
Membrane Nanoscopic Organization of D2L Dopamine Receptor Probed by Quantum Dot Tracking


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Transfections
2.3. Single Qdot Tracking via Spinning Disk Confocal Microscopy
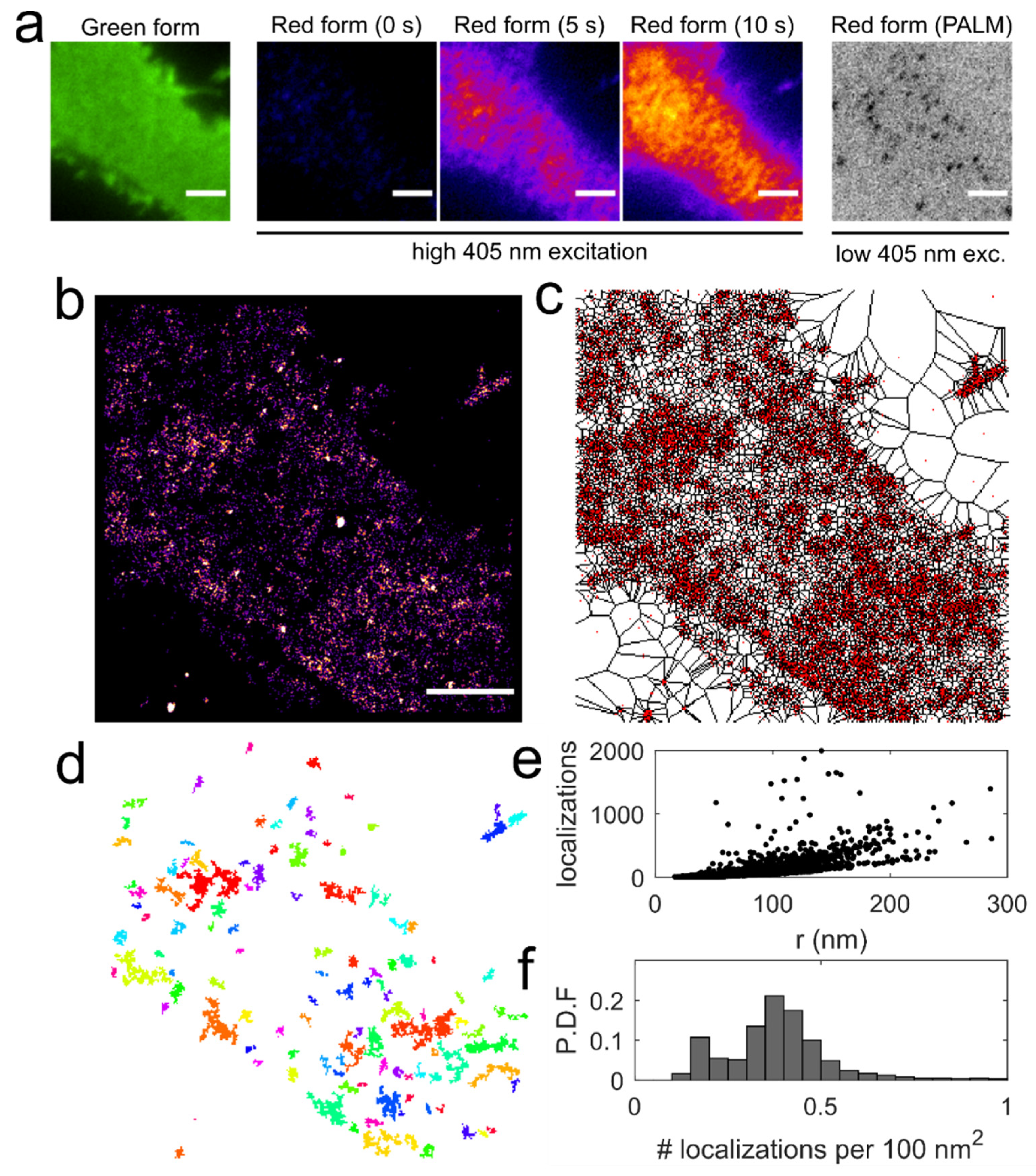
2.4. Photoactivation Localization Microscopy of mEos2-Fused PH-PLCδ
2.5. Trajectory Reconstruction and Diffusion Analysis
