
Cytochrome P450 and P-gp Mediated Herb-Drug Interactions and Molecular Docking Studies of Garcinol


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals
2.3. In-Vitro CYP 450 Incubation Studies
2.4. Time-Dependent Inhibitory Effects of Garcinol on CYP Isoforms Using IC50 Shift Assay Method
2.5. Characterization of Molecular Structures of Small Molecule and Protein Crystal Structures
2.6. In-Vivo CYP 450 Studies
2.7. Analysis of CYP Metabolites
2.8. Western Blotting Analysis
2.9. In-Vivo P-gp Pharmacokinetic Studies
2.10. Analysis of Digoxin in Rat Plasma
2.11. Data Analysis
2.12. Statistical Analysis
3. Results
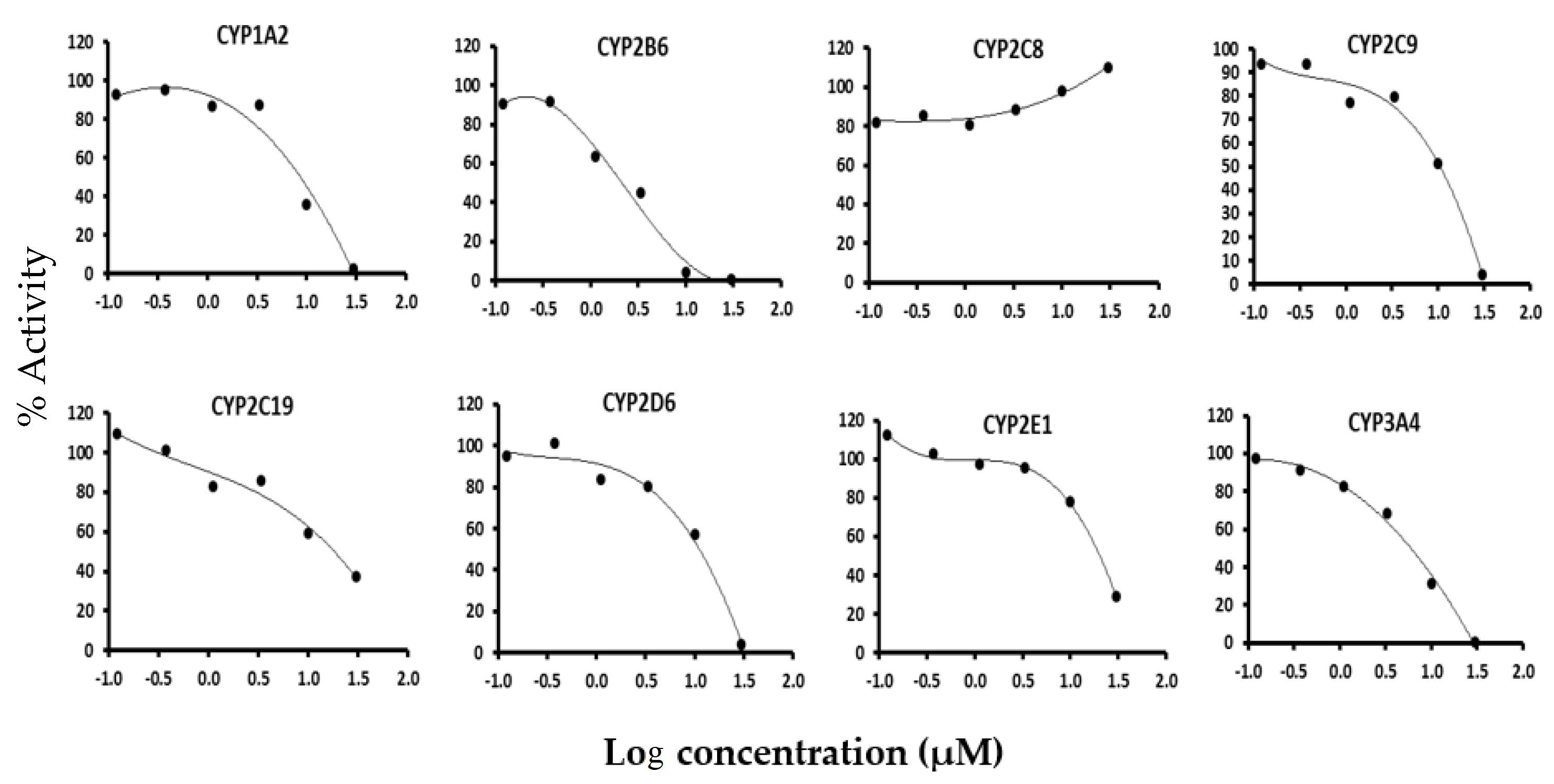
3.1. Screening for Reversible and Irreversible Inhibition of Garcinol on the Eight CYP Isoforms
3.2. Molecular Docking Studies of Garcinol on CYP2B6 and CYP3A4
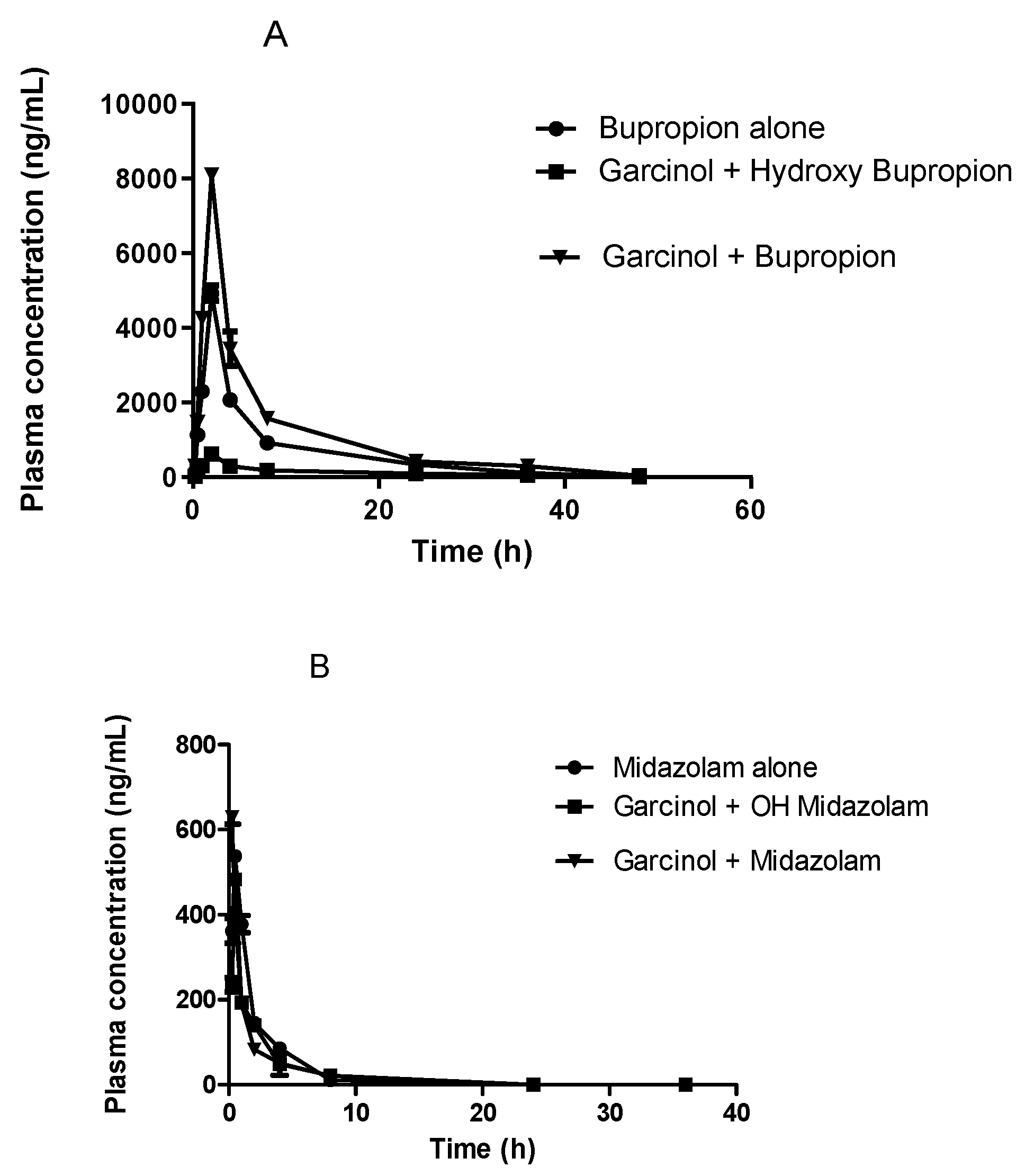
3.3. Effects of Garcinol on the Activity of CYP Isoforms In-Vivo
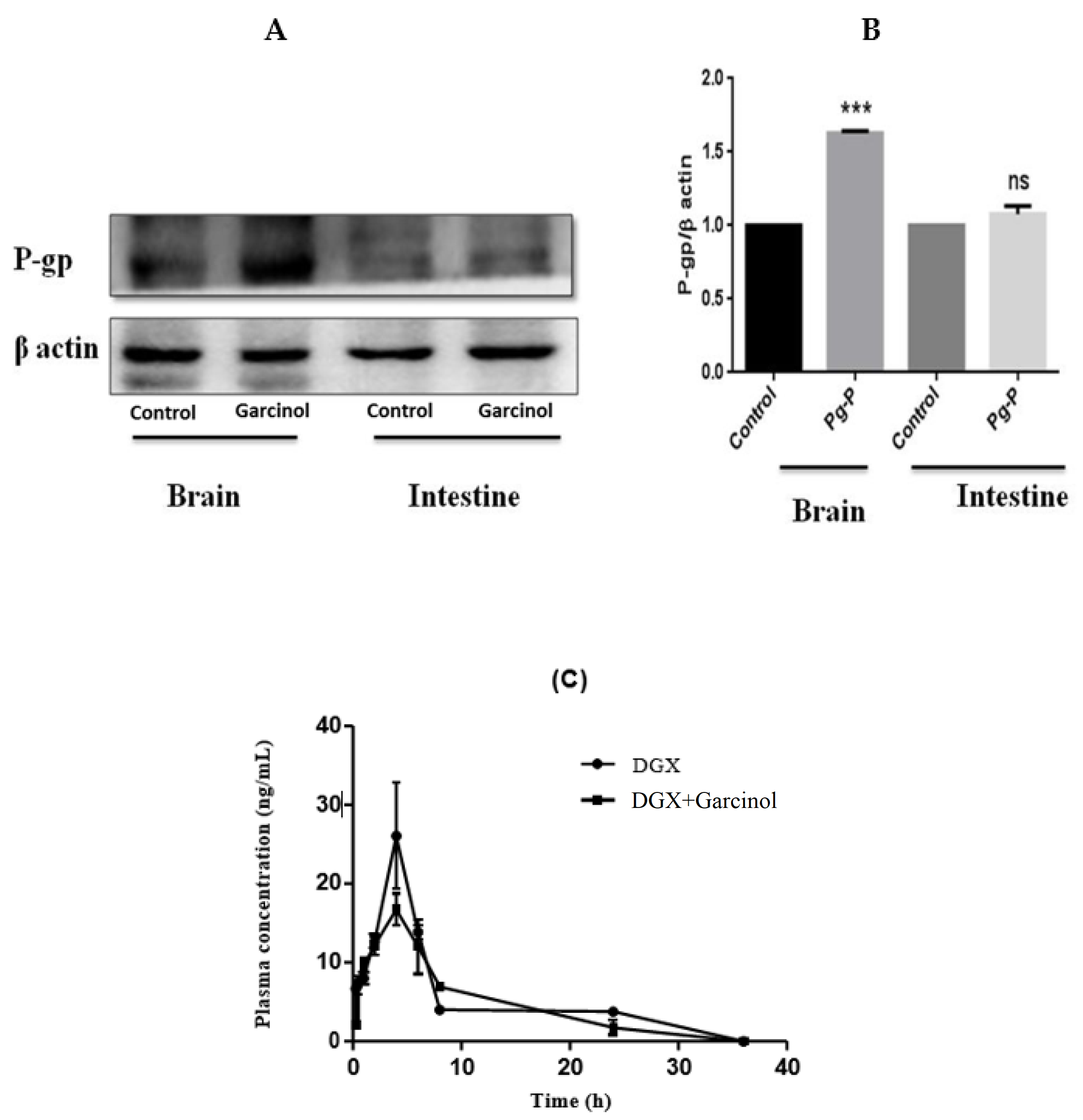
3.4. Expression of P-gp by Garcinol in Rat Brain and Intestinal Tissues
3.5. Effect of Single Dose of Garcinol on Plasma Concentration of P-gp Substrate in SD Rats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramachandran, H.; Fayaz, P.; Kusum, R. Plant profile, phytochemistry and pharmacology of garcinia indica: A review. Int. J. Pharm. Sci. Rev. Res. 2014, 27, 361–366. [Google Scholar]
- Chuah, L.O.; Ho, W.Y.; Beh, B.K.; Yeap, S.K. Updates on Antiobesity Effect of Garcinia Origin (-)-HCA. Evid Based Complement Alternat. Med. 2013, 2013, 751658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seethapathy, G.S.; Tadesse, M.; Urumarudappa, S.K.J.; Gunaga, S.V.; Vasudeva, R.; Malterud, K.E.; Shaanker, R.U.; de Boer, H.J.; Ravikanth, G.; Wangensteen, H. Authentication of Garcinia fruits and food supplements using DNA barcoding and NMR spectroscopy. Sci. Rep. 2018, 8, 10561. