
Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. GALLEX Assay
2.3. Software
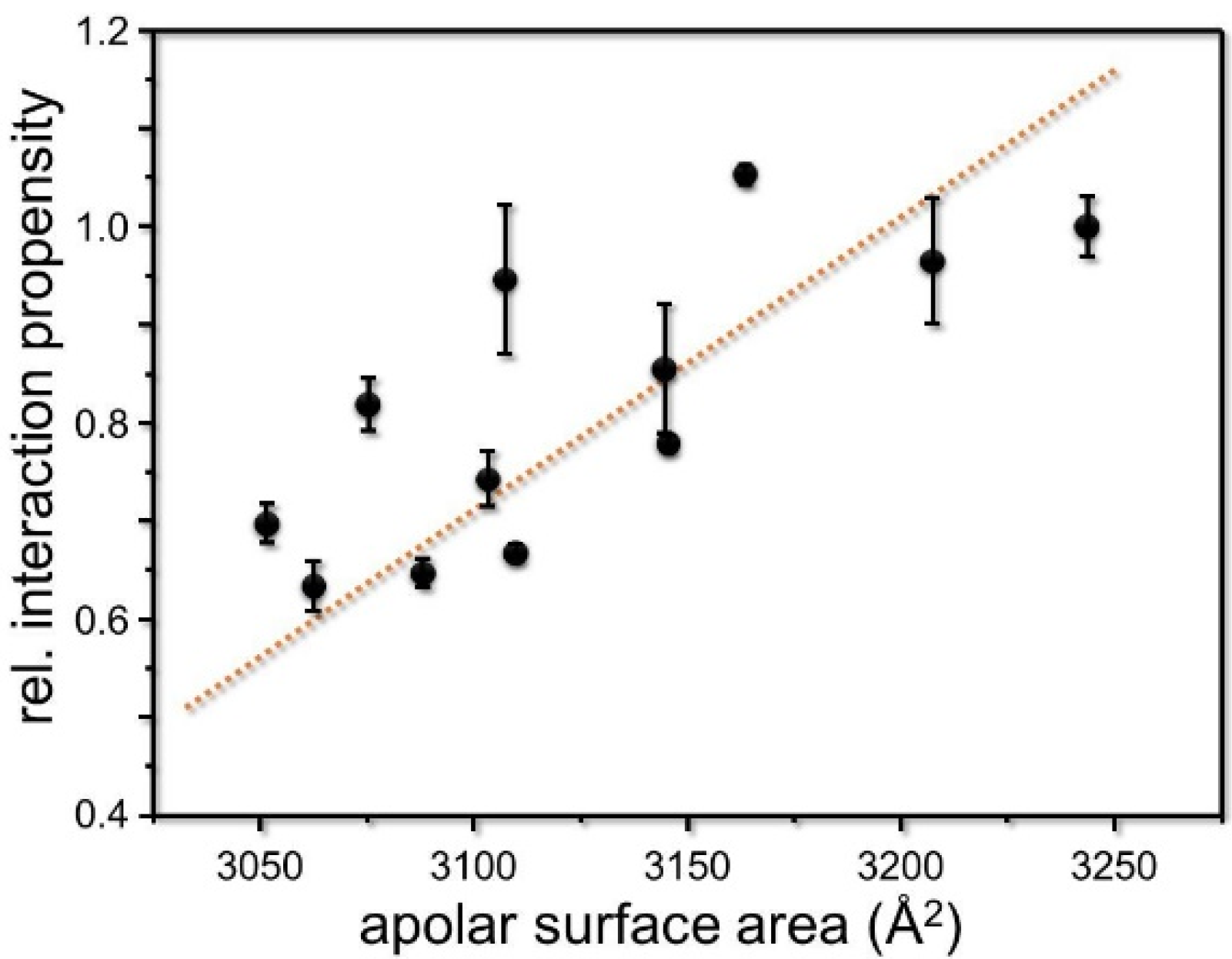
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemmon, M.A.; Flanagan, J.M.; Treutlein, H.R.; Zhang, J.; Engelman, D.M. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry 1992, 31, 12719–12725. [Google Scholar] [CrossRef]
- MacKenzie, K.R.; Prestegard, J.H.; Engelman, D.M. A transmembrane helix dimer: Structure and implications. Science 1997, 276, 131–133. [Google Scholar] [CrossRef] [Green Version]
- Brosig, B.; Langosch, D. The dimerization motif of the glycophorin a transmembrane segment in membranes: Importance of glycine residues. Protein. Sci. 1998, 7, 1052–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Senes, A.; Gerstein, M.; Engelman, D.M. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J. Mol. Biol. 2000, 296, 921–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cymer, F.; Veerappan, A.; Schneider, D. Transmembrane helix-helix interactions are modulated by the sequence context and by lipid bilayer properties. Biochim. Biophys. Acta 2012, 1818, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Teese, M.G.; Langosch, D. Role of GxxxG Motifs in Transmembrane Domain Interactions. Biochemistry 2015, 54, 5125–5135. [Google Scholar] [CrossRef]
- Schneider, D. Rendezvous in a membrane: Close packing, hydrogen bonding, and the formation of transmembrane helix oligomers. FEBS Lett. 2004, 577, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Senes, A.; Engel, D.E.; DeGrado, W.F. Folding of helical membrane proteins: The role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 2004, 14, 465–479. [Google Scholar] [CrossRef]
- Schneider, D.; Engelman, D.M. Motifs of Two Small Residues can ASSIST but are not sufficient to Mediate Transmembrane Helix Interactions. J. Mol. Biol. 2004, 343, 799–804. [Google Scholar] [CrossRef]
- Senes, A.; Ubarretxena-Belandia, I.; Engelman, D.M. The Calpha—H...O hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. USA 2001, 98, 9056–9061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbely, E.; Arkin, I.T. Experimental measurement of the strength of a C alpha—H...O bond in a lipid bilayer. J. Am. Chem. Soc. 2004, 126, 5362–5363. [Google Scholar] [CrossRef]
- Walters, R.F.S.; DeGrado, W.F. Helix-packing motifs in membrane proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 13658–13663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Jeon, T.J.; Oberai, A.; Yang, D.; Schmidt, J.J.; Bowie, J.U. Transmembrane glycine zippers: Physiological and pathological roles in membrane proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 14278–14283. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.R.; Fuchs, A.; Panitz, J.C.; Eckert, T.; Unterreitmeier, S.; Frishman, D.; Langosch, D. Ionic Interactions Promote Transmembrane Helix-Helix Association Depending on Sequence Context. J. Mol. Biol. 2010, 396, 452–461. [Google Scholar] [CrossRef]
- Herrmann, J.R.; Panitz, J.C.; Unterreitmeier, S.; Fuchs, A.; Frishman, D.; Langosch, D. Complex Patterns of Histidine, Hydroxylated Amino Acids and the GxxxG Motif Mediate High-affinity Transmembrane Domain Interactions. J. Mol. Biol. 2008, 385, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Unterreitmeier, S.; Fuchs, A.; Schaffler, T.; Heym, R.G.; Frishman, D.; Langosch, D. Phenylalanine promotes interaction of transmembrane domains via GxxxG motifs. J. Mol. Biol. 2007, 374, 705–718. [Google Scholar] [CrossRef]
