
OH− and H3O+ Diffusion in Model AEMs and PEMs at Low Hydration: Insights from Ab Initio Molecular Dynamics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
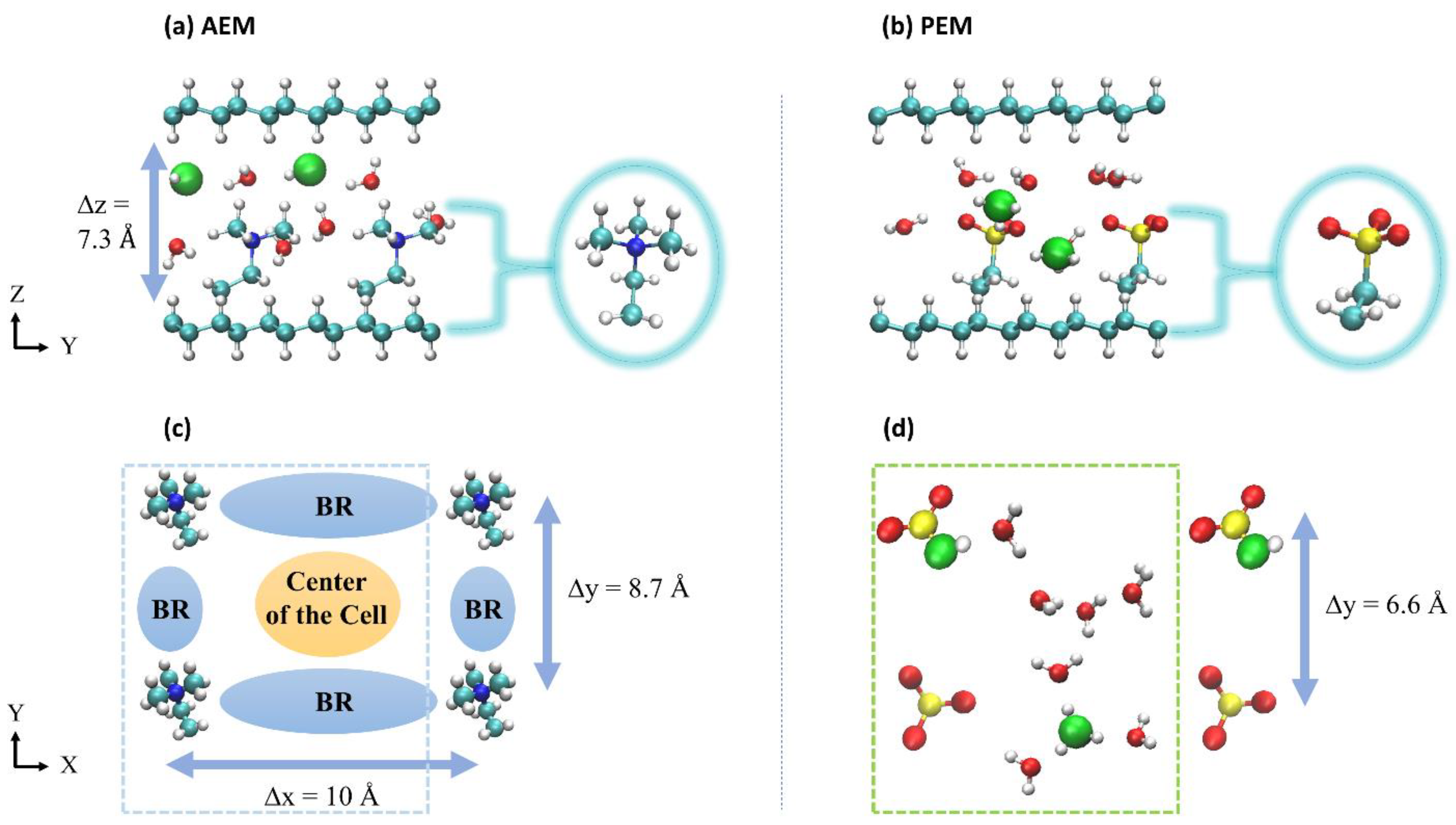
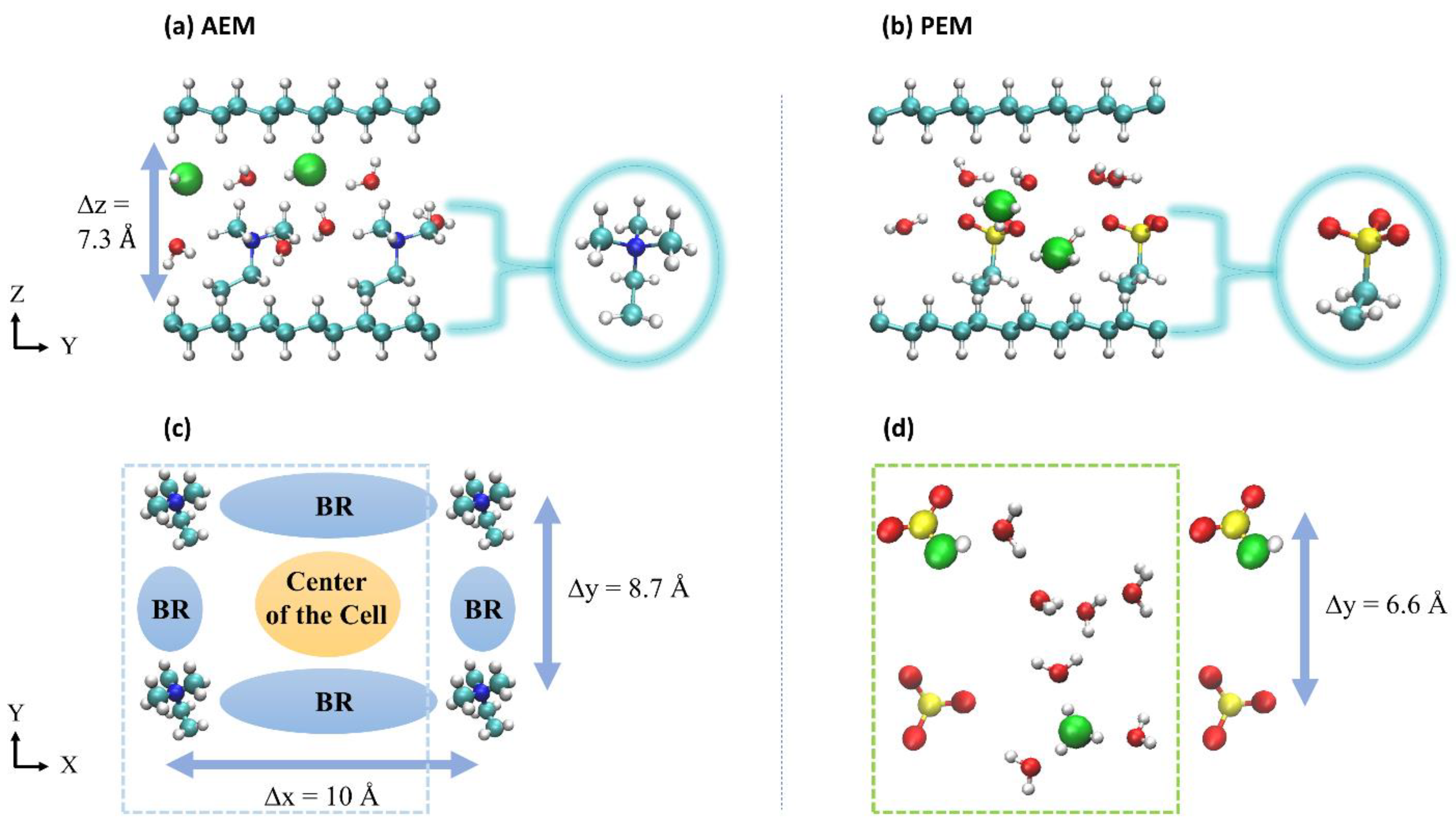
2. Description of Systems
3. Computational Method
4. Result
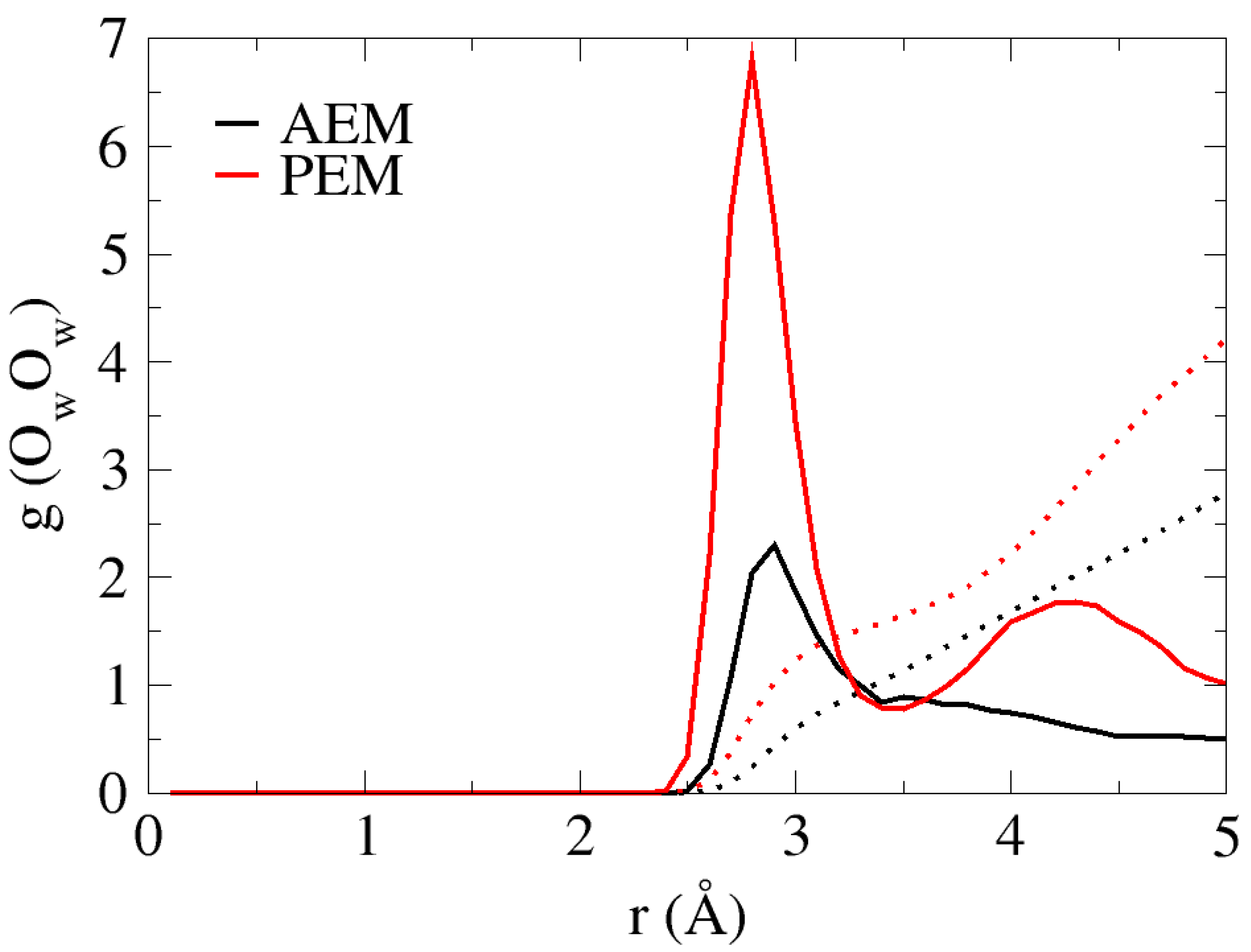
4.1. Water Distribution
4.2. Hydroxide Ion Diffusion in the AEM Model System
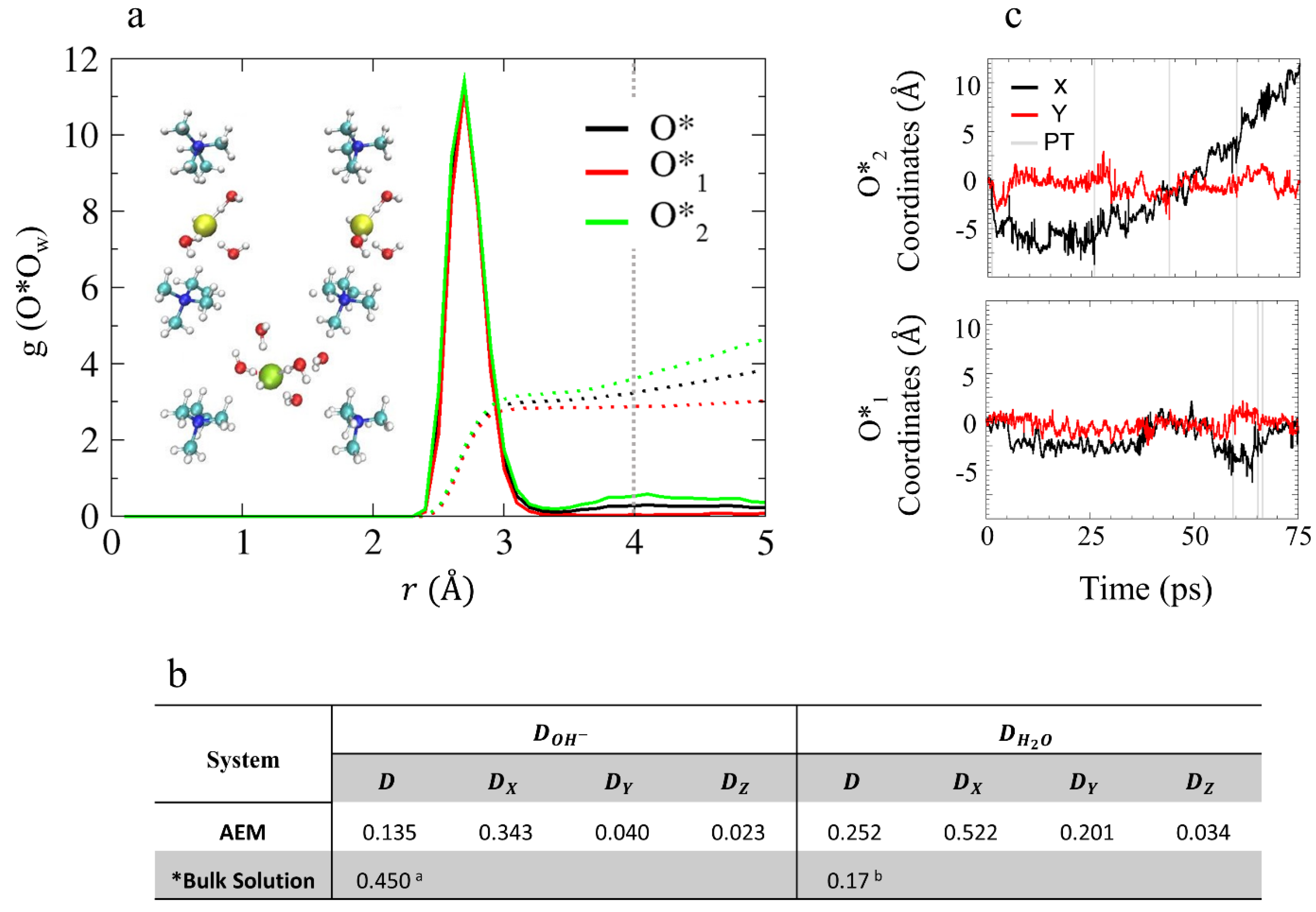
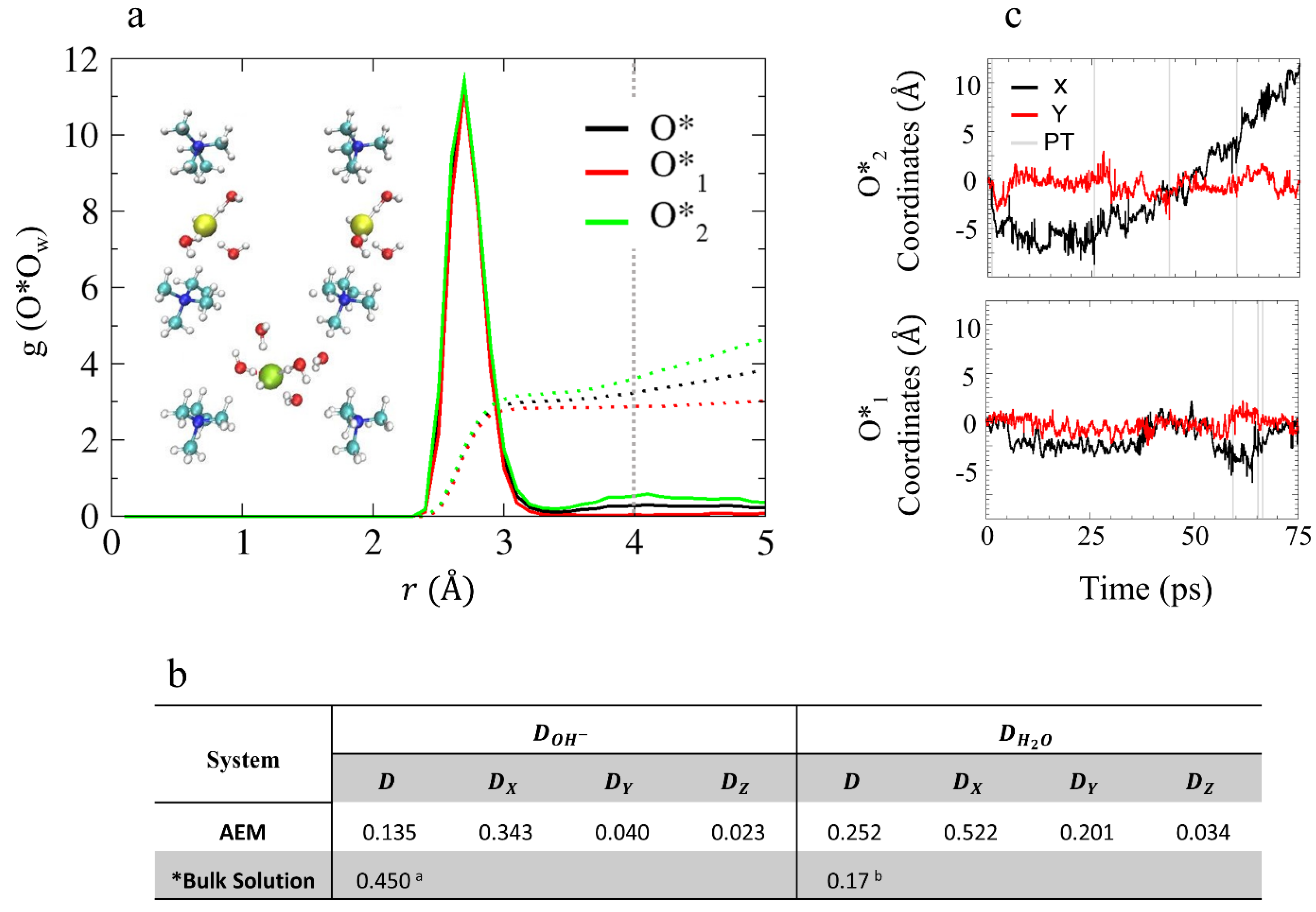
4.2.1. Solvation Structure and Dynamical Properties
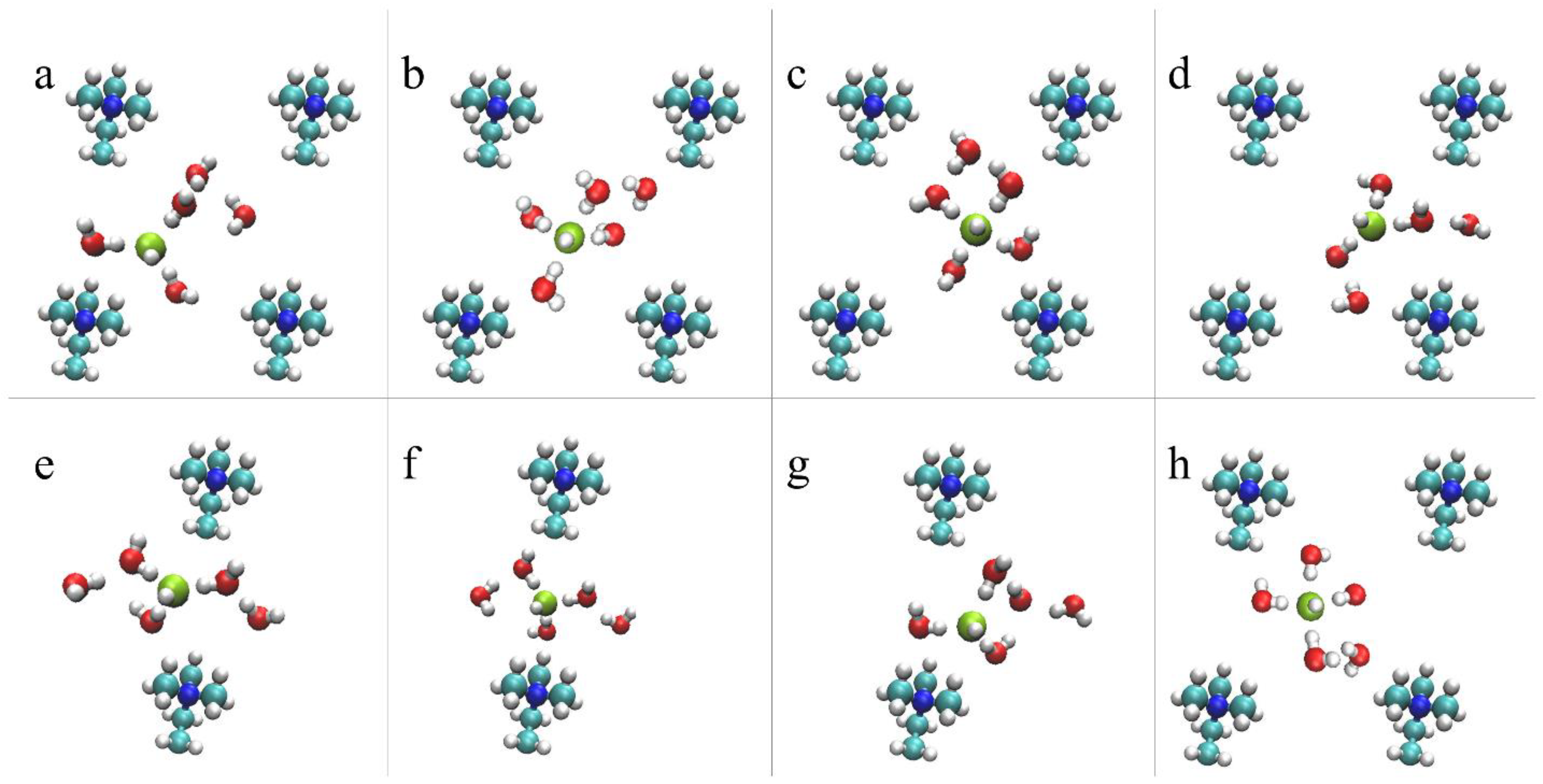
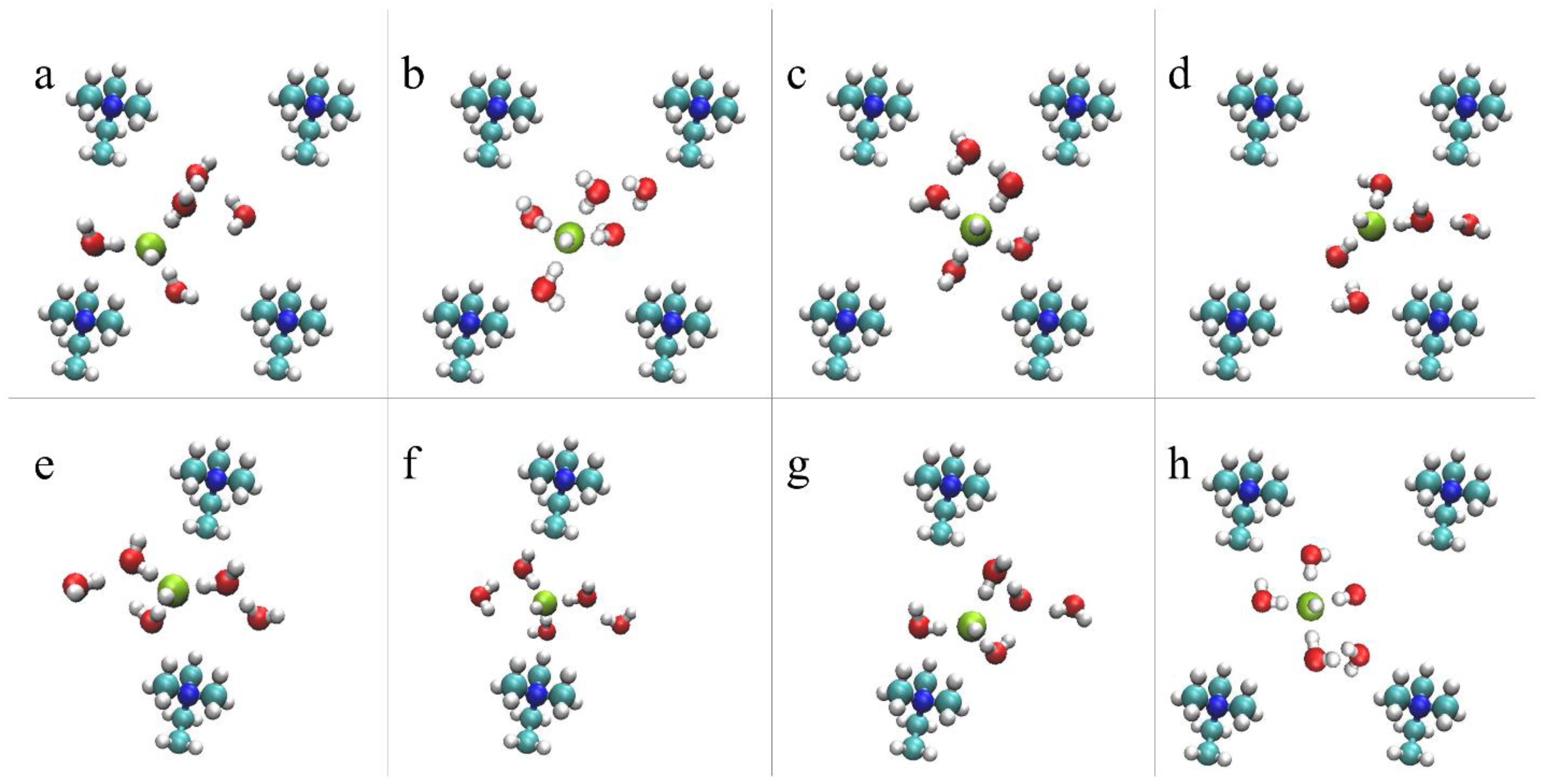
4.2.2. OH– Diffusion Mechanism
4.3. Hydronium Ion Diffusion in the PEM Model System
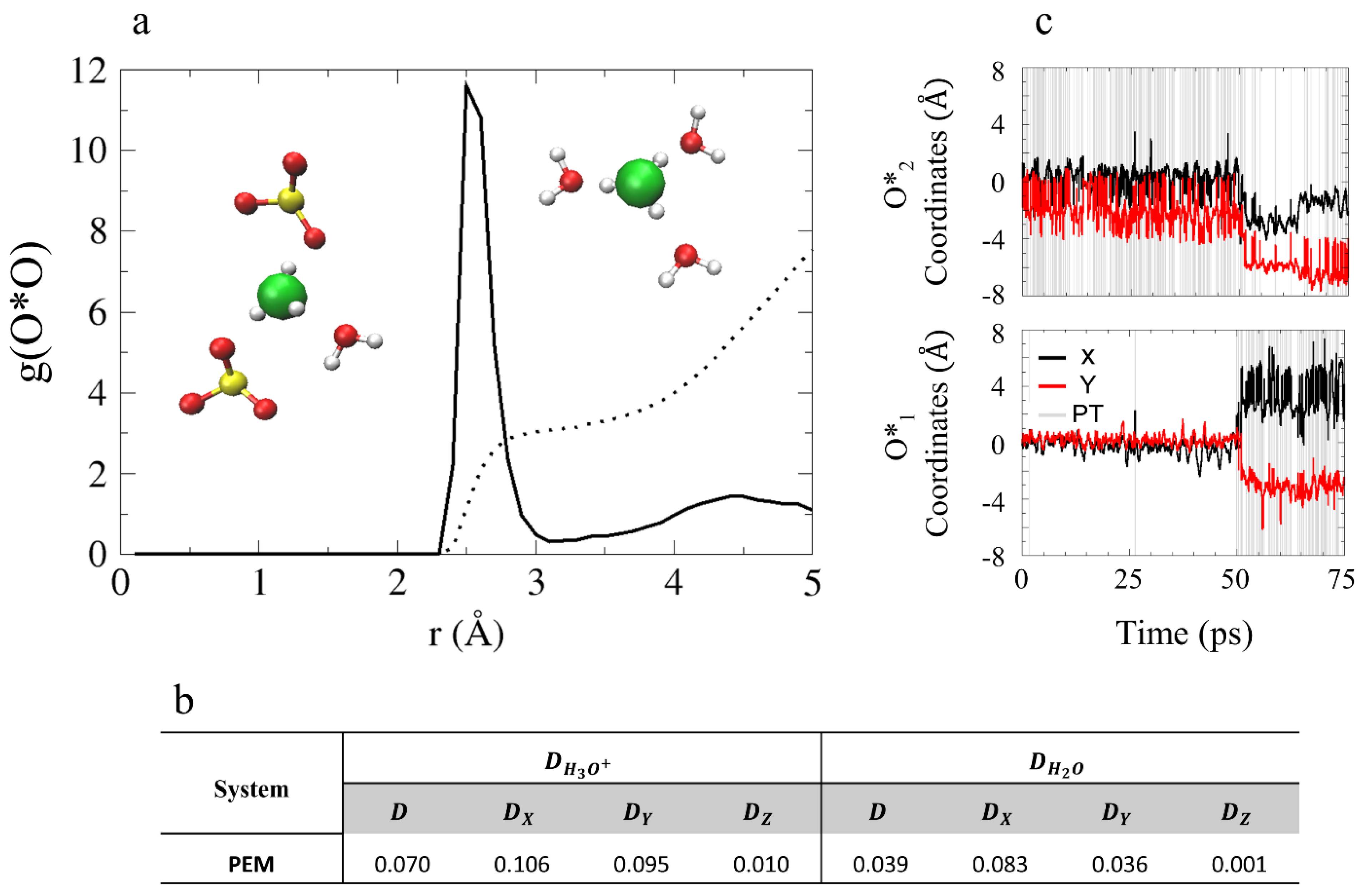
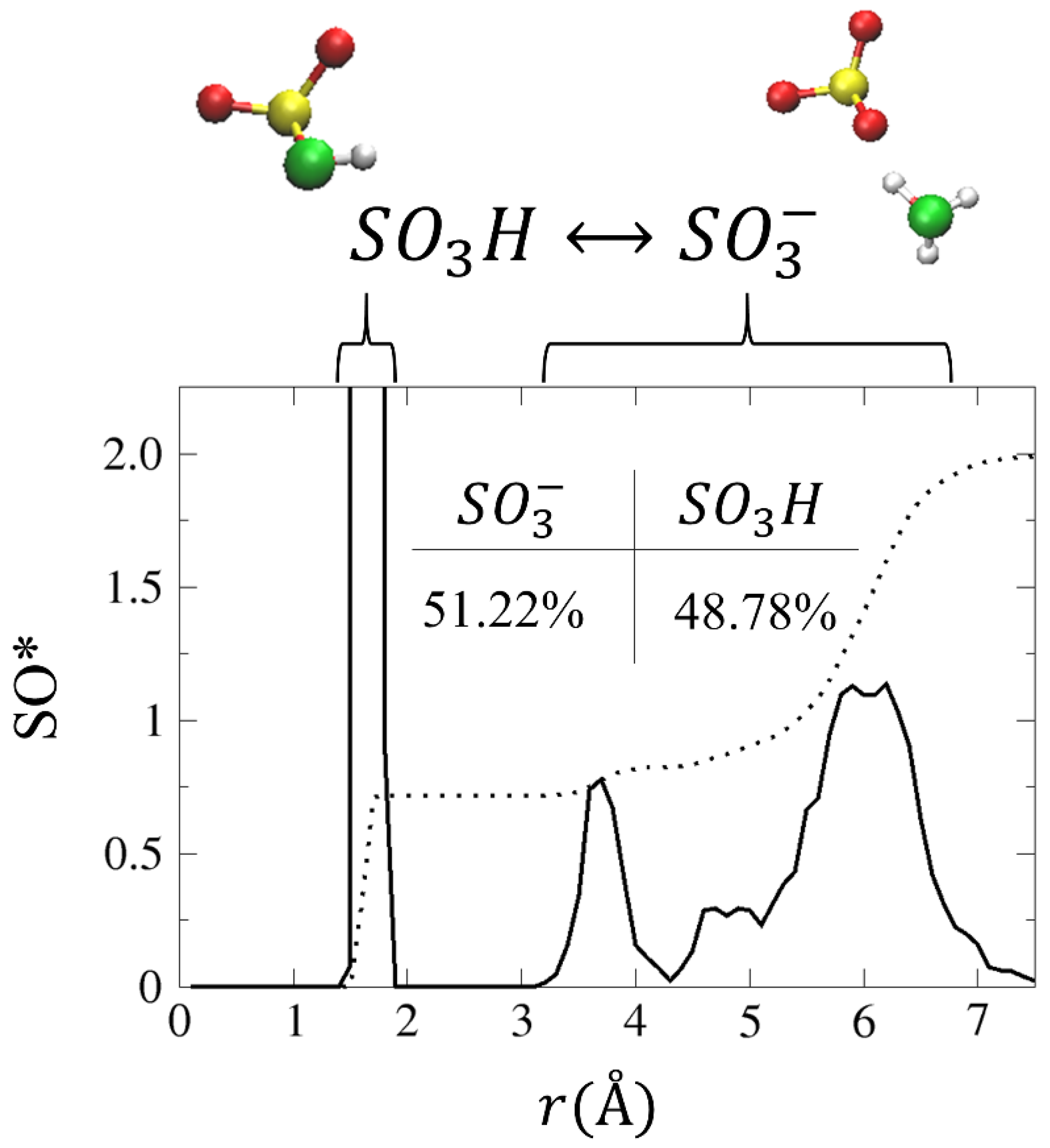
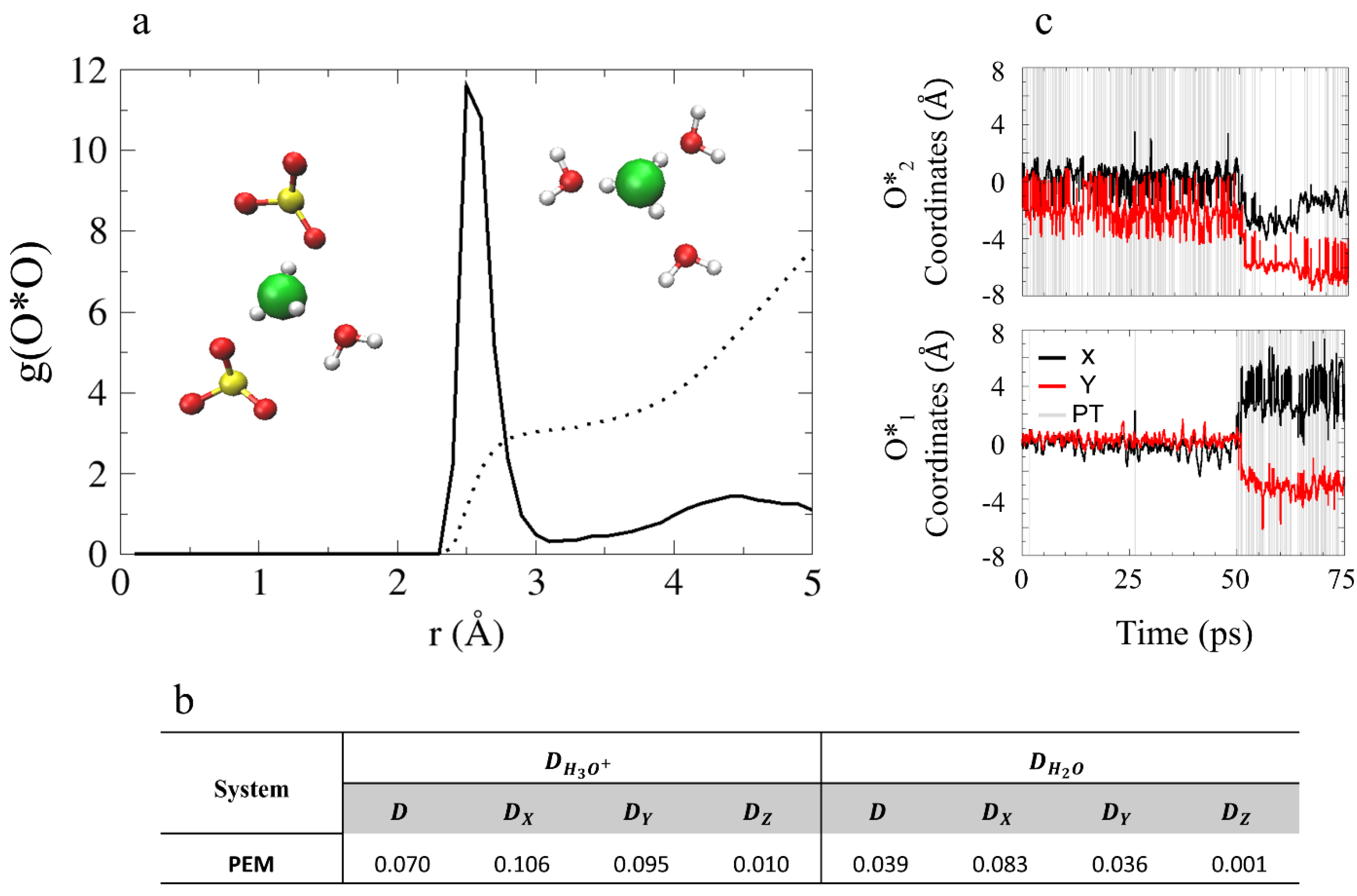
4.3.1. Solvation Structure and Dynamical Properties
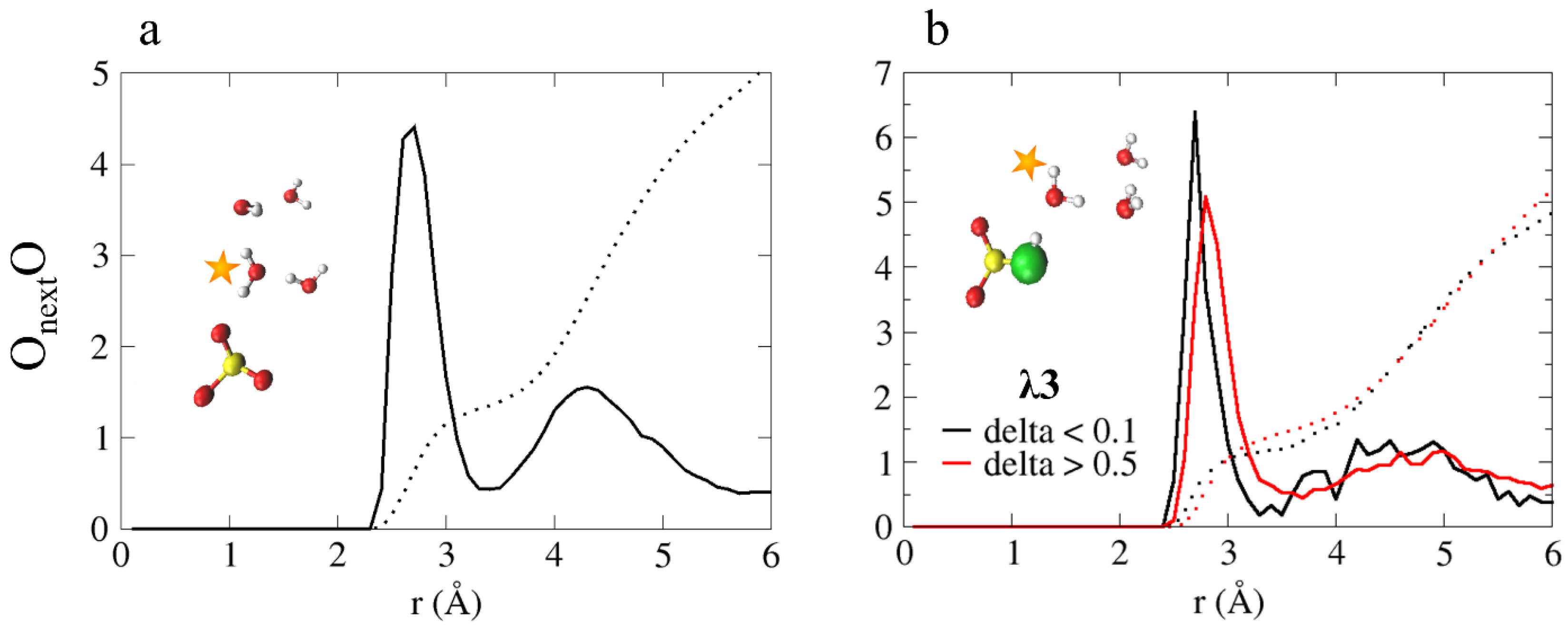
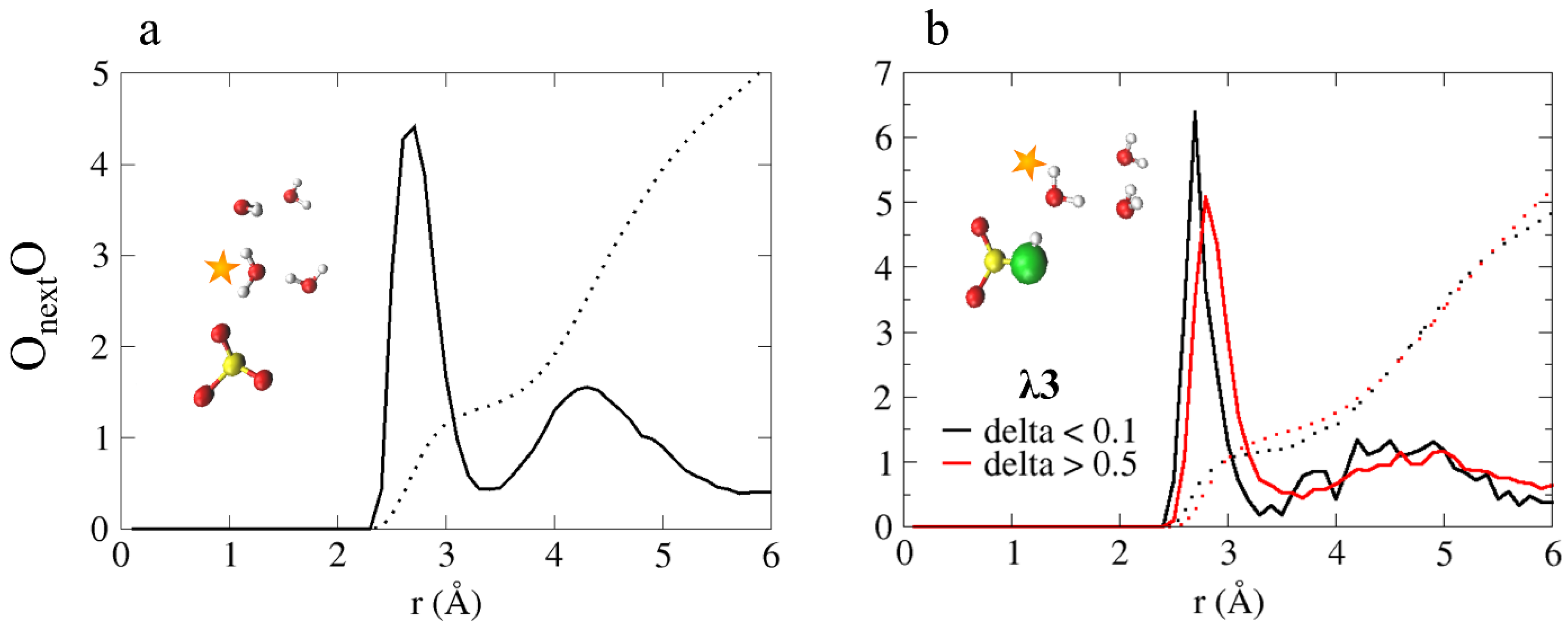
4.3.2. Diffusion Mechanism
5. Discussions and Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Pan, J.; Chen, C.; Zhuang, L.; Lu, J. Designing Advanced Alkaline Polymer Electrolytes for Fuel Cell Applications. Acc. Chem. Res. 2012, 45, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Hickner, M.A.; Herring, A.M.; Coughlin, E.B. Anion Exchange Membranes: Current Status and Moving Forward. J. Polym. Sci. PART B Polym. Phys. 2013, 51, 1727–1735. [Google Scholar] [CrossRef]
- Varcoe, J.R.; Atanassov, P.; Dekel, D.R.; Herring, A.M.; Hickner, M.A.; Kohl, P.A.; Kucernak, A.R.; Mustain, W.E.; Nijmeijer, K.; Scott, K.; et al. Anion-exchange membranes in electrochemical energy systems. Energy Environ. Sci. 2014, 7, 3135–3191. [Google Scholar] [CrossRef] [Green Version]
- Dekel, D.R.; Rasin, I.G.; Page, M.; Brandon, S. Steady state and transient simulation of anion exchange membrane fuel cells. J. Power Sour. 2018, 375, 191–204. [Google Scholar] [CrossRef]
- Dekel, D.R. Review of cell performance in anion exchange membrane fuel cells. J. Power Sources 2018, 375, 158–169. [Google Scholar] [CrossRef]
- Géraldine, M.; Wessling, M.; Nijmeijer, K. Anion Exchange Membranes for Alkaline Fuel Cells: A Review. J. Memb. Sci. 2011, 377, 1–35. [Google Scholar]