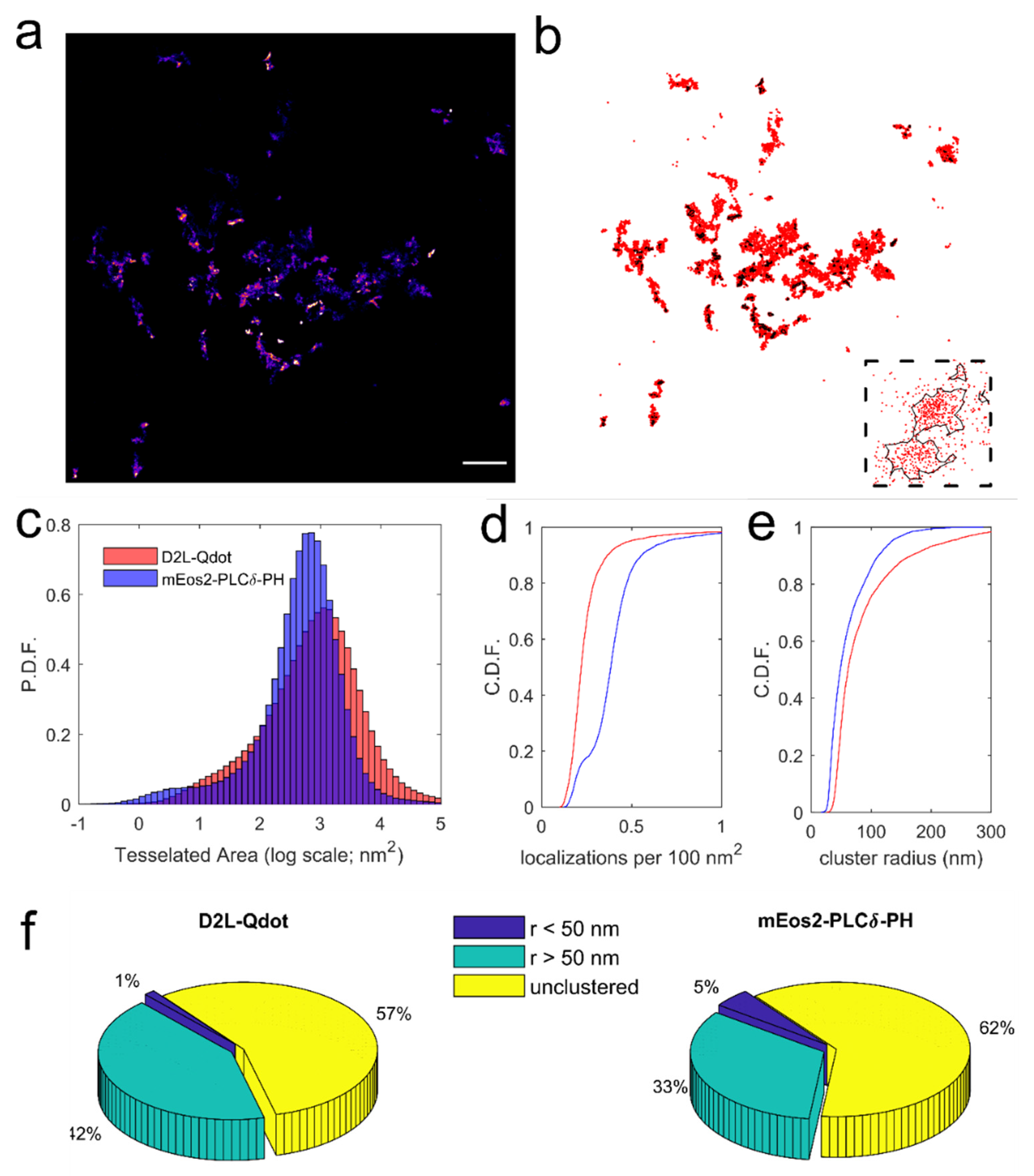
2.6. Cluster Identification Using Voronoї Tessellation
3. Results and Discussion
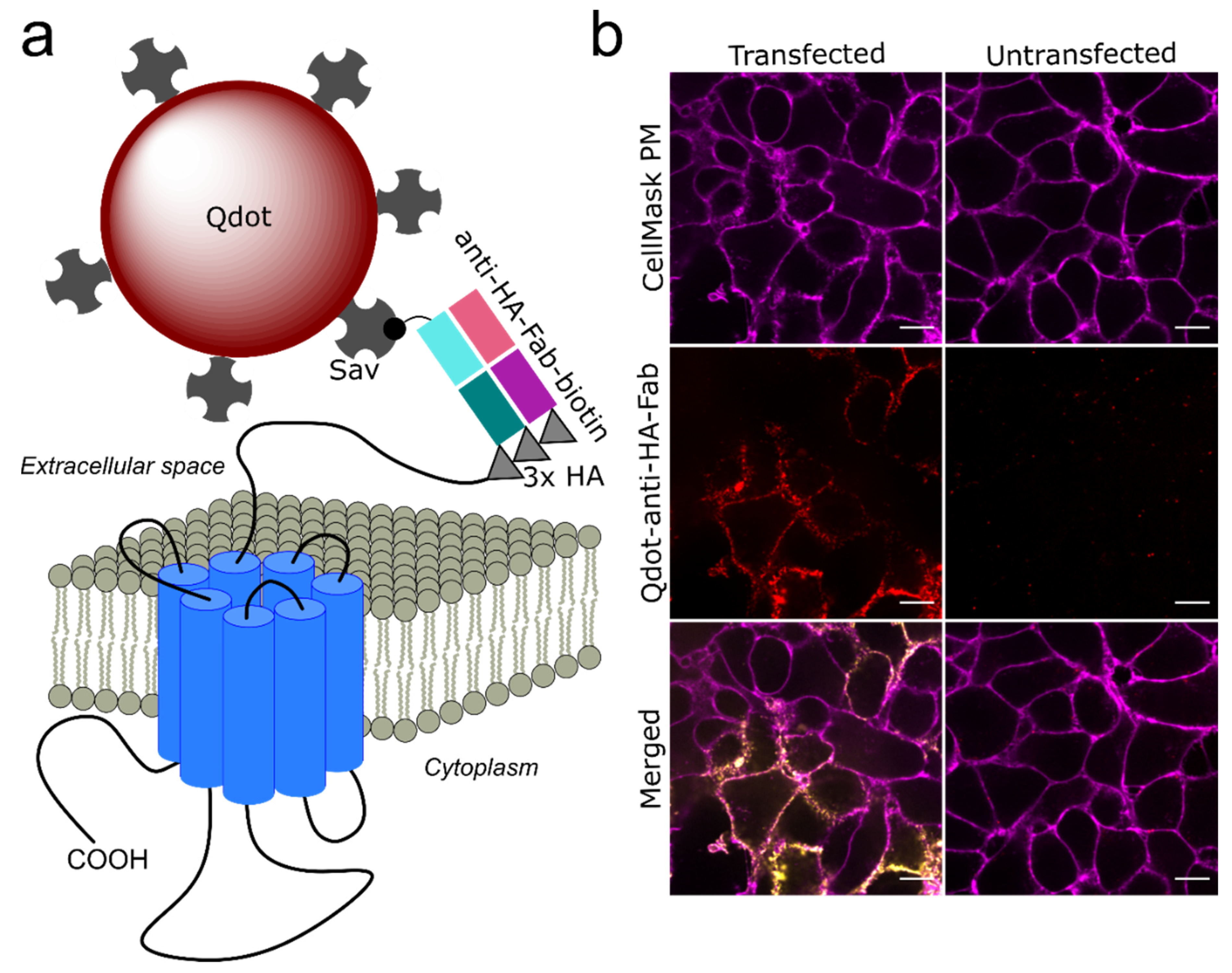
3.1. Qdot Tracking of D2L Receptors
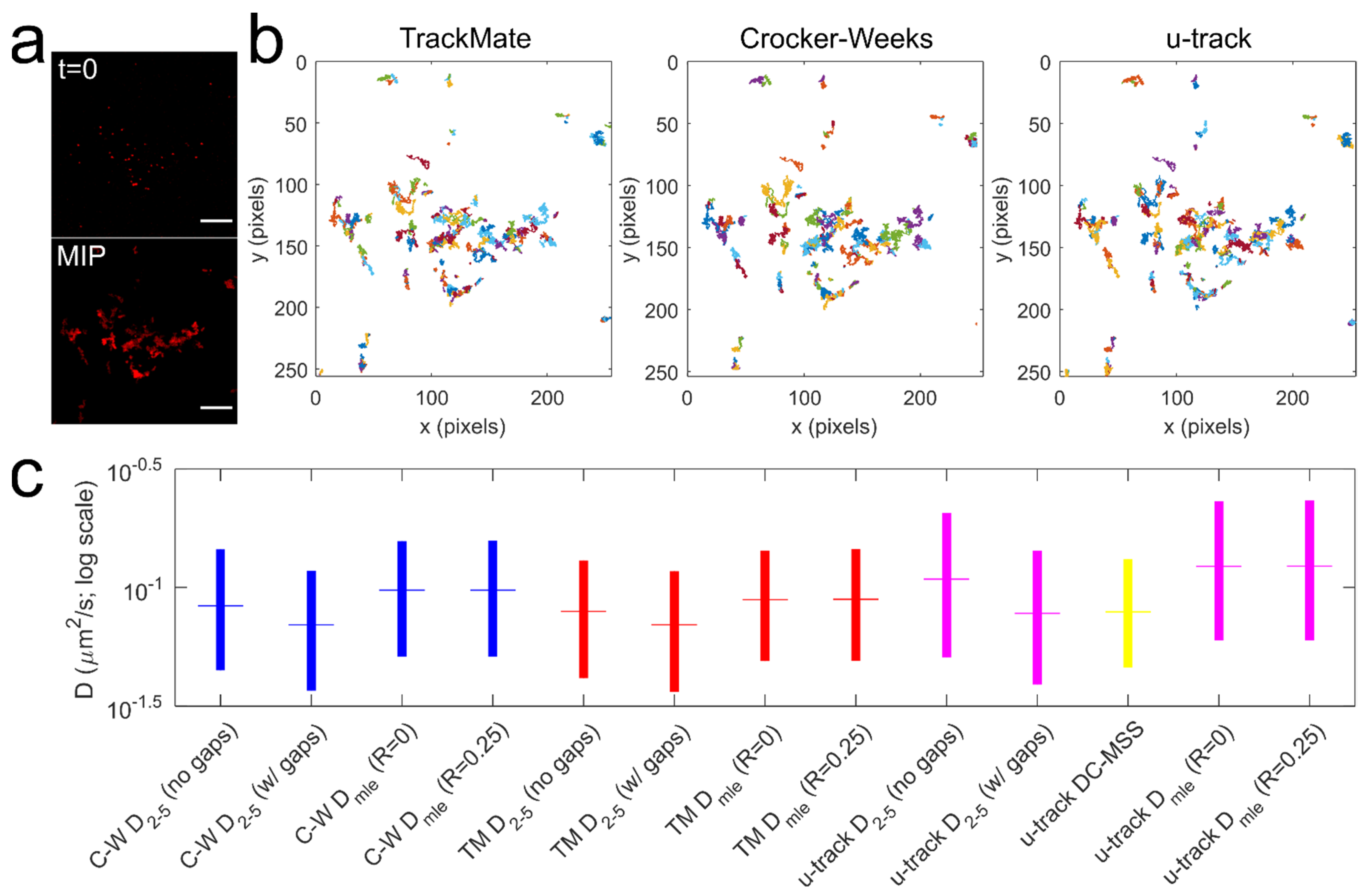
3.2. Comparison of Algorithms for Trajectory Reconstruction and Diffusion Coefficient Determination
- The D2–5 diffusion coefficient of Qdot-D2L trajectories that were rendered continuous by removing the gaps due to missing Qdot position (Qdot blinking or out-of-focus drift) was significantly faster (*** p < 0.0001, Mann-Whitney U test) than the D2–5 diffusion coefficient of Qdot-D2L trajectories that contained gaps for all trajectory reconstruction algorithms;
- The motion blur coefficient R (dynamic error in the particle’s position due to frame averaging) in the MLE algorithm had no significant effect on the DMLE diffusion coefficient for all trajectory reconstruction algorithms (Rmin = 0 versus Rmax = 0.25; p > 0.05, Mann–Whitney U test);
- The MLE algorithm consistently yielded a greater diffusion rate than the methods that rely on fitting the first few points of the MSD-time curve or its log-log form (*** p < 0.0001, Mann–Whitney U test).