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, J.; Masullo, M.; Olas, B.; Piacente, S.; Wachowicz, B. Effects of garcinol and guttiferone K isolated from Garcinia cambogia on oxidative/nitrative modifications in blood platelets and plasma. Platelets 2009, 20, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Ohia, S.E.; Opere, C.A.; LeDay, A.M.; Bagchi, M.; Bagchi, D.; Stohs, S.J. Safety and mechanism of appetite suppression by a novel hydroxycitric acid extract (HCA-SX). Mol. Cell Biochem. 2002, 238, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Ho, P.C.-L.; Wong, F.C.; Sethi, G.; Wang, L.Z.; Goh, B.C. Garcinol: Current status of its anti-oxidative, anti-inflammatory and anti-cancer effects. Cancer Lett. 2015, 362, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-H.; Chang, W.-L.; Lin-Shiau, S.-Y.; Ho, C.-T.; Lin, J.-K. Induction of apoptosis by garcinol and curcumin through cytochrome c release and activation of caspases in human leukemia HL-60 cells. J. Agric. Food Chem. 2001, 49, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Majeed, M.; Majeed, S.; Nagabhushanam, K.; Lawrence, L.; Mundkur, L. Garcinia indica extract standardized for 20% Garcinol reduces adipogenesis and high fat diet-induced obesity in mice by alleviating endoplasmic reticulum stress. J. Funct. Foods. 2020, 67, 103863. [Google Scholar] [CrossRef]
- Lee, P.S.; Teng, C.Y.; Kalyanam, N.; Ho, C.T.; Pan, M.H. Garcinol Reduces Obesity in High-Fat-Diet-Fed Mice by Modulating Gut Microbiota Composition. Mol. Nutr. Food Res. 2019, 63, e1970025. [Google Scholar]
