
O-GlcNAcylation Inhibits Endocytosis of Amyloid Precursor Protein by Decreasing Its Localization in Lipid Raft Microdomains


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Experimental Treatments
2.2. Rat Primary Hippocampal Neuron Culture
2.3. Colocalization of APP and Lipid Rafts
2.4. Lipid Raft Fractionation
2.5. Transferrin Uptake
2.6. Immunoprecipitation
2.7. Western Blotting
2.8. The Localization of Surface APP in Lipid Raft Microdomains
2.9. Rate of Endocytosis of Surface APP in Lipid Raft Microdomains
2.10. Colocalization of APP with Early Endosomes
2.11. Statistical Analysis
3. Results
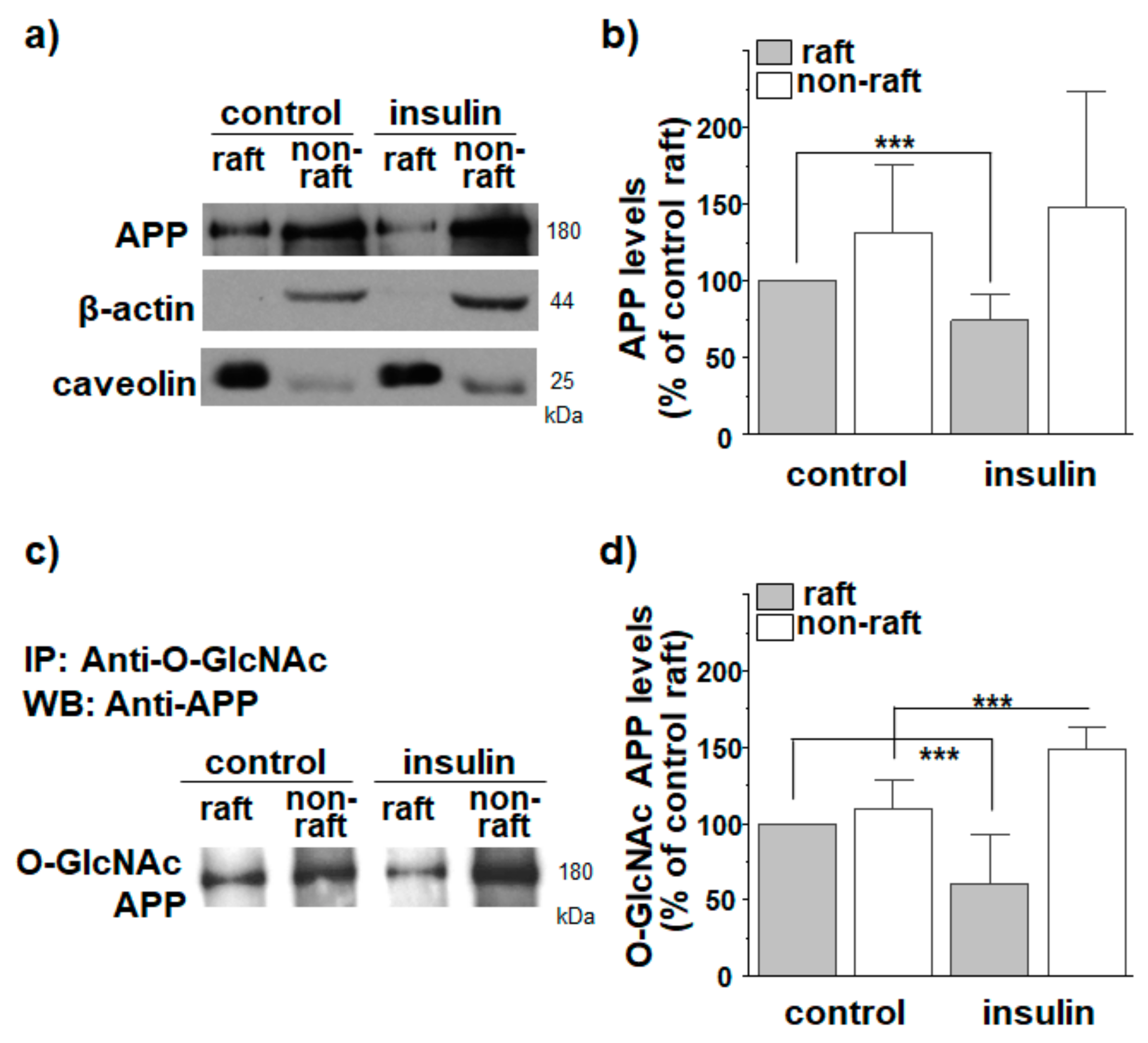
3.1. Insulin Decreases APP Localization in Lipid Rafts
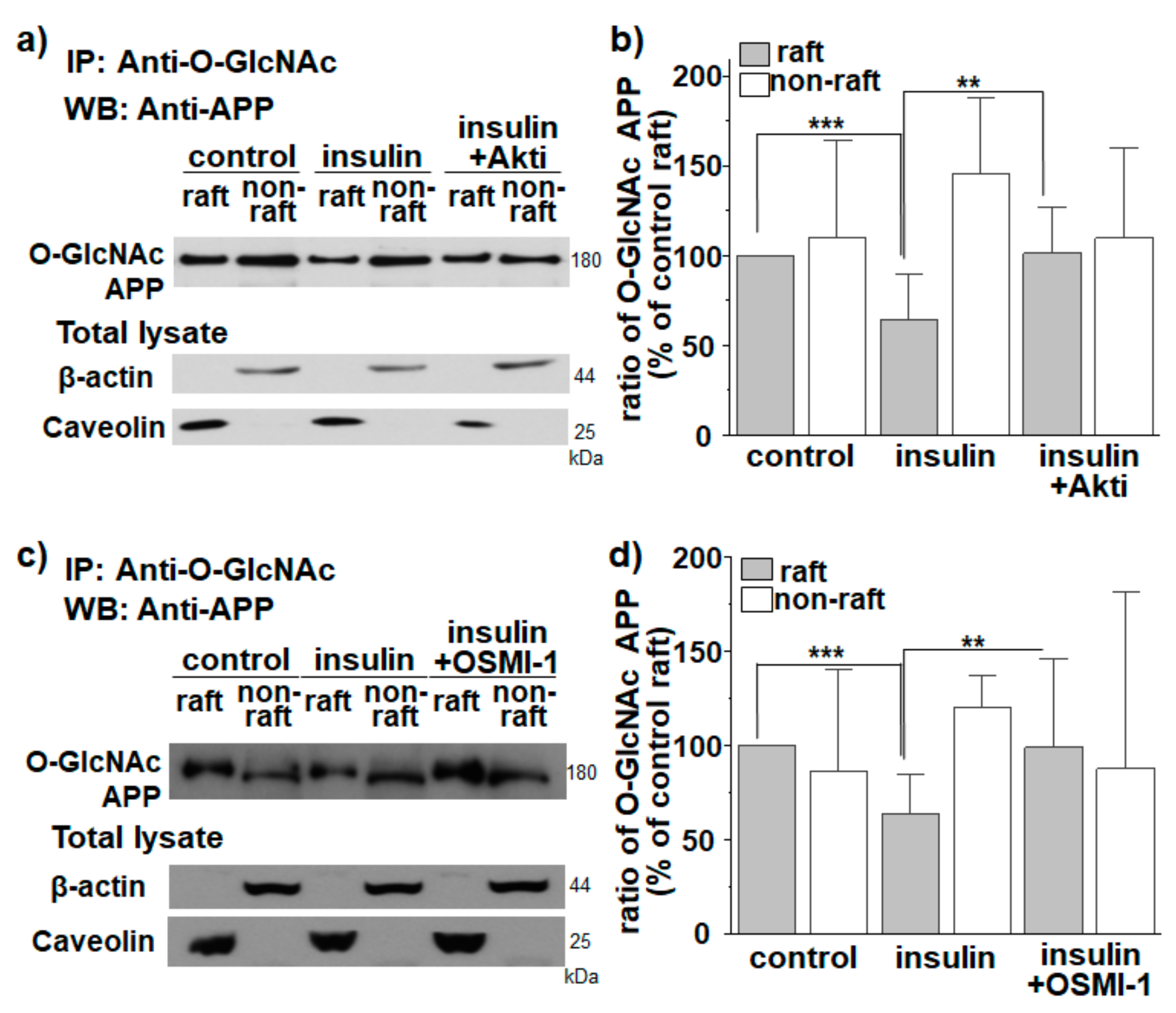
3.2. APP O-GlcNAcylation in Response to Insulin Translocates APP from Lipid Rafts to Non-Rafts
3.3. APP O-GlcNAcylation in Response to Insulin Decreases APP Internalization from Lipid Rafts
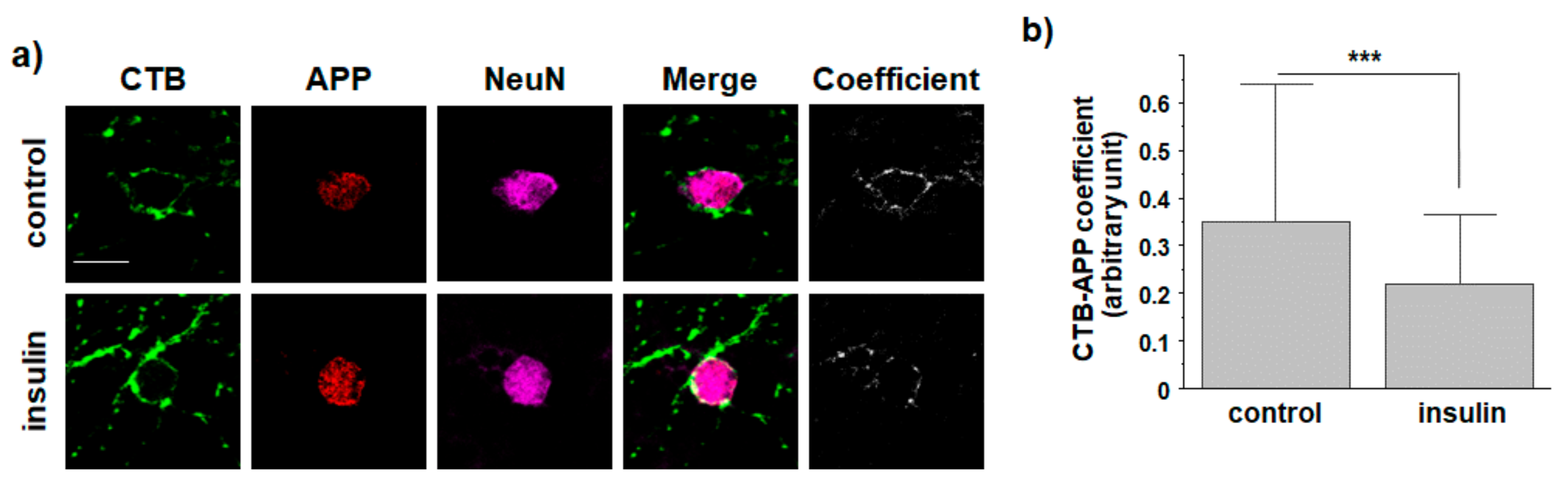
3.4. O-GlcNAcylation in Response to Insulin Promotes Translocation of Endogenous APP into Non-Rafts from Cultured Hippocampal Neurons
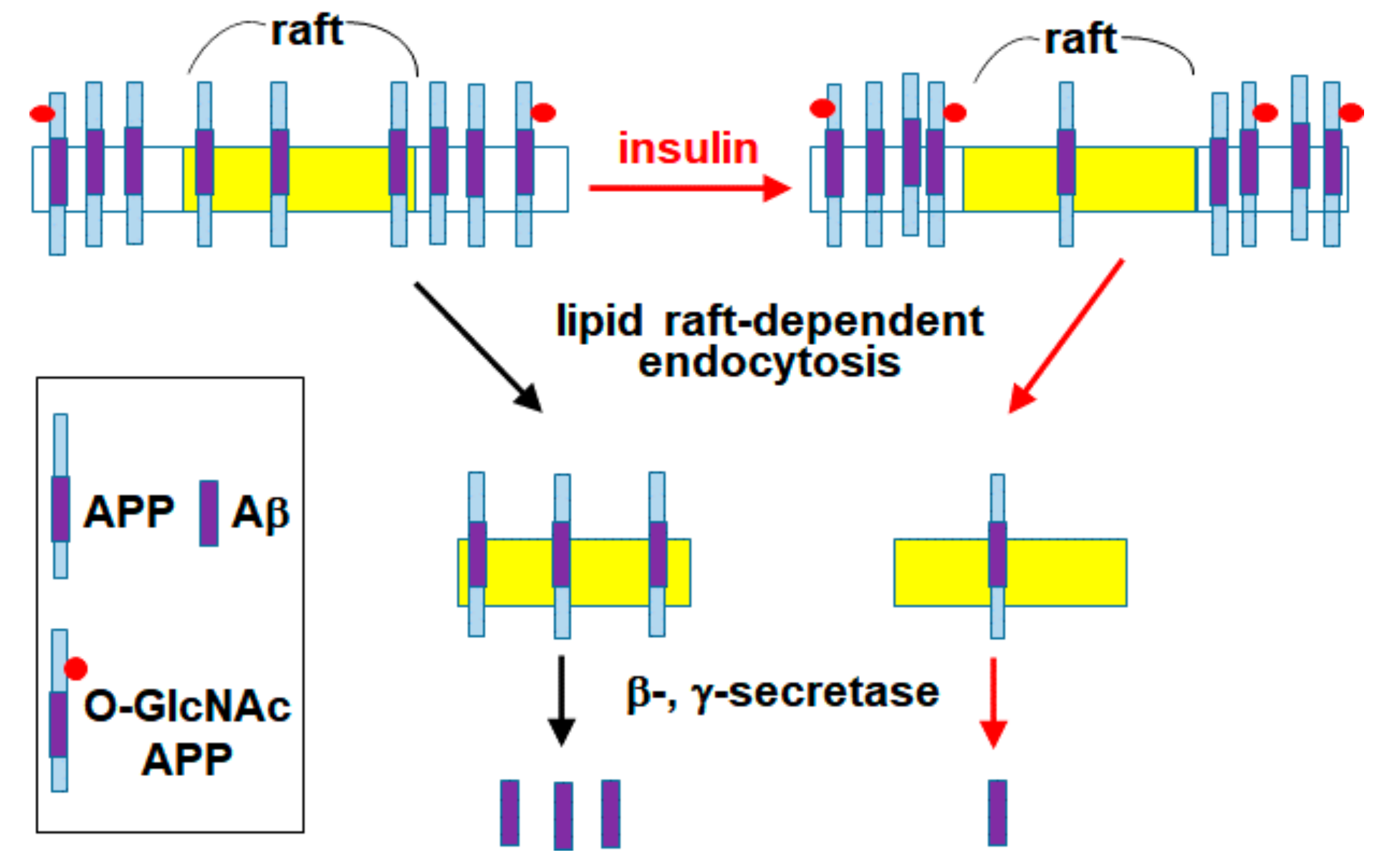
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Burns, A.; Iliffe, S. Alzheimer’s disease. Brit. Med. J. 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crous-Bou, M.; Minguillón, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimers Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757. [Google Scholar] [CrossRef]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013, 9, 63–75. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Parvathy, S.; Hussain, I.; Karran, E.H.; Turner, A.J.; Hooper, N.M. Cleavage of Alzheimer’s amyloid precursor protein by alpha-secretase occurs at the surface of neuronal cells. Biochemistry 1999, 38, 9728–9734. [Google Scholar] [CrossRef]