- Han, K.W.; Ko, K.H.; Abu-Hakmeh, K.; Bae, C.; Sohn, Y.J.; Jang, S.S. Molecular Dynamics Simulation Study of a Polysulfone-Based Anion Exchange Membrane in Comparison with the Proton Exchange Membrane. J. Phys. Chem. C 2014, 118, 12577–12587. [Google Scholar] [CrossRef]
- Miller, H.A.; Vizza, F.; Marelli, M.; Zadick, A.; Dubau, L.; Chatenet, M.; Geiger, S.; Cherevko, S.; Doan, H.; Pavlicek, R.K.; et al. Highly active nanostructured palladium-ceria electrocatalysts for the hydrogen oxidation reaction in alkaline medium. Nano Energy 2017, 33, 293–305. [Google Scholar] [CrossRef]
- Gottesfeld, S.; Dekel, D.R.; Page, M.; Bae, C.; Yan, Y.; Zelenay, P.; Kim, Y.S. Anion exchange membrane fuel cells: Current status and remaining challenges. J. Power Sour. 2018, 375, 170–184. [Google Scholar] [CrossRef]
- Hagesteijn, K.F.L.; Jiang, S.; Ladewig, B.P. A Review of the Synthesis and Characterization of Anion Exchange Membranes. J. Mater. Sci. 2018, 53, 11131–11150. [Google Scholar] [CrossRef] [Green Version]
- Arges, C.G.; Zhang, L. Anion Exchange Membranes’ Evolution toward High Hydroxide Ion Conductivity and Alkaline Resiliency. ACS Appl. Energy Mater. 2018, 1, 2991–3012. [Google Scholar] [CrossRef]
- Savage, J.; Voth, G.A. Proton Solvation and Transport in Realistic Proton Exchange Membrane Morphologies. J. Phys. Chem. C 2016, 120, 3176–3186. [Google Scholar] [CrossRef]
- Zhang, N.; Song, Y.; Ruan, X.; Yan, X.; Liu, Z.; Shen, Z.; Wu, X.; He, G. Structural characteristics of hydrated protons in the conductive channels: Effects of confinement and fluorination studied by molecular dynamics simulation. Phys. Chem. Chem. Phys. 2016, 18, 24198–24209. [Google Scholar] [CrossRef] [PubMed]
- Sepehr, F.; Paddison, S.J. Primary hydration and proton transfer of electrolyte acids: An ab initio study. Solid State Ion. 2017, 306, 2–12. [Google Scholar] [CrossRef]
- Shin, D.W.; Guiver, M.D.; Lee, Y.M. Hydrocarbon-Based Polymer Electrolyte Membranes: Importance of Morphology on Ion Transport and Membrane Stability. Chem. Rev. 2017, 117, 4759–4805. [Google Scholar] [CrossRef]