- The diffusion rate of Qdot-D2L determined via widely used u-track package was only not statistically different from D2–5 determined for u-track trajectories that contained gaps due to missing Qdot positions and CW trajectories that were rendered continuous (p > 0.05, Mann-Whitney U test).
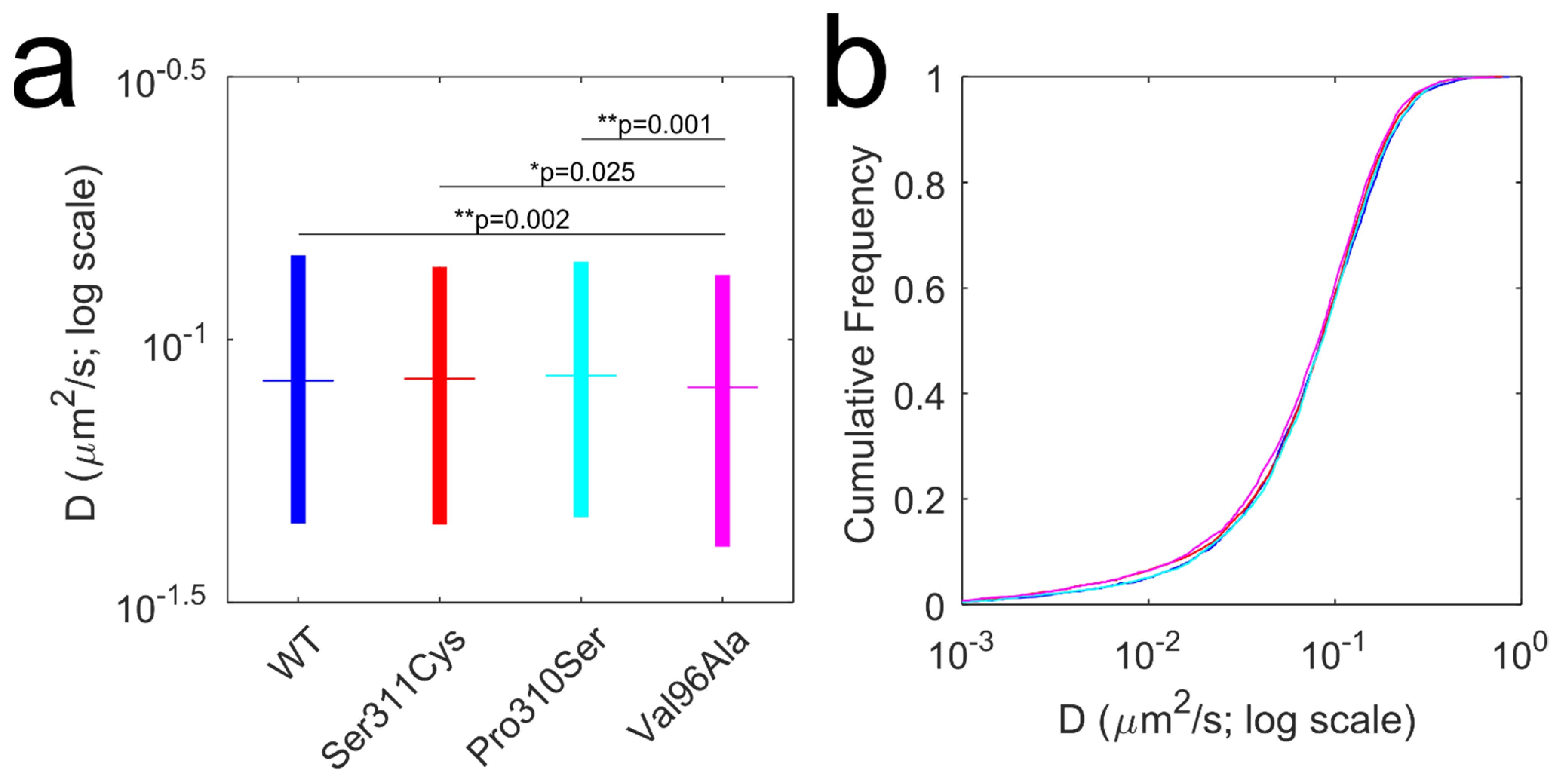
3.3. Diffusion Dynamics of D2L Variants
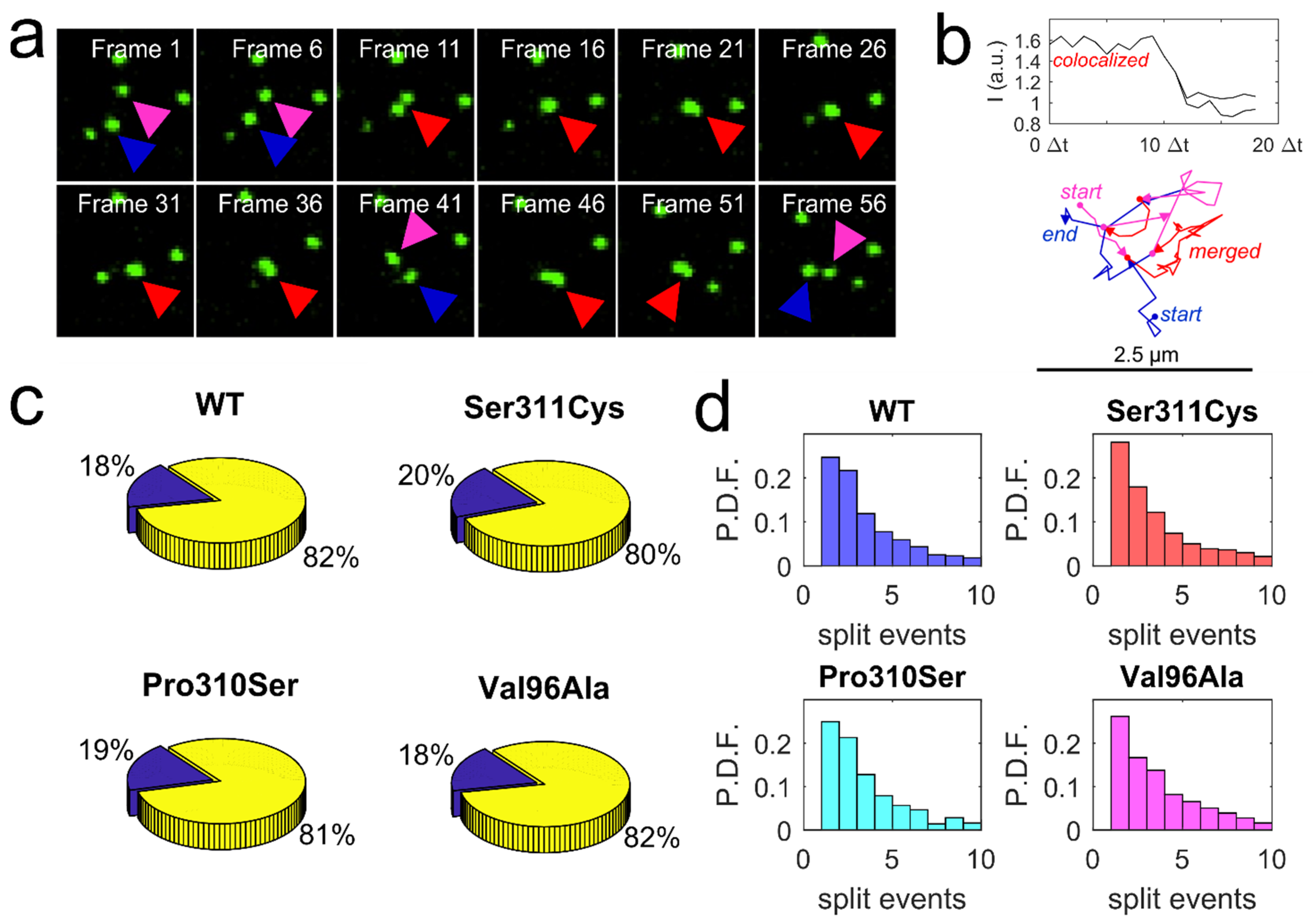
3.4. Application of Voronoї Tessellation to Qdot-D2L Trajectories
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-Function of the G Protein–Coupled Receptor Superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [Green Version]
- Calebiro, D.; Grimes, J. G Protein–Coupled Receptor Pharmacology at the Single-Molecule Level. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 73–87. [Google Scholar] [CrossRef]
- Eichel, K.; Jullié, D.; Barsi-Rhyne, B.; Latorraca, N.R.; Masureel, M.; Sibarita, J.-B.; Dror, R.O.; von Zastrow, M. Catalytic activation of β-arrestin by GPCRs. Nature 2018, 557, 381–386. [Google Scholar] [CrossRef]
