
First-Principles Density Functional Theory Calculations of Bilayer Membranes Heterostructures of Ti3C2T2 (MXene)/Graphene and AgNPs


Abstract
:1. Introduction
2. Computational Method
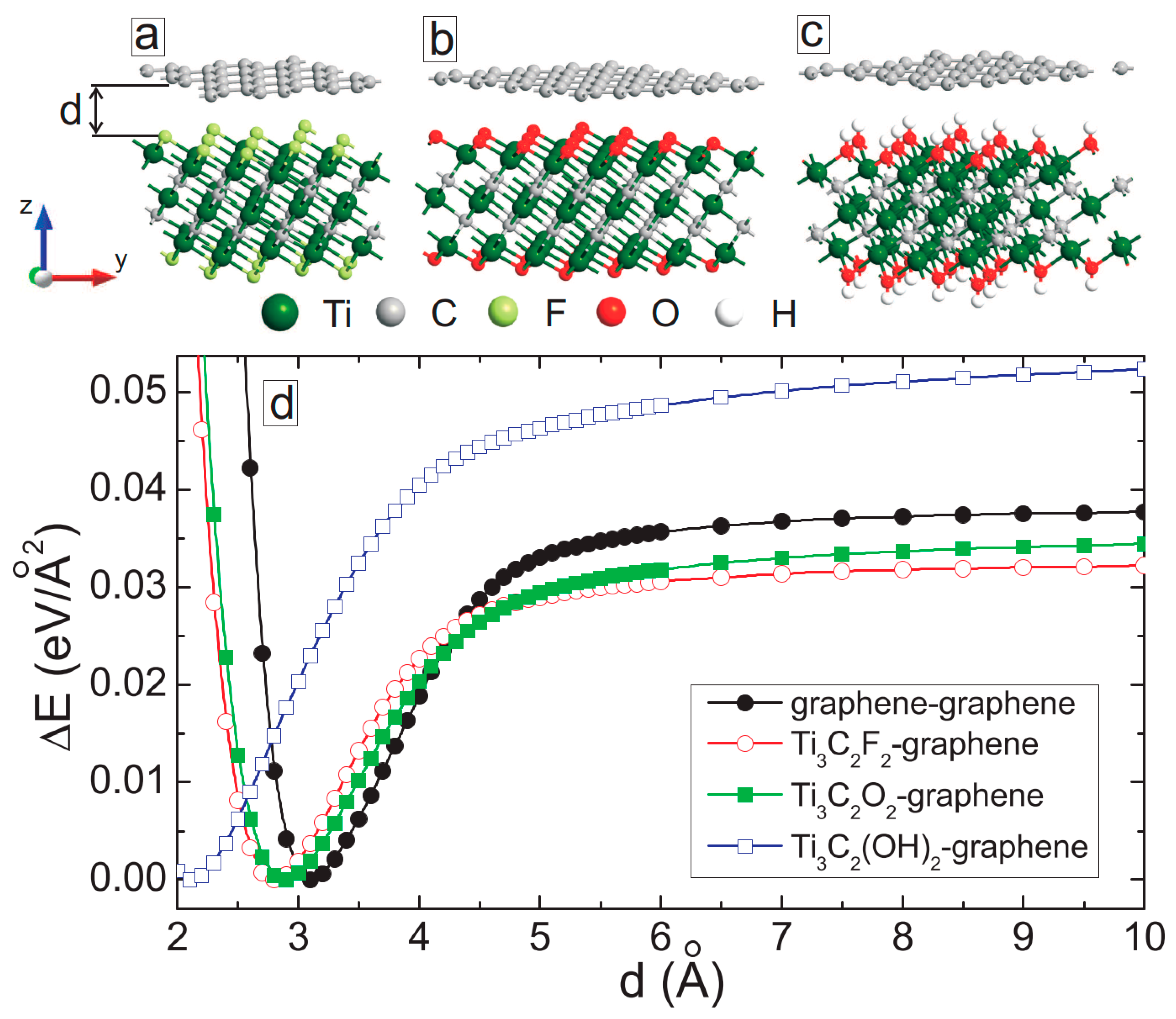
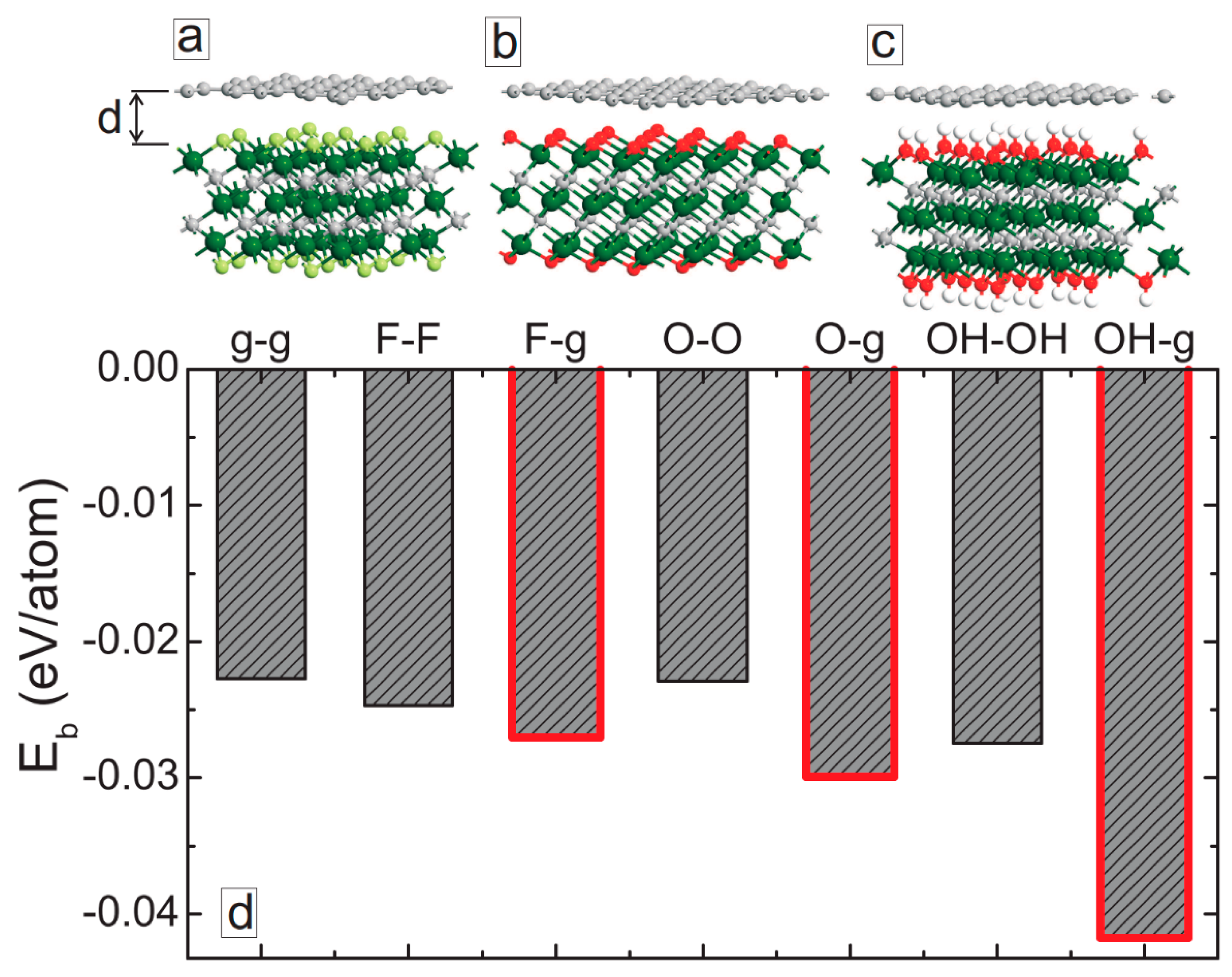
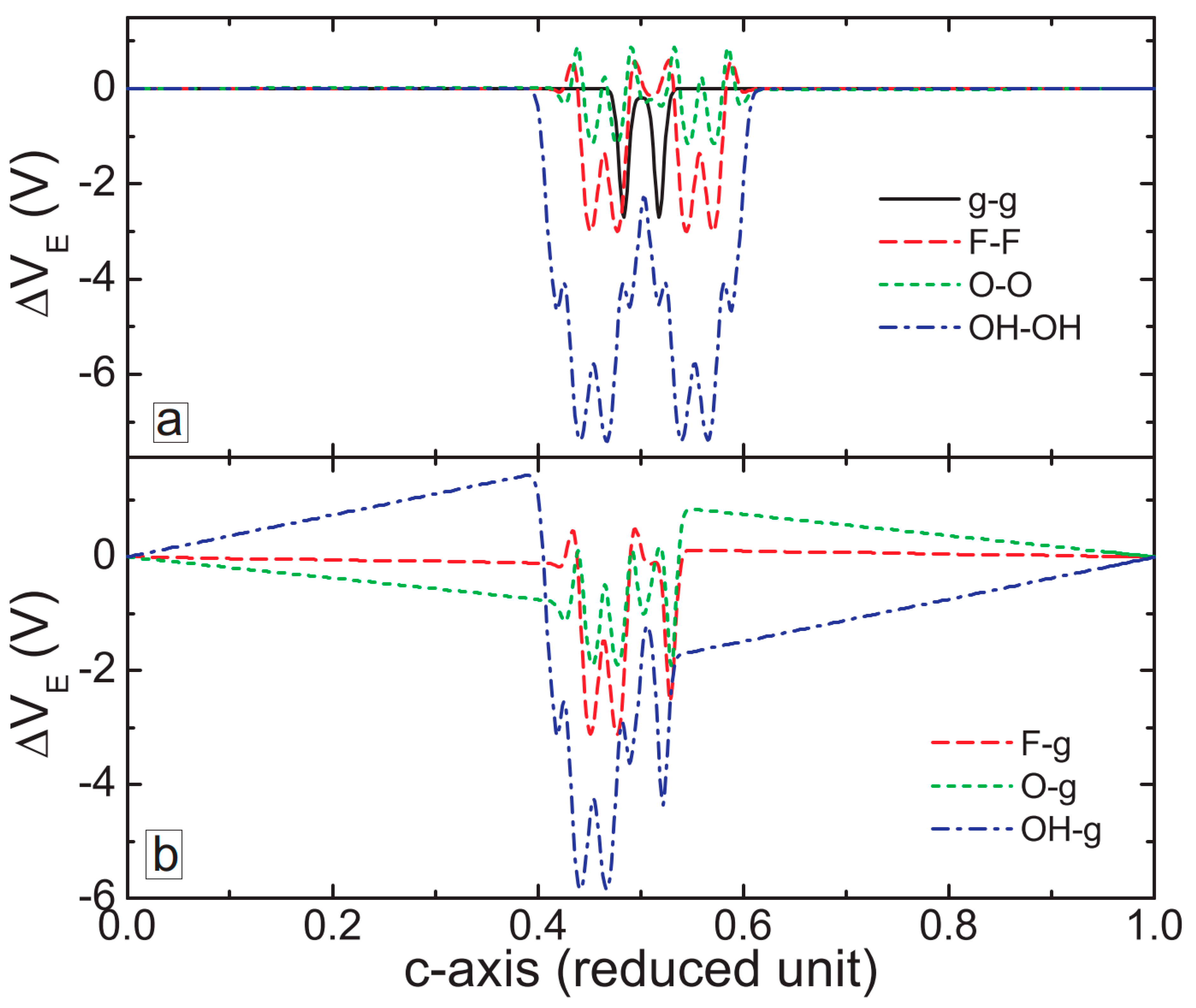
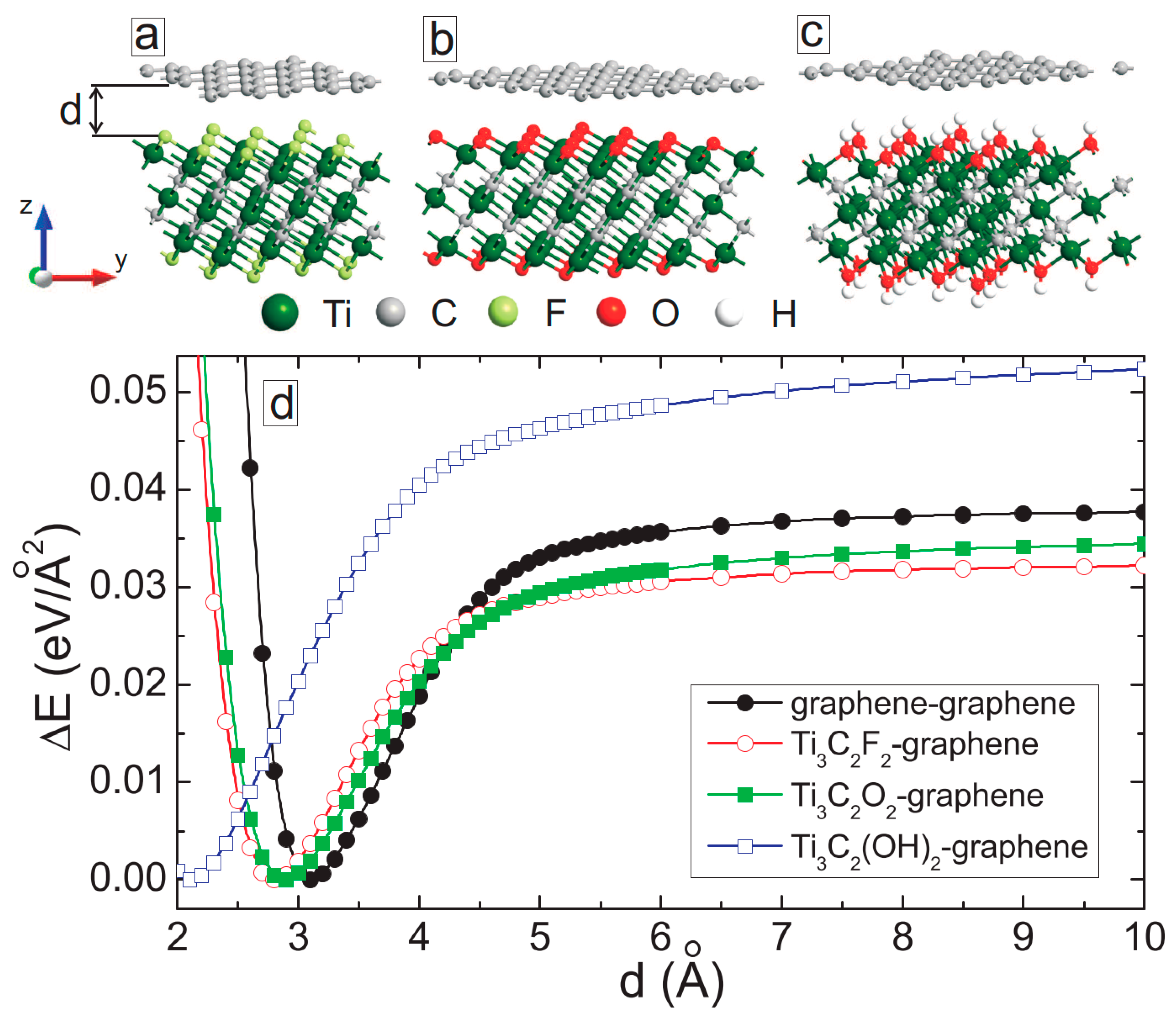
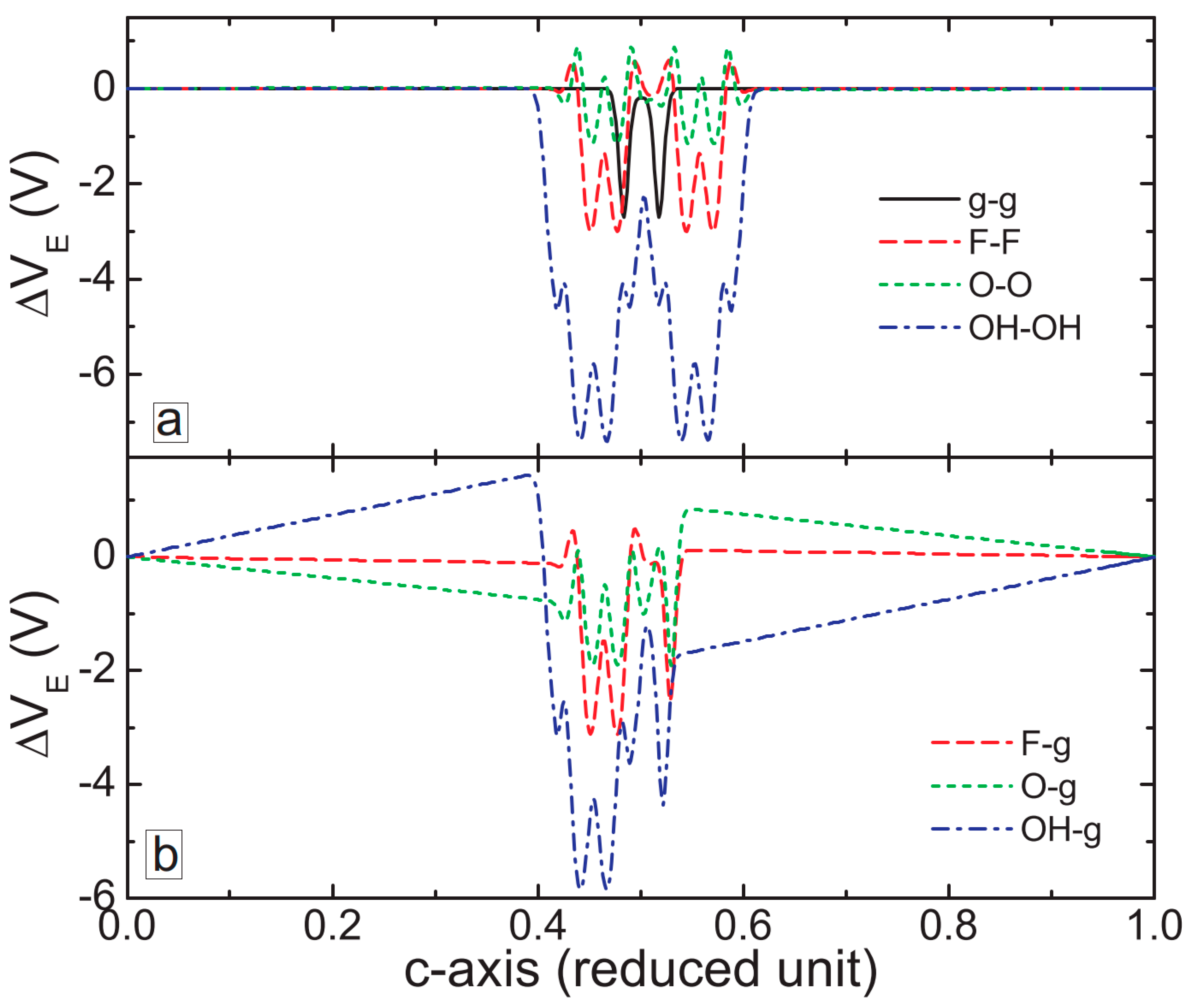
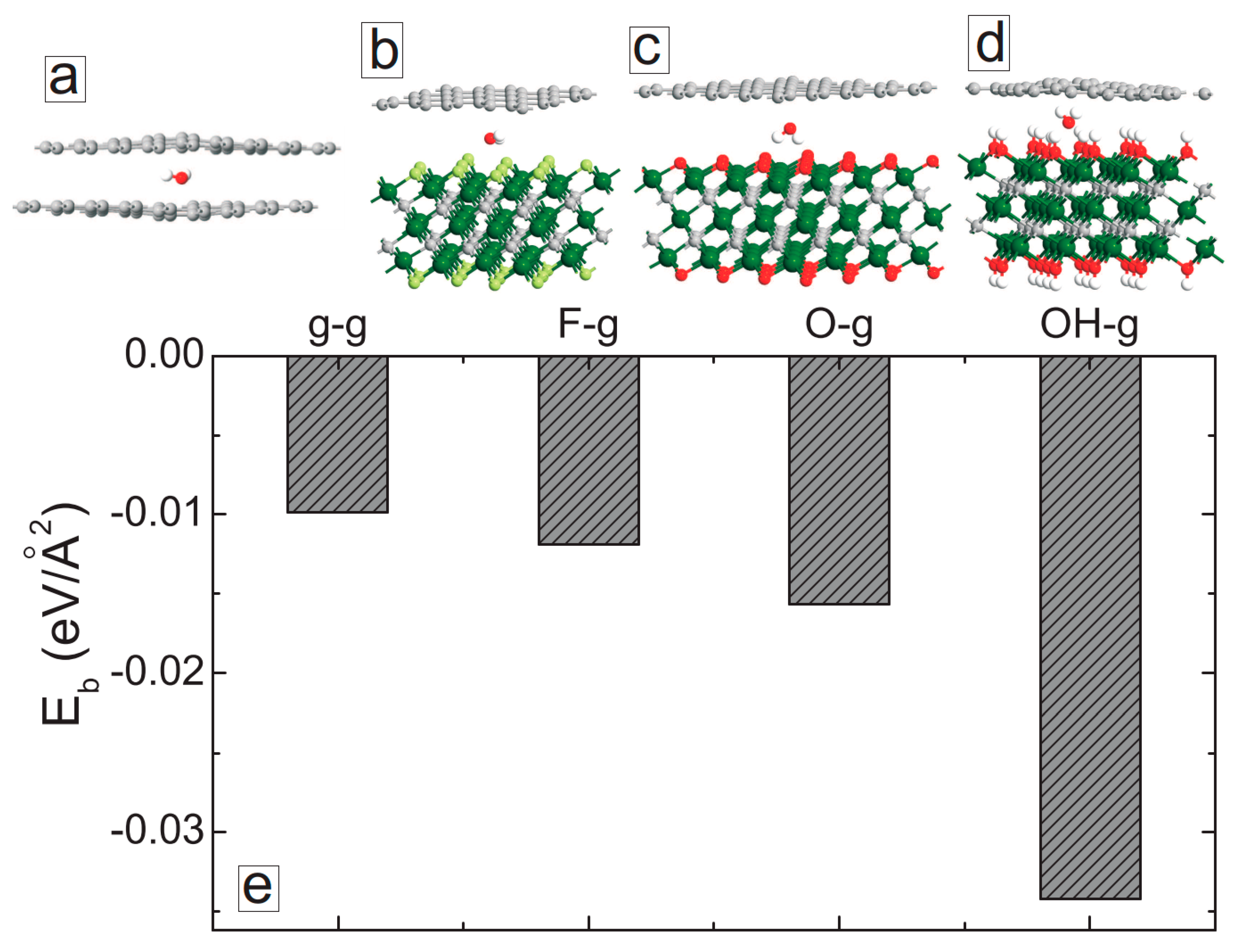
3. MXene/Graphene Bilayer
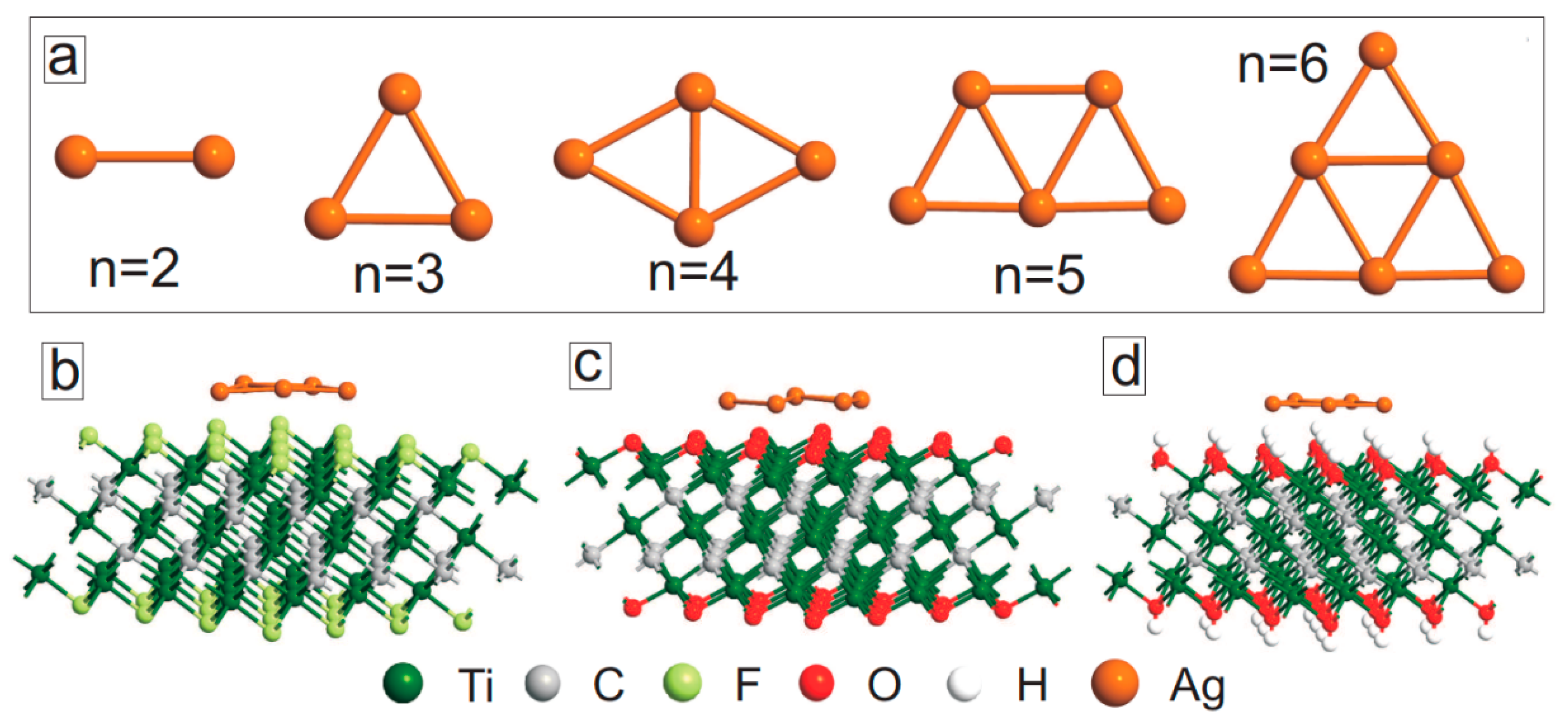
4. Ag Nanocluster-Decorated MXene
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th Anniversary Article: MXenes: A New Family of Two-Dimensional Materials. Adv. Mater. 2014, 26, 992–1005. [Google Scholar] [CrossRef]
- Lei, J.-C.; Zhang, X.; Zhou, Z. Recent advances in MXene: Preparation, properties, and applications. Front. Phys. 2015, 10, 276–286. [Google Scholar] [CrossRef]
- Naguib, M.; Come, J.; Dyatkin, B.; Presser, V.; Taberna, P.L.; Simon, P.; Barsoum, M.W.; Gogotsi, Y. MXene: A promising transition metal carbide anode for lithium-ion batteries. Electrochem. Commun. 2012, 16, 6164. [Google Scholar] [CrossRef] [Green Version]
- Lukatskaya, M.R.; Mashtalir, O.; Ren, C.E.; Dall’Agnese, Y.; Rozier, P.; Taberna, P.L.; Naguib, M.; Simon, P.; Barsoum, M.W.; Gogotsi, Y. Cation intercalation and high volumetric capacitance of two-dimensional titanium carbide. Science 2013, 341, 15021505. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Dall’Agnese, Y.; Naguib, M.; Gogotsi, Y.; Barsoum, M.W.; Zhuang, H.L.L.; Kent, P.R.C. Prediction and characterization of MXene nanosheet anodes for non-lithium-ion batteries. ACS Nano 2014, 8, 96069615. [Google Scholar] [CrossRef]