- Nanjappan, S.; Paul, D.; Bolla, L. Assessing herb–drug interactions of herbal products with therapeutic agents for metabolic diseases: Analytical and regulatory perspectives. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 283–322. [Google Scholar]
- Surendran, S.; Dhurjad, P.; Nanjappan, S. Phytotherapeutics: The Rising Role of Drug Transporters in Herb-Drug Interactions with Botanical Supplements. In Evidence Based Validation of Traditional Medicines; Mandal, S.C., Chakraborty, R., Sen, S., Eds.; Springer: Singapore, 2021; pp. 469–494. [Google Scholar]
- Shi, S.; Klotz, U. Drug interactions with herbal medicines. Clin. Pharmacokinet. 2012, 51, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Mitra, A.K. MDR-and CYP3A4-mediated drug–herbal interactions. Life Sci. 2006, 78, 2131–2145. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, T.; Kato, M.; Takano, J.; Sugiyama, Y. Predicting drug-drug interactions involving the inhibition of intestinal CYP3A4 and P-glycoprotein. Curr. Drug Metab. 2010, 11, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, T.; Kato, M.; Watanabe, T.; Mitsui, T.; Sugiyama, Y. Method for predicting the risk of drug–drug interactions involving inhibition of intestinal CYP3A4 and P-glycoprotein. Xenobiotica 2009, 39, 430–443. [Google Scholar] [CrossRef]
- Benet, L.Z. The drug transporter-metabolism alliance: Uncovering and defining the interplay. Mol. Pharm. 2009, 6, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, Y.; Huang, S.-M. Scientific and Regulatory Perspectives on Metabolizing Enzyme—Transporter Interplay and Its Role in Drug Interactions: Challenges in Predicting Drug Interactions. Mol. Pharm. 