- Knops, J.; Suomensaari, S.; Lee, M.; McConlogue, L.; Seubert, P.; Sinha, S. Cell-type and amyloid precursor protein-type specific inhibition of A beta release by bafilomycin A1, a selective inhibitor of vacuolar ATPases. J. Biol. Chem. 1995, 270, 2419–2422. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R. BACE1: The beta-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Wahle, T.; Prager, K.; Raffler, N.; Haass, C.; Famulok, M.; Walter, J. GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol. Cell. Neurosci. 2005, 29, 453–461. [Google Scholar] [CrossRef]
- Perez, R.G.; Soriano, S.; Hayes, J.D.; Ostaszewski, B.; Xia, W.; Selkoe, D.J.; Chen, X.; Stokin, G.B.; Koo, E.H. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Biol. Chem. 1999, 274, 18851–18856. [Google Scholar] [CrossRef] [Green Version]
- Claeysen, S.; Cochet, M.; Donneger, R.; Dumuis, A.; Bockaert, J.; Giannoni, P. Alzheimer culprits: Cellular crossroads and interplay. Cell. Signal. 2012, 24, 1831–1840. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef]
- Marcelli, S.; Corbo, M.; Iannuzzi, F.; Negri, L.; Blandini, F.; Nistico, R.; Feligioni, M. The Involvement of Post-Translational Modifications in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 313–335. [Google Scholar] [CrossRef]
- Ruan, H.B.; Singh, J.P.; Li, M.D.; Wu, J.; Yang, X. Cracking the O-GlcNAc code in metabolism. Trends. Endocrinol. Metab. 2013, 24, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Nagel, A.K.; Ball, L.E. O-GlcNAc transferase and O-GlcNAcase: Achieving target substrate specificity. Amino Acids 2014, 46, 2305–2316. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Tjian, R. O-glycosylation of eukaryotic transcription factors: Implications for mechanisms of transcriptional regulation. Cell 1988, 55, 125–133. [Google Scholar] [CrossRef]