- Xiong, D.; Li, X.; Bai, Z.; Lu, S. Recent Advances in Layered Ti3C2Tx MXene for Electrochemical Energy Storage. Small 2018, 14, 1703419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.M.; Guo, J.X.; Zhang, Q.R.; Xiang, J.Y.; Liu, B.Z.; Zhou, A.G.; Liu, R.; Tian, Y. Unique lead adsorption behavior of activated hydroxyl group in two-dimensional titanium carbide. J. Am. Chem. Soc. 2014, 136, 41134116. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.Z.; Zhu, M.S.; Zhang, W.C.; Zhen, X.; Pei, Z.X.; Xue, Q.; Zhi, C.; Shi, P. Ultrathin MXene-micropattern-based field-Effect transistor for probing neural activity. Adv. Mater. 2016, 28, 3333–3339. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Li, Y.C.; Yu, X.F.; Cheng, J.B. MXenes: Reusable materials for NH3 sensor or capturer by controlling the charge injection. Sens. Actuators B Chem. 2016, 235, 103–109. [Google Scholar] [CrossRef]
- Lorencova, L.; Bertok, T.; Dosekova, E.; Holazova, A.; Paprckova, D.; Vikartovska, A.; Sasinkova, V.; Filip, J.; Kasak, P.; Jerigova, M.; et al. Electrochemical performance of Ti3C2Tx MXene in aqueous media: Towards ultrasensitive H2O2 sensing. Electrochim. Acta 2017, 235, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Ling, Z.; Ren, C.E.; Zhao, M.Q.; Yang, J.; Giammarco, J.M.; Qiu, J.S.; Barsoum, M.W.; Gogotsi, Y. Flexible and conductive MXene films and nanocomposites with high capacitance. Proc. Natl. Acad. Sci. USA 2014, 111, 16676–16681. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.Q.; Ren, C.E.; Ling, Z.; Lukatskaya, M.R.; Zhang, C.F.; Van Aken, K.L.; Barsoum, M.W.; Gogotsi, Y. Flexible MXene/Carbon nanotube composite paper with high volumetric capacitance. Adv. Mater. 