- Trigg, E.B.; Gaines, T.W.; Maréchal, M.; Moed, D.E.; Rannou, P.; Wagener, K.B.; Stevens, M.J.; Winey, K.I. Self-assembled highly ordered acid layers in precisely sulfonated polyethylene produce efficient proton transport. Nat. Mater. 2018, 17, 725–731. [Google Scholar] [CrossRef]
- Linga, X.; Bonna, M.; Domkea, K.F.; Parekha, S.H. Correlated Interfacial Water Transport and Proton Conductivity in Perfluorosulfonic acid Membranes. Proc. Natl. Acad. Sci. USA 2019, 116, 8715–8720. [Google Scholar] [CrossRef] [Green Version]
- Roget, S.A.; Kramer, P.L.; Thomaz, J.E.; Fayer, M.D. Bulk-like and Interfacial Water Dynamics in Nafion Fuel Cell Membranes Investigated with Ultrafast Nonlinear IR Spectroscopy. J. Phys. Chem. B 2019, 123, 9408–9417. [Google Scholar] [CrossRef]
- Paddison, S.J.; Elliott, J.A. Molecular Modeling of the Short-Side-Chain Perfluorosulfonic Acid Membrane. J. Phys. Chem. A 2005, 109, 7583–7593. [Google Scholar] [CrossRef]
- Paddison, S.J.; Kreuer, K.-D.; Maier, J. About the choice of the protogenic group in polymer electrolyte membranes: Ab initio modelling of sulfonic acid, phosphonic acid, and imidazole functionalized alkanes. Phys. Chem. Chem. Phys. 2006, 8, 4530–4542. [Google Scholar] [CrossRef]
- Hayes, R.L.; Paddison, S.J.; Tuckerman, M.E. Proton Transport in Triflic Acid Hydrates Studied via Path Integral Car−Parrinello Molecular Dynamics. J. Phys. Chem. B 2009, 113, 16574–16589. [Google Scholar] [CrossRef] [PubMed]
- Ludueña, G.A.; Kühne, T.D.; Sebastiani, D. Mixed Grotthuss and Vehicle Transport Mechanism in Proton Conducting Polymers from Ab initio Molecular Dynamics Simulations. Chem. Mater. 2011, 23, 1424–1429. [Google Scholar] [CrossRef]
- Hayes, R.L.; Paddison, S.J.; Tuckerman, M.E. Proton Transport in Triflic Acid Pentahydrate Studied via Ab Initio Path Integral Molecular Dynamics. J. Phys. Chem. A 2011, 115, 6112–6124. [Google Scholar] [CrossRef]