- Dehennaut, V.; Leprince, D.; Lefebvre, T. O-GlcNAcylation, an Epigenetic Mark Focus on the Histone Code, TET Family Proteins, and Polycomb Group Proteins. Front. Endocrinol. Lausanne 2014, 5, 155. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.A.; Hanover, J.A. O-GlcNAc and the epigenetic regulation of gene expression. J. Biol. Chem. 2014, 289, 34440–34448. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.P.; Zhang, K.; Wu, J.; Yang, X. O-GlcNAc signaling in cancer metabolism and epigenetics. Cancer Lett. 2015, 356, 244–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, S.A.; Dias, W.B.; Thiruneelakantapillai, L.; Lane, M.D.; Hart, G.W. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-Linked beta-N-acetylglucosamine in 3T3-L1 adipocytes. J. Biol. Chem. 2010, 285, 5204–5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, P.J.; Coleman, P.D. Reduction of O-linked N-acetylglucosamine-modified assembly protein-3 in Alzheimer’s disease. J. Neurosci. 1998, 18, 2399–2411. [Google Scholar] [CrossRef] [Green Version]
- Cha, M.Y.; Cho, H.J.; Kim, C.; Jung, Y.O.; Kang, M.J.; Murray, M.E.; Hong, H.S.; Choi, Y.J.; Choi, H.; Kim, D.K.; et al. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 6492–6504. [Google Scholar] [CrossRef] [Green Version]
- Griffith, L.S.; Mathes, M.; Schmitz, B. Beta-amyloid precursor protein is modified with O-linked N-acetylglucosamine. J. Neurosci. Res. 1995, 41, 270–278. [Google Scholar] [CrossRef]
- Jacobsen, K.T.; Iverfeldt, K. O-GlcNAcylation increases non-amyloidogenic processing of the amyloid-beta precursor protein (APP). Biochem. Biophys. Res. Commun. 2011, 404, 882–886. [Google Scholar] [CrossRef]
- Chun, Y.S.; Park, Y.; Oh, H.G.; Kim, T.W.; Yang, H.O.; Park, M.K.; Chung, S. O-GlcNAcylation promotes non-amyloidogenic processing of amyloid-β protein precursor via inhibition of endocytosis from the plasma membrane. J. Alzheimers Dis. 2015, 44, 261–275. [Google Scholar] [CrossRef]
- Kwon, O.H.; Cho, Y.Y.; Kim, T.W.; Chung, S. O-GlcNAcylation of Amyloid-β Protein Precursor by Insulin Signaling Reduces Amyloid-β Production. J. Alzheimers Dis. 2019, 69, 1195–1211. [Google Scholar] [CrossRef]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef] [Green Version]