2015, 27, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.P.; Wang, L.B.; Li, Z.Y.; Zhou, A.G.; Hu, Q.K.; Cao, X.X. Preparation of MXene-Cu2O nanocomposite and effect on thermal decomposition of ammonium perchlorate. Solid State Sci. 2014, 35, 62–65. [Google Scholar] [CrossRef]
- Rakhi, R.B.; Nayuk, P.; Xia, C.; Alshareef, H.N. Novel amperometric glucose biosensor based on MXene nanocomposite. Sci. Rep. 2016, 6, 36422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, C.E.; Hatzell, K.B.; Alhabeb, M.; Ling, Z.; Mahmoud, K.A.; Gogotsi, Y. Charge- and Size-Selective Ion Sieving Through Ti3C2Tx MXene Membranes. J. Phys. Chem. Lett. 2015, 6, 4026–4031. [Google Scholar] [CrossRef] [PubMed]
- Osti, N.C.; Naguib, M.; Ostadhossein, A.; Xie, Y.; Kent, P.R.C.; Dyatkin, B.; Rother, G.; Heller, W.T.; van Duin, A.C.T.; Gogotsi, Y.; et al. Effect of Metal Ion Intercalation on the Structure of MXene and Water Dynamics on its Internal Surfaces. ACS Appl. Mater. Interfaces 2016, 8, 8859–8863. [Google Scholar] [CrossRef]
- Berdiyorov, G.R.; Madjet, M.E.; Mahmoud, K.A. Ionic sieving through Ti3C2(OH)2 MXene: First-principles calculations. Appl. Phys. Lett. 2016, 108, 113110. [Google Scholar] [CrossRef]
- Rasool, K.; Mahmoud, K.A.; Johnson, D.J.; Helal, M.; Berdiyorov, G.R.; Gogotsi, Y. Efficient antibacterial membrane based on two-dimensional Ti3C2Tx (MXene) nanosheets. Sci. Rep. 2017, 7, 1598. [Google Scholar] [CrossRef]