- Clark, J.K.; Paddison, S.J.; Eikerling, M.; Dupuis, M.; Thomas, J.; Zawodzinski, A. A Comparative Ab Initio Study of the Primary Hydration and Proton Dissociation of Various Imide and Sulfonic Acid Ionomers. J. Phys. Chem. A 2012, 116, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Paddison, S.J. Hydration and Proton Transfer in Highly Sulfonated Poly (phenylene sulfone) Ionomers: An Ab Initio Study. J. Phys. Chem. A 2013, 117, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Devanathan, R.; Idupulapati, N.; Baer, M.D.; Mundy, C.J.; Dupuis, M. Ab Initio Molecular Dynamics Simulation of Proton Hopping in a Model Polymer Membrane. J. Phys. Chem. 2013, 117, 16522–16529. [Google Scholar] [CrossRef] [PubMed]
- Ensing, W.; Hunger, J.; Ottosson, N.; Bakker, H.J. On the Orientational Mobility of Water Molecules in Proton and Sodium Terminated Nafion Membranes. J. Phys. Chem. C 2013, 117, 12930–12935. [Google Scholar] [CrossRef]
- Savage, J.; Tse, Y.-L.S.; Voth, G.A. Proton Transport Mechanism of Perfluorosulfonic Acid Membranes. J. Phys. Chem. C 2014, 118, 17436–17445. [Google Scholar] [CrossRef]
- Habenicht, B.F.; Paddison, S.J.; Tuckerman, M.E. Ab initio molecular dynamics simulations investigating proton transfer in perfluorosulfonic acid functionalized carbon nanotubes. Phys. Chem. Chem. Phys. 2010, 12, 8728–8732. [Google Scholar] [CrossRef]
- Habenicht, B.F.; Paddison, S.J.; Tuckerman, M.E. The effects of the hydrophobic environment on proton mobility in perfluorosulfonic acid systems: An ab initio molecular dynamics study. J. Mater. Chem. 2010, 20, 6342–6351. [Google Scholar] [CrossRef]
- Modestino, M.A.; Paul, D.K.; Dishari, S.; Petrina, S.A.; Allen, F.I.; Hickner, M.A.; Karan, K.; Segalman, R.A.; Weber, A.Z. Self-Assembly and Transport Limitations in Confined Nafion Films. Macromolecules 2013, 46, 867–873. [Google Scholar] [CrossRef]
- Yudan, Z.; Jian, Z.; Xiaohua, L.; Xiaojing, G.; Linghong, L. Molecular Simulations on Nanoconfined Water Molecule Behaviors for Nanoporous Material Applications. Microfluid Nanofluid 2013, 15, 191–205. [Google Scholar]
- Kusoglu, A.; Kushner, D.; Paul, D.K.; Karan, K.; Hickner, M.A.; Weber, A.Z. Impact of Substrate and Processing on Confinement of Nafion Thin Films. Adv. Funct. Mater. 2014, 24, 4763–4774. [Google Scholar] [CrossRef]