2009, 6, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Valicherla, G.R.; Mishra, A.; Lenkalapelly, S.; Jillela, B.; Francis, F.M.; Rajagopalan, L.; Srivastava, P. Investigation of the Inhibition of Eight Major Human Cytochrome P450 Isozymes by a Probe Substrate Cocktail in vitro with Emphasis on CYP2E1. Xenobiotica 2019, 49, 1396–1402. [Google Scholar] [CrossRef]
- Shah, M.B.; Liu, J.; Zhang, Q.; Stout, C.D.; Halpert, J.R. Halogen—π interactions in the cytochrome P450 active site: Structural insights into human CYP2B6 substrate selectivity. ACS Chem. Biol. 2017, 12, 1204–1210. [Google Scholar] [CrossRef] [Green Version]
- Sevrioukova, I.F.; Poulos, T.L. Dissecting cytochrome P450 3A4–ligand interactions using ritonavir analogues. Biochemistry 2013, 52, 4474–4481. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, H.; Yano, I.; Ito, T.; Hashida, T.; Futami, T.; Nohara, R.; Sasayama, S.; Inui, K. Effect of clarithromycin on renal excretion of digoxin: Interaction with P-glycoprotein. Clin. Pharmacol. Ther. 1998, 64, 123–128. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Ichikawa, H.; Garodia, P.; Weerasinghe, P.; Sethi, G.; Bhatt, I.D.; Pandey, M.K.; Shishodia, S.; Nair, M.G. From traditional Ayurvedic medicine to modern medicine: Identification of therapeutic targets for suppression of inflammation and cancer. Expert Opin. Ther. Targets. 2006, 10, 87–118. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.-L.; Takahashi, K.; Tanaka, K.; Tougou, K.; Qiu, F.; Komatsu, K.; Takahashi, K.; Azuma, J. Curcuma drugs and curcumin regulate the expression and function of P-gp in Caco-2 cells in completely opposite ways. Int. J. Pharm. 2008, 358, 224–229. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| CYP Isoform | Substrates | Concentration (µM) | Metabolites | Transition (m/z) | Polarity | CE (eV) |
