, W. Nuclear Magnetic Resonance Spectroscopy; LibreTexts: Davis, CA, USA, 2019. [Google Scholar]
- Parmer, J.; Dickinson, L.; Chien, J.; Porter, R. Polymer-polymer miscibility determination via CP-MAS NMR in blends of deuteriated and protonated polymers. Macromol. 1987, 20, 2308–2310. [Google Scholar] [CrossRef]
- Zatoń, M.; Rozière, J.; Jones, D.J. Current understanding of chemical degradation mechanisms of perfluorosulfonic acid membranes and their mitigation strategies: A review. Sustain. Energy Fuels 2017, 1, 409–438. [Google Scholar] [CrossRef]
- Kinumoto, T.; Inaba, M.; Nakayama, Y.; Ogata, K.; Umebayashi, R.; Tasaka, A.; Iriyama, Y.; Abe, T.; Ogumi, Z. Durability of perfluorinated ionomer membrane against hydrogen peroxide. J. Power Sources 2006, 158, 1222–1228. [Google Scholar] [CrossRef]
- Parrondo, J.; Arges, C.; Niedzwiecki, M.; Anderson, E.; Ayers, K.; Ramani, V. Degradation of anion exchange membranes used for hydrogen production by ultrapure water electrolysis. RSC Adv. 2014, 4, 9875–9879. [Google Scholar] [CrossRef]
- McMurry, J. Organic Chemistry; Cengage Learning: Boston, MA, USA, 2011. [Google Scholar]
- Kong, X.; Terskikh, V.; Khade, R.; Yang, L.; Rorick, A.; Zhang, Y.; He, P.; Huang, Y.; Wu, G. Solid-state ¹⁷O NMR spectroscopy of paramagnetic coordination compounds. Angew. Chem. 2015, 54, 4753–4757. [Google Scholar] [CrossRef]
- Moore, T. Characterization of Integrated Circuit Packaging Materials; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Xiong, Y.; Lu, X. Metallic Nanostructures: From Controlled Synthesis to Applications; Springer International Publishing: New York, NY, USA, 2014. [Google Scholar]
- Wadekar, S.; Vidic, R. Comparison of ceramic and polymeric nanofiltration membranes for treatment of abandoned coal mine drainage. Desalination 2018, 440, 135–145. [Google Scholar] [CrossRef]
- Ayesh, A.; Salah, B.; Al-Sulaiti, L. Production and characterization of flexible semiconducting polymer-nanoparticle composites for x-ray sensors. Radiat. Phys. Chem. 2019, 167, 108233. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific. EDX Analysis with SEM: How Does it Work? Available online: https://www.thermofisher.com/blog/microscopy/edx-analysis-with-sem-how-does-it-work/ (accessed on 1 November 2019).
- Saxena, P. IIT Chemistry-I; Krishna Prakashan: Uttar Pradesh, India, 1995. [Google Scholar]
- Russ, J.; Frs, M.; Kiessling, R.; Charles, J. Fundamentals of Energy Dispersive X-ray Analysis: Butterworths Monographs in Materials; Elsevier Science: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Taylor, P.; Yurdakul, E.; Ceylan, H. Concrete Pavement Mixtures Design and Analysis: The Application of Portable X-ray Fluorescence Technique to Assess Concrete Mix Proportions; National Concrete Pavement Technology Center: Ames, IA, USA, 2012. [Google Scholar]
- Verma, H. Atomic and Nuclear Analytical Methods: XRF, Mössbauer, XPS, NAA and Ion-Beam Spectroscopic Techniques; Springer: Berlin, Germany, 2007. [Google Scholar]
- Shugar, A.; Mass, J. Handheld XRF for Art and Archaeology; Leuven University Press: Leuven, Belgium, 2012. [Google Scholar]
- Crain, E. Crain’s Petrophysical Handbook; Spectrum 2000 Mindware: Rocky Mountain House, AB, Canada, 2010. [Google Scholar]
- Elias, G.; Marguí, E.; Díez, S.; Fontàs, C. Polymer inclusion membrane as an effective sorbent to facilitate mercury storage and detection by x-ray fluorescence in natural waters. Anal. Chem. 2018, 90, 4756–4763. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific. The Power Behind the Analyzer: X-ray Tubes. Available online: https://www.thermofisher.com/blog/metals/the-power-behind-the-analyzer-x-ray-tubes (accessed on 24 January 2020).