- Page, K.A.; Kusoglu, A.; Stafford, C.M.; Kim, S.; Kline, R.J.; Weber, A.Z. Confinement-Driven Increase in Ionomer Thin-Film Modulus. Nano Lett. 2014, 14, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.K., II; Habenichtb, B.F.; Paddison, S.J. Ab Initio Molecular Dynamics Simulations of Aqueous Triflic Acid Confined in Carbon Nanotubes. Phys. Chem. Chem. Phys. 2014, 16, 16465–16479. [Google Scholar] [CrossRef] [PubMed]
- Kusoglu, A.; Weber, A.Z. New Insights into Perfluorinated Sulfonic-Acid Ionomers. Chem. Rev. 2017, 117, 987–1104. [Google Scholar] [CrossRef] [PubMed]
- Crothers, A.R.; Radke, C.J.; Weber, A.Z. Impact of Nano- and Mesoscales on Macroscopic Cation Conductivity in Perfluorinated-Sulfonic-Acid Membranes. J. Phys. Chem. C 2017, 121, 28262–28274. [Google Scholar] [CrossRef] [Green Version]
- Tuckerman, M.E.; Laasonen, K.; Sprik, M.; Parrinello, M. Ab Initio Molecular Dynamics Simulation of the Solvation and Transport of Hydronium and Hydroxide Ions in Water. J. Chem. Phys. 1995, 103, 150–161. [Google Scholar] [CrossRef]
- Tuckerman, M.; Laasonen, K.; Sprik, M.; Parrinello, M. Ab Initio Molecular Dynamics Simulation of the Solvation and Transport of H3O+ and OH- Ions in Water. J. Phys. Chem. 1995, 99, 5749–5752. [Google Scholar] [CrossRef]
- Marx, D.; Tuckerman, M.E.; Hutter, J.; Parrinello, M. The nature of the hydrated excess proton in water. Nat. Cell Biol. 1999, 397, 601–604. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Marx, D.; Parrinello, M. The nature and transport mechanism of hydrated hydroxide ions in aqueous solution. Nat. Cell Biol. 2002, 417, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Tuckerman, M.E.; Chandra, A.; Marx, D. Structure and Dynamics of OH-(aq). Acc. Chem. Res. 2006, 39, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Tuckerman, M.E.; Marx, D. Connecting Solvation Shell Structure to Proton Transport Kinetics in Hydrogen–Bonded Networks via Population Correlation Functions. Phys. Rev. Lett. 2007, 99, 145901. [Google Scholar] [CrossRef] [Green Version]