|---|---|---|---|---|---|---|
| CYP1A2 | Tacrine | 50 | 1-Hydroxy Tacrine | 215.3 → 182.1 | ES+ | 43 |
| CYP2B6 | Bupropion | 50 | Hydroxy Bupropion | 256.1 → 238.0 | ES+ | 7 |
| CYP2C8 | Paclitaxel | 50 | 6-Hydroxy Paclitaxel | 870.0 → 286.0 | ES+ | 17 |
| CYP2C9 | Diclofenac | 50 | 4-Hydroxy Diclofenac | 312.2 → 230.2 | ES+ | 12 |
| CYP2C19 | S-Mephenytoin | 300 | 4-Hydroxy Mephenytoin | 235.2 → 150.2 | ES+ | 25 |
| CYP2D6 | Dextromethorphan | 50 | Dextrorphan | 258.2 → 157 | ES+ | 25 |
| CYP2E1 | Chlorzoxazone | 2000 | 6-Hydroxy Chlorzoxazone | 183.0 → 120.0 | ES- | −28 |
| CYP3A4 | Midazolam | 20 | 1-Hydroxy Midazolam | 342.2 → 203.0 | ES+ | 18 |
| Internal Standard | - | - | Telmisartan | 515.4 → 276.1 | ES+ | 50 |
| CYP Isoform | Substrates | Inhibitors | IC50 (µM) Garcinol |
|---|---|---|---|
| CYP1A2 | Tacrine | α-naphthoflavone | 7.6 * |
| CYP2B6 | Bupropion | Ticlopidine | 2.1 * |
| CYP2C8 | Paclitaxel | Quercetin | >30 |
| CYP2C9 | Diclofenac | Sulphaphenazole | 8.0 * |
| CYP2C19 | S-Mephenytoin | N-Benzyl nirvanol | 16.4 # |
| CYP2D6 | Dextromethorphan | Quinidine | 9.5 * |
| CYP2E1 | Chlorzoxazone | 4-Methyl Pyrazole | 19 # |
| CYP3A4 | Midazolam | Ketoconazole | 5.1 * |
| Parameters | Control Group (Bupropion Alone) | Bupropion with Garcinol | Hydroxy Bupropion with Garcinol |
|---|---|---|---|
| Cmax (ng/mL) | 4929.05 ±198.13 | 8095.72 ± 120.63 | 632.02 ± 30.55 |
| AUC0-∞ (h*ng/mL) | 29,335.73 ± 2034.86 | 48,768.38 ±1279.63 | 6200.97 ± 106.97 |
| AUClast (h*ng/mL) | 29,212.34 ± 2057.52 | 48,245.49 ± 1243.61 | 6057.86 ± 101.41 |
| Tmax (h) | 1.5 ± 2.31 | 2.5 ± 3.52 | 1 ± 1.93 |
| t1/2 (h) | 6.11 ± 1.03 | 7.93 ± 0.08 | 10.23 ± 0.04 |
| Cl (ml/h/kg) | 341.99 ± 24.16 | 205.14 ± 5.31 | NA |