- Melnyk, R.A.; Kim, S.; Curran, A.R.; Engelman, D.M.; Bowie, J.U.; Deber, C.M. The affinity of GXXXG motifs in transmembrane helix-helix interactions is modulated by long-range communication. J. Biol. Chem. 2004, 279, 16591–16597. [Google Scholar] [CrossRef] [Green Version]
- Gurezka, R.; Langosch, D. In vitro selection of membrane-spanning leucine zipper protein-protein interaction motifs using POSSYCCAT. J. Biol. Chem. 2001, 276, 45580–45587. [Google Scholar] [CrossRef] [Green Version]
- Dawson, J.P.; Weinger, J.S.; Engelman, D.M. Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol. 2002, 316, 799–805. [Google Scholar] [CrossRef]
- Zhou, F.X.; Merianos, H.J.; Brunger, A.T.; Engelman, D.M. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. USA 2001, 98, 2250–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratkowski, H.; Lear, J.D.; De Grado, W.F. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. USA 2001, 98, 880–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.M.; Hecht, K.; Deber, C.M. Aromatic and cation-pi interactions enhance helix-helix association in a membrane environment. Biochemistry 2007, 46, 9208–9214. [Google Scholar] [CrossRef] [PubMed]
- Sal-Man, N.; Gerber, D.; Bloch, I.; Shai, Y. Specificity in transmembrane helix-helix interactions mediated by aromatic residues. J. Biol. Chem. 2007, 282, 19753–19761. [Google Scholar] [CrossRef] [Green Version]
- Sal-Man, N.; Gerber, D.; Shai, Y. The composition rather than position of polar residues (QxxS) drives aspartate receptor transmembrane domain dimerization in vivo. Biochemistry 2004, 43, 2309–2313. [Google Scholar] [CrossRef]
- Steindorf, D.; Schneider, D. In vivo selection of heterotypically interacting transmembrane helices: Complementary helix surfaces, rather than conserved interaction motifs, drive formation of transmembrane hetero-dimers. Biochim. Et Biophys. Acta Biomembr. 2017, 1859, 245–256. [Google Scholar] [CrossRef]
- Hubert, P.; Sawma, P.; Duneau, J.-P.; Khao, J.; Henin, J.; Bagnard, D.; Sturgis, J. Single-spanning transmembrane domains in cell growth and cell-cell interactions. Cell Adhes. Migr. 2010, 4, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Kirrbach, J.; Krugliak, M.; Ried, C.L.; Pagel, P.; Arkin, I.T.; Langosch, D. Self-interaction of transmembrane helices representing pre-clusters from the human single-span membrane proteins. Bioinformatics 2013, 29, 1623–1630. [Google Scholar] [CrossRef] [Green Version]
- Ried, C.L.; Kube, S.; Kirrbach, J.; Langosch, D. Homotypic interaction and amino acid distribution of unilaterally conserved transmembrane helices. J. Mol. Biol. 2012, 420, 251–257. [Google Scholar] [CrossRef]
- Langosch, D.; Arkin, I.T. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 2009, 18, 1343–1358. [Google Scholar] [CrossRef] [Green Version]
- Cymer, F.; Sanders, C.R.; Schneider, D. Analyzing oligomerization of individual transmembrane helices and of entire membrane proteins in E. coli: A hitchhiker’s guide to GALLEX. Methods Mol. Biol. 2013, 932, 259–276. [Google Scholar] [PubMed]