- Acharya, N.; Kulshrestha, V.; Awasthi, K.; Jain, A.; Singh, M.; Vijay, Y. Hydrogen separation in doped and blend polymer membranes. Int. J. Hydrogen Energy 2008, 33, 327–331. [Google Scholar] [CrossRef]
- Panchuk, V.; Rabdano, N.; Goidenko, A.; Grebenyuk, A.; Irkaev, S.; Semenov, V. Determination of the oxidation state of iron by x-ray fluorescence spectroscopy using chemometric approaches. J. Anal. Chem. 2017, 72, 662–670. [Google Scholar] [CrossRef]
- Weidner, J. Separators and Membranes for Batteries, Capacitors, Fuel Cells, and Other Electrochemical Systems; Electrochemical Society: Pennington, NJ, USA, 2009. [Google Scholar]
- Hüfner, S. Photoelectron Spectroscopy: Principles and Applications; Springer: New York, NY, USA, 2003. [Google Scholar]
- Aulisa, E.; Gilliam, D. A Practical Guide to Geometric Regulation for Distributed Parameter Systems; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Shard, A. Detection limits in XPS for more than 6000 binary systems using Al and Mg Kα X-rays. Surf. Interface Anal. 2014, 46, 175–185. [Google Scholar] [CrossRef]
- Du, X.; Meng, J.; Xu, R.; Shi, Q.; Zhang, Y. Polyol-grafted polysulfone membranes for boron removal: Effects of the ligand structure. J. Membr. Sci. 2015, 476, 205–215. [Google Scholar] [CrossRef]
- Van der Heide, P. X-ray Photoelectron Spectroscopy: An Introduction to Principles and Practices; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Futter, G.A.; Latz, A.; Jahnke, T. Physical modeling of chemical membrane degradation in polymer electrolyte membrane fuel cells: Influence of pressure, relative humidity and cell voltage. J. Power Sources 2019, 410–411, 78–90. [Google Scholar] [CrossRef]
- Chen, C.; Levitin, G.; Hess, D.; Fuller, T. XPS investigation of Nafion® membrane degradation. J. Power Sources 2007, 169, 288–295. [Google Scholar] [CrossRef]
- Riviere, J.; Myhra, S. Handbook of Surface and Interface Analysis: Methods for Problem-Solving; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Stojilovic, N. Why can’t we see hydrogen in x-ray photoelectron spectroscopy? J. Chem. Edu. 2012, 89, 1331–1332. [Google Scholar] [CrossRef]















| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Agglomeration of particles in membranes | Polybenzimidazole (PBI) with titanium oxides particles | Cut samples in liquid nitrogen. Gold coated samples. | Low agglomeration effect | [31] |
| Film thickness | Polyacrylic acid (PAA)/polysulfone (PSf) | Stained samples by sodium hydroxide. Immersed samples in uranyl nitrate for 15 min. Washed samples with distilled water. Cut samples to 60-100 nm in thickness by ultramicrotome. | PAA film of 20 nm. | [29] |
| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Membrane purity | Polyetherimide | Samples fractured in liquid nitrogen. Carbon tape to hold the samples. | Only polyetherimide peaks were detected indicating a pure sample. | [15] |
| Polymer Crystallinity | Polyethersulfone with aluminosilicate particles | Not reported. | Intensity increased indicating a more crystallized structure with better mechanical properties. | [66] |
| Polymer chain distance (d-space) | 6H,12H-5,11-methanodibenzo[b,f][1,5]diazocine [PIM-EA(Me2)-TB] | Not reported. | Increase in d-space indicated transformation from glassy to rubbery phase. | [69] |
| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Polymer-filler interaction and particle size measurements | Sulfonated poly(aryl ether ketone) (SPEEK) with phosphomolybdic acid (PMoA) filler | Fixed wavelength of 1.5Å. Vacuum operation at room temperature. Data calibrated using positron-emission tomography (PET). | No detection of –SO3H group indicated no nano-phase separation. Broad peak showed amorphous SPEEK structure. Particle radius of 524 Å. | [80] |
| Pore size measurements | Polymer of intrinsic microporosity (PIM) with amidoxime groups | Not reported. | Pore size distribution from 3.9 to 5.9 Å. | [78] |
| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Membrane purity | Polyetherimide | Spinning carbon (13C) to generate the magnetic field. | Only peaks of carboxylic acid, carboxylic-amide-carbon, phenyl-carbon-oxygen, and carbon-aromatic ring-amine were detected indicating a pure polyetherimide. | [19] |
| Polymer-filler interaction | Polysulfone with functionalized carbon nanotubes | Dissolved samples in deuterated chloroform. | Peaks of NH2 protons and amino-benzo-crown ether demonstrated the functionalization of carbon nanotubes in the polymer. | [121] |
| Polymer miscibility | Polysulfone and polyvinyl methyl ether(PVME) | Not reported | Increase in polymer intensity in the mixture indicated a good mixing. | [123] |
| Membrane degradation | Perfluorinated ionomer (Nafion® 117) | Solid-state 19F NMR. Inserted samples in rotors filled with alcohol or water. | Detection of F−, SO4−2, and OH− indicated membrane decomposition | [125] |
| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Membrane purity | Polyetherimide | Cut samples in liquid nitrogen. Coated samples by gold. | No additional elements to polyetherimide were detected indicating a pure sample. | [15] |