| Cmax (ng/mL) | 537.63 ± 10.24 | 828.69 ± 15.65 | 482.85 ± 14.34 |
| AUC0-∞ (h*ng/mL) | 1023.21 ± 58.29 | 1138.22 ± 77.38 | 607.65 ± 34.26 |
| AUClast (h*ng/mL) | 998.54 ± 53.44 | 1237.81 ± 18.59 | 737.45 ± 66.95 |
| Tmax (h) | 0.25 ±3.2 | 0.5 ±1.23 | 0.25 ± 1.92 |
| t1/2 (h) | 1.51 ± 0.18 | 3.12 ± 0.88 | 2.15 ± 0.36 |
| Cl (ml/h/kg) | 9795.11 ± 577.02 | 11,996.63 ± 1083.16 | NA |
| Groups | Cmax (ng/mL) | AUClast (h*ng/mL) | AUC0-∞ (h*ng/mL) | t1/2 (h) | Cl (ml/h/kg) |
|---|---|---|---|---|---|
| DGX | 26.075 ± 5.23 | 172.08 ± 12.26 | 254.06 ± 19.23 | 15.06 ± 0.35 | 984.06 ± 56.87 |
| DGX + Garcinol | 16.75 ± 4.22 | 151.39 ± 0.58 | 172.27 ± 12.58 | 7.318 ± 1.75 | 1474.76 ± 85.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolla, L.; Srivastava, P.; Ravichandiran, V.; Nanjappan, S.K. Cytochrome P450 and P-gp Mediated Herb-Drug Interactions and Molecular Docking Studies of Garcinol. Membranes 2021, 11, 992. https://doi.org/10.3390/membranes11120992
Bolla L, Srivastava P, Ravichandiran V, Nanjappan SK. Cytochrome P450 and P-gp Mediated Herb-Drug Interactions and Molecular Docking Studies of Garcinol. Membranes. 2021; 11(12):992. https://doi.org/10.3390/membranes11120992
Chicago/Turabian StyleBolla, Lavanya, Pratima Srivastava, Velayutham Ravichandiran, and Satheesh Kumar Nanjappan. 2021. "Cytochrome P450 and P-gp Mediated Herb-Drug Interactions and Molecular Docking Studies of Garcinol" Membranes 11, no. 12: 992. https://doi.org/10.3390/membranes11120992
APA StyleBolla, L., Srivastava, P., Ravichandiran, V., & Nanjappan, S. K. (2021). Cytochrome P450 and P-gp Mediated Herb-Drug Interactions and Molecular Docking Studies of Garcinol. Membranes, 11(12), 992. https://doi.org/10.3390/membranes11120992