- Schneider, D.; Engelman, D.M. GALLEX, a Measurement of Heterologous Association of Transmembrane Helices in a Biological Membrane. J. Biol. Chem. 2003, 278, 3105–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treptow, N.A.; Shuman, H.A. Genetic evidence for substrate and periplasmic-binding-protein recognition by the MalF and MalG proteins, cytoplasmic membrane components of the Escherichia coli maltose transport system. J. Bacteriol. 1985, 163, 654–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyansky, A.A.; Chugunov, A.O.; Volynsky, P.E.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PREDDIMER: A web server for prediction of transmembrane helical dimers. Bioinformatics 2014, 30, 889–890. [Google Scholar] [CrossRef] [Green Version]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Fraczkiewicz, R.; Braun, W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Cunningham, F.; Poulsen, B.E.; Ip, W.; Deber, C.M. Beta-branched residues adjacent to GG4 motifs promote the efficient association of glycophorin A transmembrane helices. Biopolymers 2011, 96, 340–347. [Google Scholar] [CrossRef]
- Whittington, D.A.; Waheed, A.; Ulmasov, B.; Shah, G.N.; Grubb, J.H.; Sly, W.S.; Christianson, D.W. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9545–9550. [Google Scholar] [CrossRef] [Green Version]
- Alterio, V.; Hilvo, M.; di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [Green Version]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic anhydrase IX: Regulation and role in cancer. Subcell Biochem. 2014, 75, 199–219. [Google Scholar] [PubMed]
- Waheed, A.; Sly, W.S. Carbonic anhydrase XII functions in health and disease. Gene 2017, 623, 33–40. [Google Scholar] [CrossRef]
- Escher, C.; Cymer, F.; Schneider, D. Two GxxxG-like motifs facilitate promiscuous interactions of the human ErbB transmembrane domains. J. Mol. Biol. 2009, 389, 10–16. [Google Scholar] [CrossRef]
- Anderson, S.M.; Mueller, B.K.; Lange, E.J.; Senes, A. Combination of Calpha-H Hydrogen Bonds and van der Waals Packing Modulates the Stability of GxxxG-Mediated Dimers in Membranes. J. Am. Chem. Soc. 2017, 139, 15774–15783. [Google Scholar] [CrossRef]
- Xiao, Y.; Zeng, B.; Berner, N.; Frishman, D.; Langosch, D.; Teese, M.G. Experimental determination and data-driven prediction of homotypic transmembrane domain interfaces. Comput. Struct. Biotechnol. J. 2020, 18, 3230–3242. [Google Scholar] [CrossRef]
- MacKenzie, K.R.; Engelman, D.M. Structure-based prediction of the stability of transmembrane helix-helix interactions: The sequence dependence of glycophorin a dimerization. Proc. Natl. Acad. Sci. USA 1998, 95, 3583–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.; Wimley, W.C.; Hristova, K. Transmembrane helix dimerization: Beyond the search for sequence motifs. Biochim. Biophys. Acta 2012, 1818, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Hogel, P.; Gotz, A.; Kuhne, F.; Ebert, M.; Stelzer, W.; Rand, K.D.; Scharnagl, C.; Langosch, D. Glycine Perturbs Local and Global Conformational Flexibility of a Transmembrane Helix. Biochemistry 2018, 57, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Deber, C.M. A measure of helical propensity for amino acids in membrane environments. Nat. Struct. Biol. 1994, 1, 558. [Google Scholar] [CrossRef]
- Padmanabhan, S.; Baldwin, R.L. Tests for helix-stabilizing interactions between various nonpolar side chains in alanine-based peptides. Protein. Sci. 1994, 3, 1992–1997. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cymer, F.; Schneider, D. Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain. Membranes 2021, 11, 512. https://doi.org/10.3390/membranes11070512
Cymer F, Schneider D. Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain. Membranes. 2021; 11(7):512. https://doi.org/10.3390/membranes11070512
Chicago/Turabian StyleCymer, Florian, and Dirk Schneider. 2021. "Small Residues Inhibit Homo-Dimerization of the Human Carbonic Anhydrase XII Transmembrane Domain" Membranes 11, no. 7: 512. https://doi.org/10.3390/membranes11070512