- Abdul Rasheed, P.; Pandey, R.P.; Rasool, K.; Mahmoud, K.A. Ultra-sensitive electrocatalytic detection of bromate in drinking water based on Nafion/Ti3C2Tx (MXene) modified glassy carbon electrode. Sens. Actuators B Chem. 2018, 265, 652–659. [Google Scholar] [CrossRef]
- Xu, S.; Wei, G.; Li, J.; Han, W.; Gogotsi, Y. Flexible MXene graphene electrodes with high volumetric capacitance for integrated co-cathode energy conversion/storage devices. J. Mater. Chem. A 2017, 5, 17442–17451. [Google Scholar] [CrossRef]
- Ma, Z.; Zhou, X.; Deng, W.; Lei, D.; Liu, Z. 3D Porous MXene (Ti3C2)/Reduced Graphene Oxide Hybrid Films for Advanced Lithium Storage. ACS Appl. Mater. Interfaces 2018, 10, 36343643. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, N.; Ma, Y.; Wang, S.; Liu, W.; Luo, C.; Zhang, H.; Cheng, F.; Rao, J.; Hu, X.; et al. Highly Self-Healable 3D Microsupercapacitor with MXene—Graphene Composite Aerogel. ACS Nano 2018, 12, 4224–4232. [Google Scholar] [CrossRef]
- Al Aani, S.; Wright, C.J.; Ali Atieh, M.; Hilal, N. Engineering nanocomposite membranes: Addressing current challenges and future opportunities. Desalination 2017, 401, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rasool, K.; Pandey, R.P.; Abdul Rasheed, P.; Buczek, S.; Gogotsi, Y.; Mahmoud, K.A. Water treatment and environmental remediation applications of two-dimensional metal carbides (MXenes). Mater. Today 2019, 30, 80–102. [Google Scholar] [CrossRef]
- Liang, L.; Li, Q.; Yan, X.; Feng, Y.; Wang, Y.; Zhang, H.-B.; Zhou, X.; Liu, C.; Shen, C.; Xie, X. Multifunctional Magnetic Ti3C2Tx MXene/Graphene Aerogel with Superior Electromagnetic Wave Absorption Performance. ACS Nano 2021, 15, 6622–6632. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Choi, J.S.; Lee, M.J.; Park, B.H.; Bukhvalov, D.; Son, Y.-W.; Yoon, D.; Cheong, H.; Yun, J.-N.; Jung, Y.; et al. Between Scylla and Charybdis: Hydrophobic Graphene-Guided Water Diffusion on Hydrophilic Substrates. Sci. Rep. 2013, 3, 2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Wu, C.; Wang, Y.; Tomsia, A.P.; Li, M.; Saiz, E.; Fang, S.; Baughman, R.H.; Jiang, L.; Cheng, Q. Super-tough MXene-functionalized graphene sheets. Nat. Commun. 2020, 11, 2077. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; An, X.; Huang, L.; Wang, X.; Feng, W.; Qiu, S.; Wang, Q.; Sun, C. A DFT study of Ti3C2O2 MXenes quantum dots supported on single layer graphene: Electronic structure an hydrogen evolution performance. Front. Phys. 2021, 16, 53506. [Google Scholar] [CrossRef]
- Al-Hamadani, Y.A.J.; Jun, B.-M.; Yoon, M.; Taheri-Qazvini, N.; Snyder, S.A.; Jang, M.; Heo, J.; Yoon, Y. Applications of MXene-based membranes in water purification: A review. Chemosphere 2020, 254, 126821. [Google Scholar] [CrossRef]
- Ding, L.; Wei, Y.Y.; Wang, Y.J.; Chen, H.B.; Caro, J.; Wang, H.H. A Two-Dimensional Lamellar Membrane: MXene Nanosheet Stacks. Angew. Chem. Int. Ed. 2017, 56, 1825–1829. [Google Scholar] [CrossRef]
- Pandey, R.P.; Rasool, K.; Madhavan, V.E.; Aissa, B.; Gogotsi, Y.; Mahmoud, K.A. Ultrahigh-flux and fouling-resistant membranes based on layered silver/MXene (Ti3C2Tx) nanosheets. J. Mater. Chem. A 2018, 6, 3522–3533. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, H.; Zou, G.; Fernandez, C.; Liu, B.; Zhang, Q.; Hu, J.; Peng, Q. Self-Reduction Synthesis of New MXene/Ag Composites with Unexpected Electrocatalytic Activity. ACS Sustain. Chem. Eng. 2016, 4, 6763–6771. [Google Scholar] [CrossRef]