- Joseph D’Ercole, A.; Ye, P. Expanding the mind: Insulin-like growth factor I and brain development. Endocrinology 2008, 149, 5958–5962. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.J.; Khatoon, S.; An, W.L.; Nordlinder, M.; Tanaka, T.; Braak, H.; Tsujio, I.; Takeda, M.; Alafuzoff, I.; Winblad, B. Role of protein kinase B in Alzheimer’s neurofibrillary pathology. Acta Neuropathol. 2003, 105, 381–392. [Google Scholar] [CrossRef]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Chua, L.M.; Lim, M.L.; Chong, P.R.; Hu, Z.P.; Cheung, N.S.; Wong, B.S. Impaired neuronal insulin signaling precedes Abeta42 accumulation in female AbetaPPsw/PS1DeltaE9 mice. J. Alzheimers Dis. 2012, 29, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; D’Ercole, J. Insulin-like growth factor I (IGF-I) regulates IGF binding protein-5 gene expression in the brain. Endocrinology 1998, 139, 65–71. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, J.; Meichsner, S.; Kölsch, H.; Lewczuk, P.; Maier, W.; Kornhuber, J.; Jessen, F.; Lütjohann, D. Cerebral and extracerebral cholesterol metabolism and CSF markers of Alzheimer’s disease. Biochem. Pharm. 2013, 86, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Allinquant, B.; Clamagirand, C.; Potier, M.C. Role of cholesterol metabolism in the pathogenesis of Alzheimer’s disease. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Hancock, J.F. Lipid rafts: Contentious only from simplistic standpoints. Nat. Rev. Mol. Cell Biol. 2006, 7, 456–462. [Google Scholar] [CrossRef] [Green Version]
- Helms, J.B.; Zurzolo, C. Zurzolo, Lipids as targeting signals: Lipid rafts and intracellular trafficking. Traffic 2004, 5, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Kenworthy, A.K.; Sanders, C.R. Cholesterol as a co-solvent and a ligand for membrane proteins. Protein. Sci. 2014, 23, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef] [Green Version]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [Green Version]