| Restoration of a fouled membrane by chemical cleaning | Polypiperazine-amide | Not reported | Reduction in sulfur content due to chemical cleaning showed membrane restoration. | [131] |
| Filler chemical formula | Poly(vinyl alcohol) and bismuth(III) oxide fillers | Not reported | The calculated formula matched bismuth(III) oxide. | [132] |
| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Chemical formula of filler | Polycarbonate and iron chloride filler | Not reported | Elemental composition of iron and chloride gives chemical formula of FeCl3 that matched the added filler. | [139] |
| Mercury extraction from natural waters | Trioctylmethylammonium thiosalicylate (TOMATS) | Cut sample to disks of 1 to 3 cm in diameter. Palladium target X-ray tube with beryllium window. SPECTRA EDX software for intensity measurements. | Mercury extraction by the membrane was monitored by detecting the amounts of mercury in the polymer. | [140] |
| Study | Membrane | Methodology | Conclusion | Ref. |
|---|---|---|---|---|
| Surface chemical composition | polyol-grafted polysulfone | Monochromatic Al Kα X-ray source. Measurements at 45° take-off angle. Survey scan from 0 to 1000 eV then high-resolution scans of C1s regions. | O1s intensity increased while C1s intensity decreased confirming the grafting of hydroxyl groups on the membrane surface. | [148] |
| Membrane degradation | Nafion® 112 | Monochromator with Al Kα source. Step of 0.025 eV with 100ms. | Polymer backbone was decomposed due to detection of fluoride and sulfate ions. | [151] |
| Technique | Performed studies | Advantages | Limitations |
|---|---|---|---|
| SEM | Surface topography. Pore size. Particle size Membrane Thickness | Magnification of up to 1 million. | Samples needs to be conductive. Not accurate for measurements less 10 nm. |
| TEM | Nanoparticles. Nanofilms. Nanopores. Membrane thickness. | Higher resolution than SEM. | Images does not show topography data. Staining the sample may be required. Thick samples (> 100μm) are difficult to be analyzed in TEM. |
| AFM | Nano-profiling. Surface roughness. Pore size distribution. Membrane stiffness. | No sample preparation. Similar resolution to TEM. Measurements of mechanical properties. | Requires more processing time. Lower depth of field. |
| XRD | Membrane purity. Compounds chemical formula. Crystal structure. Polymer chain distance. | No sample preparation. Detection of wide range of crystalline compounds. | Heavy elements are less sensitive to XRD. Less accuracy for small crystals. Peaks overlap for some compounds. |
| SAXS WAXS | Crystal structure. Polymer-filler interaction. Particle size distribution. Pore size measurements. | No sample preparation. Suitable for semi- and non-crystalline materials. More accurate average measurements for particle size and pore size. | Scattering intensity can be weak for some systems. |
| FTIR | Functional groups. Polymer-solvent compatibility. Polymer-filler interaction. Miscibility of polymer blends. Membrane degradation. | Detection of variety of compounds. High sensitivity of parts per million (ppm). Fast analysis time (in seconds). | Cannot analyze aqueous samples. Cannot detect molecules of two identical atoms. |
| Raman | Functional groups. Crystal structure. Polymer chain orientation. Polymer blends. Membrane fouling. | No sample preparation. More sensitive to functional groups with better intensity peaks. | Release of fluorescent light of some samples may cause background noise. Polar molecules have lower Raman signal. |
| NMR | Functional groups. Polymer-blend miscibility. Membrane decomposition. | Less background interference. Detection of polar molecules. | Liquid samples. Paramagnetic elements have less NMR signal. |
| EDS | Elemental composition. Chemical formula of fillers. Membrane fouling. | Fast analysis time. | Samples needs to be conductive. Limitation in detecting light elements. Cannot quantify ions. |
| XRF | Elemental composition. Chemical formula of fillers. Membrane degradation. | No sample preparation. In-situ analysis. | Very low sensitivity to hydrogen, carbon and oxygen. Cannot quantify ions. Unsuitable for thin film measurements. |
| XPS | Elemental composition. Functional groups. Formula of chemical compounds. Membrane degradation. | High sensitivity. Quantification of ions. Measurements of thin films of nm. | Solid samples. Cannot detect hydrogen and helium. Peaks Overlap for some elements. Long processing time. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqaheem, Y.; Alomair, A.A. Microscopy and Spectroscopy Techniques for Characterization of Polymeric Membranes. Membranes 2020, 10, 33. https://doi.org/10.3390/membranes10020033
Alqaheem Y, Alomair AA. Microscopy and Spectroscopy Techniques for Characterization of Polymeric Membranes. Membranes. 2020; 10(2):33. https://doi.org/10.3390/membranes10020033
Chicago/Turabian StyleAlqaheem, Yousef, and Abdulaziz A. Alomair. 2020. "Microscopy and Spectroscopy Techniques for Characterization of Polymeric Membranes" Membranes 10, no. 2: 33. https://doi.org/10.3390/membranes10020033
APA StyleAlqaheem, Y., & Alomair, A. A. (2020). Microscopy and Spectroscopy Techniques for Characterization of Polymeric Membranes. Membranes, 10(2), 33. https://doi.org/10.3390/membranes10020033