- Li, C.; Zhuo, Y.; Xiao, X.; Li, S.; Han, K.; Lu, M.; Zhang, J.; Chen, S.; Gu, H. Facile Electrochemical Microbiosensor Based on In Situ Self-Assembly of Ag Nanoparticles Coated on Ti3C2Tx for In Vivo Measurements of Chloride Ions in the PD Mouse Brain. Anal. Chem. 2021, 93, 7647–7656. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zhou, Z.; Shen, P. Are MXenes Promising Anode Materials for Li Ion Batteries? Computational Studies on Electronic Properties and Li Storage Capability of Ti3C2 and Ti3C2X2 (X = F, OH) Monolayer. J. Am. Chem. Soc. 2012, 134, 16909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, Z.; Zhao, X.; Tang, Q.; Zhou, Z. Computational studies on structural and electronic properties of functionalized MXene monolayers and nanotubes. J. Mater. Chem. A 2015, 3, 4960–4966. [Google Scholar] [CrossRef]
- Xie, Y.; Kent, P.R.C. Hybrid density functional study of structural and electronic properties of functionalized Tin+1Xn (X = C, N) monolayers. Phys. Rev. B 2013, 87, 235441. [Google Scholar] [CrossRef] [Green Version]
- Berdiyorov, G.R. Effect of surface functionalization on the electronic transport properties of Ti3C2 MXene. Eurphys. Lett. 2015, 111, 67002. [Google Scholar] [CrossRef]
- Berdiyorov, G.R. Effect of lithium and sodium ion adsorption on the electronic transport properties of Ti3C2 MXene. Appl. Surf. Sci. 2015, 359, 153–157. [Google Scholar] [CrossRef]
- Berdiyorov, G.R. Optical properties of functionalized Ti3C2T2 (T = F, O, OH) MXene: First-principles calculations. AIP Adv. 2016, 6, 055105. [Google Scholar] [CrossRef] [Green Version]