- Vetrivel, K.S.; Cheng, H.; Kim, S.H.; Chen, Y.; Barnes, N.Y.; Parent, A.T.; Sisodia, S.S.; Thinakaran, G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J. Biol. Chem. 2005, 280, 25892–25900. [Google Scholar] [CrossRef] [Green Version]
- Frick, M.; Bright, N.A.; Riento, K.; Bray, A.; Merrified, C.; Nichols, B.J. Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr. Biol. 2007, 17, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Lajoie, P.; Nabi, I.R. Lipid rafts, caveolae, and their endocytosis. Int. Rev. Cell Mol. Biol. 2010, 282, 135–163. [Google Scholar]
- Beel, A.J.; Sakakura, M.; Barrett, P.J.; Sanders, C.R. Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships? Biochim. Biophys. Acta 2010, 1801, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Thinakaran, G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim. Biophys. Acta 2010, 1801, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.Y.; Kwon, O.H.; Park, M.K.; Kim, T.W.; Chung, S. Elevated cellular cholesterol in Familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of β-amyloid precursor protein. PLoS ONE 2019, 14, e0210535. [Google Scholar] [CrossRef]
- Cho, Y.Y.; Kwon, O.H.; Chung, S. Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains. Molecules 2020, 25, 5490. [Google Scholar] [CrossRef]
- McIntire, L.B.; Landman, N.; Kang, M.S.; Finan, G.M.; Hwang, J.C.; Moore, A.Z.; Park, L.S.; Lin, C.S.; Kim, T.W. Phenotypic assays for β-amyloid in mouse embryonic stem cell-derived neurons. Chem. Biol. 2013, 20, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnapen, D.J.; Chinnapen, H.; Saslowsky, D.; Lencer, W.I. Rafting with cholera toxin: Endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007, 266, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohl, C.R.; Abrahamyan, L.G.; Wood, C. Human Ubc9 is involved in intracellular HIV-1 Env stability after trafficking out of the trans-Golgi network in a Gag dependent manner. PLoS ONE 2013, 8, e69359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhungana, S.; Merrick, B.A.; Tomer, K.B.; Fessler, M.B. Quantitative proteomics analysis of macrophage rafts reveals compartmentalized activation of the proteasome and of proteasome-mediated ERK activation in response to lipopolysaccharide. Mol. Cell Proteom. 2009, 8, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wakim, B.; Li, M.; Halligan, B.; Tint, G.S.; Patel, S.B. Quantifying raft proteins in neonatal mouse brain by ‘tube-gel’ protein digestion label-free shotgun proteomics. Proteome Sci. 2007, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; McMahon, H.T. McMahon, Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Raghu, H.; Sodadasu, P.K.; Malla, R.R.; Gondi, C.S.; Estes, N.; Rao, J.S. Localization of uPAR and MMP-9 in lipid rafts is critical for migration, invasion and angiogenesis in human breast cancer cells. BMC Cancer 2010, 10, 647. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Vetrivel, K.S.; Drisdel, R.C.; Meckler, X.; Gong, P.; Leem, J.Y.; Li, T.; Carter, M.; Chen, Y.; Nguyen, P.; et al. S-palmitoylation of gamma-secretase subunits nicastrin and APH-1. J. Biol. Chem. 2009, 284, 1373–1384. [Google Scholar] [CrossRef] [Green Version]
- Vetrivel, K.S.; Meckler, X.; Chen, Y.; Nguyen, P.D.; Seidah, N.G.; Vassar, R.; Wong, P.C.; Fukata, M.; Kounnas, M.Z.; Thinakaran, G. Alzheimer disease Abeta production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J. Biol. Chem. 2009, 284, 3793–3803. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Nam, D.W.; Park, S.Y.; Song, H.; Hong, H.S.; Boo, J.H.; Jung, E.S.; Kim, Y.; Baek, J.Y.; Kim, K.S.; et al. O-linked β-N-acetylglucosaminidase inhibitor attenuates β-amyloid plaque and rescues memory impairment. Neurobiol. Aging 2013, 34, 275–285. [Google Scholar] [CrossRef]