- Marx, D.; Chandra, A.; Tuckerman, M.E. Aqueous Basic Solutions: Hydroxide Solvation, Structural Diffusion, and Comparison to the Hydrated Proton. Chem. Rev. 2010, 110, 2174–2216. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Tuckerman, M.E. On the connection between proton transport, structural diffusion, and reorientation of the hydrated hydroxide ion as a function of temperature. Chem. Phys. Lett. 2011, 511, 177–182. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Chandra, A.; Marx, D. A Statistical Mechanical Theory of Proton Transport Kinetics in Hydrogen-Bonded Networks Based on Population Correlation Functions with Applications to Acids and Bases. J. Chem. Phys. 2010, 133, 124108–124129. [Google Scholar] [CrossRef] [PubMed]
- Hassanali, A.; Giberti, F.; Cuny, J.; Kühne, T.D.; Parrinello, M. Proton transfer through the water gossamer. Proc. Natl. Acad. Sci. USA 2013, 110, 13723–13728. [Google Scholar] [CrossRef] [Green Version]
- Agmon, N.; Bakker, H.J.; Campen, R.K.; Henchman, R.H.; Pohl, P.; Roke, S.; Thämer, M.; Hassanali, A. Protons and Hydroxide Ions in Aqueous Systems. Chem. Rev. 2016, 116, 7642–7672. [Google Scholar] [CrossRef]
- Hub, J.S.; Wolf, M.G.; Caleman, C.; van Maaren, P.J.; Groenhoff, G.; Spoel van Der, D. Thermodynamics of Hydronium and Hydroxide Surface Solvation. Chem. Sci. 2014, 5, 1745–1749. [Google Scholar] [CrossRef] [Green Version]
- Tse, Y.-L.S.; Chen, C.; Lindberg, G.E.; Kumar, R.; Voth, G.A. Propensity of Hydrated Excess Protons and Hydroxide Anions for the Air–Water Interface. J. Am. Chem. Soc. 2015, 137, 12610–12616. [Google Scholar] [CrossRef]
- Tuckerman, M.E. Ab Initio Molecular Dynamics: Basic Concepts, Current Trends and Novel Applications. J. Phys. Condens. Matter 2002, 14, R1297–R1355. [Google Scholar] [CrossRef]
- Zelovich, T.; Long, Z.; Hickner, M.; Paddison, S.J.; Bae, C.; Tuckerman, M.E. Ab initio Molecular Dynamics Study of Hydroxide Diffusion Mechanisms in Nano-Confined Structural mimics of Anion Exchange Membranes. J. Phys. Chem. C 2019, 123, 4638–4653. [Google Scholar] [CrossRef]
- Zelovich, T.; Vogt-Maranto, L.; Hickner, M.A.; Paddison, S.J.; Bae, C.; Dekel, D.R.; Tuckerman, M.E. Hydroxide Ion Diffusion in Anion-Exchange Membranes at Low Hydration: Insights from Ab Initio Molecular Dynamics. Chem. Mater. 2019, 31, 5778–5787. [Google Scholar] [CrossRef]