- Hope, M.A.; Forse, A.C.; Griffith, K.J.; Lukatskaya, M.R.; Ghidiu, M.; Gogotsi, Y.; Grey, C.P. NMR reveals the surface functionalisation of Ti3C2 MXene. Phys. Chem. Chem. Phys. 2016, 18, 5099–5102. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- QuantumATK. Synopsys QuantumATK. 2019. Available online: https://www.synopsys.com/silicon/quantumatk.html (accessed on 1 January 2019).
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellen-dorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G. QuantumATK: An integrated platform of electronic and atomic-scale modelling tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef]
- Smidstrup, S.; Stradi, D.; Wellendorff, J.; Khomyakov, P.A.; Vej-Hansen, U.G.; Lee, M.-E.; Ghosh, T.; Jonsson, E.; Jonsson, H.; Stokbro, K. First-principles Green’s-function method for surface calculations: A pseudopotential localized basis set approach. Phys. Rev. B 2017, 96, 195309. [Google Scholar] [CrossRef] [Green Version]
- Manz, T.A.; Limas, N.G. Introducing DDEC6 atomic population analysis: Part 1. Charge partitioning theory and methodology. RSC Adv. 2016, 6, 47771–47801. [Google Scholar] [CrossRef] [Green Version]
- Limas, N.G.; Manz, T.A. Introducing DDEC6 atomic population analysis: Part 2. Computed results for a wide range of periodic and nonperiodic materials. RSC Adv. 2016, 6, 45727–45747. [Google Scholar] [CrossRef]
- Madjet, M.E.; El-Mellouhi, F.; Carignano, M.A.; Berdiyorov, G.R. Atomic partial charges on CH3NH3PbI3 from first-principles electronic structure calculations. J. Appl. Phys. 2016, 119, 165501. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | Number of Atoms | Area (Å2) | Mean Absolute Strain (%) |