- Kizuka, Y.; Nakano, M.; Kitazume, S.; Saito, T.; Saido, T.C.; Taniguchi, N. Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem. J. 2016, 473, 21–30. [Google Scholar] [CrossRef]
- Simons, K.; Gerl, M.J. Revitalizing membrane rafts: New tools and insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688–699. [Google Scholar] [CrossRef]
- Munro, S. Lipid rafts: Elusive or illusive? Cell 2003, 115, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.J. The challenge of lipid rafts. J. Lipid Res. 2008, 50, S323–S328. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, M.; Suzuki, K.G.N.; Murata, M.; Matsumori, N. Evidence of lipid rafts based on the partition and dynamic behavior of sphingomyelins. Chem. Phys. Lipids 2018, 215, 84–95. [Google Scholar] [CrossRef]
- Charollais, J.; Van Der Goot, F.G. Palmitoylation of membrane proteins (Review). Mol. Membr. Biol. 2009, 26, 55–66. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef] [Green Version]
- Pietrzik, C.U.; Yoon, I.S.; Jaeger, S.; Busse, T.; Weggen, S.; Koo, E.H. FE65 constitutes the functional link between the low-density lipoprotein receptor-related protein and the amyloid precursor protein. J. Neurosci. 2004, 24, 4259–4265. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.; Liu, X.; Südhof, T.C. Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2008, 28, 14392–14400. [Google Scholar] [CrossRef] [Green Version]
- Kienlen-Campard, P.; Tasiaux, B.; Van Hees, J.; Li, M.; Huysseune, S.; Sato, T.; Fei, J.Z.; Aimoto, S.; Courtoy, P.J.; Smith, S.O.; et al. Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GXXXG motifs. J. Biol. Chem. 2008, 283, 7733–7744. [Google Scholar] [CrossRef] [Green Version]