- Zelovich, T.; Tuckerman, M.E. Water Layering Affects Hydroxide Diffusion in Functionalized Nanoconfined Environments. J. Phys. Chem. Lett. 2020, 11, 5087–5091. [Google Scholar] [CrossRef] [PubMed]
- Zelovich, T.; Winey, K.I.; Tuckerman, M.E. Hydronium ion diffusion in model proton exchange membranes at low hydration: Insights from ab initio molecular dynamics. J. Mater. Chem. A 2021, 9, 2448–2458. [Google Scholar] [CrossRef]
- Marino, M.G.; Kreuer, K.D. Alkaline Stability of Quaternary Ammonium Cations for Alkaline Fuel Cell Membranes and Ionic Liquids. ChemSusChem 2015, 8, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Sepehr, F.; Liu, H.; Luo, X.; Bae, C.; Tuckerman, M.E.; Hickner, M.A.; Paddison, S.J. Mesoscale Simulations of Anion Exchange Membranes Based on Quaternary Ammonium Tethered Triblock Copolymers. Macromolecules 2017, 50, 4397–4405. [Google Scholar] [CrossRef]
- Marx, D.; Hutter, J. Ab Initio Molecular Dynamics: Theory and Implementation, in Modern Methods and Algorithms of Quantum Chemistry; John von Institute for Computing: Juelich, Germany, 2000. [Google Scholar]
- Hutter, D.M.J.; Alavi, A.; Deutsch, T.; Bernasconi, M.; Goedecker, S.; Parrinello, M. CPMD, IBM Corporation 1990–2009 and MPI für Festkörperforschung 1997–2001. 2009. Available online: www.cpmd.org (accessed on 20 March 2021).
- Martyna, G.J.; Tuckerman, M.E.; Klein, M.L. Nose-Hoover Chains: The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Aeberhard, P.C.; Arey, J.S.; Lin, I.-C.; Rothlisberger, U. Accurate DFT Descriptions for Weak Interactions of Molecules Containing Sulfur. J. Chem. Theory Comput. 2009, 5, 23–28. [Google Scholar] [CrossRef]
- Lin, I.-C.; Coutinho-Neto, M.D.; Felsenheimer, C.; von Lilienfeld, O.A.; Tavernelli, I.; Rothlisberger, U. Library of Dispersion-Corrected Atom-Centered Potentials for Generalized Gradient Approximation Functionals: Elements H, C, N, O, He, Ne, Ar, and Kr. Phys. Rev. B 2007, 75, 205131–205135. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenness, G.R.; Karalti, O.; Jordan, K.D. Benchmark calculations of water–acene interaction energies: Extrapolation to the water–graphene limit and assessment of dispersion–corrected DFT methods. Phys. Chem. Chem. Phys. 2010, 12, 6375–6381. [Google Scholar] [CrossRef]