|---|---|---|---|
| graphene/Ti3C2F2 | 122 | 101.3 | 0.09 |
| graphene/Ti3C2O2 | 162 | 132.6 | 0.03 |
| graphene/Ti3C2(OH)2 | 159 | 111.4 | 0.07 |
| Systems | No Ag Cluster | Ag2 | Ag3 | Ag4 | Ag5 | Ag6 |
|---|---|---|---|---|---|---|
| Ti3C2F2 | 2.013 | 4.995 | 4.981 | 5.012 | 4.995 | 5.011 |
| Ti3C2O2 | 2.298 | 4.441 | 4.017 | 4.039 | 4.042 | 4.043 |
| Ti3C2(OH)2 | 0.486 | 4.155 | 3.845 | 3.935 | 3.701 | 3.704 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berdiyorov, G.R.; Madjet, M.E.; Mahmoud, K.A. First-Principles Density Functional Theory Calculations of Bilayer Membranes Heterostructures of Ti3C2T2 (MXene)/Graphene and AgNPs. Membranes 2021, 11, 543. https://doi.org/10.3390/membranes11070543
Berdiyorov GR, Madjet ME, Mahmoud KA. First-Principles Density Functional Theory Calculations of Bilayer Membranes Heterostructures of Ti3C2T2 (MXene)/Graphene and AgNPs. Membranes. 2021; 11(7):543. https://doi.org/10.3390/membranes11070543
Chicago/Turabian StyleBerdiyorov, Golibjon. R., Mohamed E. Madjet, and Khaled. A. Mahmoud. 2021. "First-Principles Density Functional Theory Calculations of Bilayer Membranes Heterostructures of Ti3C2T2 (MXene)/Graphene and AgNPs" Membranes 11, no. 7: 543. https://doi.org/10.3390/membranes11070543
APA StyleBerdiyorov, G. R., Madjet, M. E., & Mahmoud, K. A. (2021). First-Principles Density Functional Theory Calculations of Bilayer Membranes Heterostructures of Ti3C2T2 (MXene)/Graphene and AgNPs. Membranes, 11(7), 543. https://doi.org/10.3390/membranes11070543