- Eggert, S.; Midthune, B.; Cottrell, B.; Koo, E.H. Induced dimerization of the amyloid precursor protein leads to decreased amyloid-beta protein production. J. Biol. Chem. 2009, 284, 28943–28952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Hustedt, E.J.; Brandon, S.; Sanders, C.R. Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 2013, 52, 5051–5064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, R.; Fenn, R.H.; Barren, C.; Tanzi, R.E.; Kovacs, D.M. Palmitoylated APP Forms Dimers, Cleaved by BACE1. PLoS ONE 2016, 11, e0166400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandy, J.C.; Rountree, A.E.; Bijur, G.N. Akt1 is dynamically modified with O-GlcNAc following treatments with PUGNAc and insulin-like growth factor-1. FEBS Lett. 2006, 580, 3051–3058. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Huang, X.; Sun, D.; Xin, X.; Pan, Q.; Peng, S.; Liang, Z.; Luo, C.; Yang, Y.; Jiang, H.; et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PLoS ONE 2012, 7, e37427. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, O.-H.; Cho, Y.Y.; Lee, J.H.; Chung, S. O-GlcNAcylation Inhibits Endocytosis of Amyloid Precursor Protein by Decreasing Its Localization in Lipid Raft Microdomains. Membranes 2021, 11, 909. https://doi.org/10.3390/membranes11120909
Kwon O-H, Cho YY, Lee JH, Chung S. O-GlcNAcylation Inhibits Endocytosis of Amyloid Precursor Protein by Decreasing Its Localization in Lipid Raft Microdomains. Membranes. 2021; 11(12):909. https://doi.org/10.3390/membranes11120909
Chicago/Turabian StyleKwon, Oh-Hoon, Yoon Young Cho, Jung Hee Lee, and Sungkwon Chung. 2021. "O-GlcNAcylation Inhibits Endocytosis of Amyloid Precursor Protein by Decreasing Its Localization in Lipid Raft Microdomains" Membranes 11, no. 12: 909. https://doi.org/10.3390/membranes11120909
APA StyleKwon, O.-H., Cho, Y. Y., Lee, J. H., & Chung, S. (2021). O-GlcNAcylation Inhibits Endocytosis of Amyloid Precursor Protein by Decreasing Its Localization in Lipid Raft Microdomains. Membranes, 11(12), 909. https://doi.org/10.3390/membranes11120909