- Lin, I.-C.; Seitsonen, A.P.; Tavernelli, I.; Rothlisberger, U. Structure and Dynamics of Liquid Water from ab Initio Molecular Dynamics—Comparison of BLYP, PBE, and revPBE Density Functionals with and without van der Waals Corrections. J. Chem. Theory Comput. 2012, 8, 3902–3910. [Google Scholar] [CrossRef] [PubMed]
- Berkelbach, T.C.; Lee, H.-S.; Tuckerman, M.E. Concerted Hydrogen-Bond Dynamics in the Transport Mechanism of the Hydrated Proton: A First-Principles Molecular Dynamics Study. Phys. Rev. Lett. 2009, 103, 238302. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Tuckerman, M.E. Dynamical properties of liquid water from ab initio molecular dynamics performed in the complete basis set limit. J. Chem. Phys. 2007, 126, 164501. [Google Scholar] [CrossRef]
- Zhu, Z.; Tuckerman, M.E. Ab Initio Molecular Dynamics Investigation of the Concentration Dependence of Charged Defect Transport in Basic Solutions via Calculation of the Infrared Spectrum†. J. Phys. Chem. B 2002, 106, 8009–8018. [Google Scholar] [CrossRef]
- Morrone, J.A.; Haslinger, K.E.; Tuckerman, M.E. Ab Initio Molecular Dynamics Simulation of the Structure and Proton Transport Dynamics of Methanol−Water Solutions †. J. Phys. Chem. B 2006, 110, 3712–3720. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zelovich, T.; Tuckerman, M.E. OH− and H3O+ Diffusion in Model AEMs and PEMs at Low Hydration: Insights from Ab Initio Molecular Dynamics. Membranes 2021, 11, 355. https://doi.org/10.3390/membranes11050355
Zelovich T, Tuckerman ME. OH− and H3O+ Diffusion in Model AEMs and PEMs at Low Hydration: Insights from Ab Initio Molecular Dynamics. Membranes. 2021; 11(5):355. https://doi.org/10.3390/membranes11050355
Chicago/Turabian StyleZelovich, Tamar, and Mark E. Tuckerman. 2021. "OH− and H3O+ Diffusion in Model AEMs and PEMs at Low Hydration: Insights from Ab Initio Molecular Dynamics" Membranes 11, no. 5: 355. https://doi.org/10.3390/membranes11050355
APA StyleZelovich, T., & Tuckerman, M. E. (2021). OH− and H3O+ Diffusion in Model AEMs and PEMs at Low Hydration: Insights from Ab Initio Molecular Dynamics. Membranes, 11(5), 355. https://doi.org/10.3390/membranes11050355