- Ferguson, S.S.G.; Downey, W.E.; Colapietro, A.-M.; Barak, L.S.; Ménard, L.; Caron, M.G. Role of β-Arrestin in Mediating Agonist-Promoted G Protein-Coupled Receptor Internalization. Science 1996, 271, 363. [Google Scholar] [CrossRef]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.F.; Els-Heindl, S.; Beck-Sickinger, A.G.; Wieneke, R.; Tampé, R. Photoinduced receptor confinement drives ligand-independent GPCR signaling. Science 2021, 371, eabb7657. [Google Scholar] [CrossRef] [PubMed]
- Asher, W.B.; Geggier, P.; Holsey, M.D.; Gilmore, G.T.; Pati, A.K.; Meszaros, J.; Terry, D.S.; Mathiasen, S.; Kaliszewski, M.J.; McCauley, M.D.; et al. Single-molecule FRET imaging of GPCR dimers in living cells. Nat. Methods 2021, 18, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Möller, J.; Isbilir, A.; Sungkaworn, T.; Osberg, B.; Karathanasis, C.; Sunkara, V.; Grushevskyi, E.O.; Bock, A.; Annibale, P.; Heilemann, M.; et al. Single-molecule analysis reveals agonist-specific dimer formation of µ-opioid receptors. Nat. Chem. Biol. 2020, 16, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, M.; Hiroshima, M.; Togashi, Y.; Abe, M.; Yamashita, T.; Shichida, Y.; Murata, M.; Ueda, M.; Sako, Y. Single-molecule diffusion-based estimation of ligand effects on G protein–coupled receptors. Sci. Signal. 2018, 11, eaao1917. [Google Scholar] [CrossRef] [Green Version]
- Halls, M.L.; Yeatman, H.R.; Nowell, C.J.; Thompson, G.L.; Gondin, A.B.; Civciristov, S.; Bunnett, N.W.; Lambert, N.A.; Poole, D.P.; Canals, M. Plasma membrane localization of the μ-opioid receptor controls spatiotemporal signaling. Sci. Signal. 2016, 9, ra16. [Google Scholar] [CrossRef]
- Sungkaworn, T.; Jobin, M.-L.; Burnecki, K.; Weron, A.; Lohse, M.J.; Calebiro, D. Single-molecule imaging reveals receptor–G protein interactions at cell surface hot spots. Nature 2017, 550, 543–547. [Google Scholar] [CrossRef]
- Jullié, D.; Stoeber, M.; Sibarita, J.-B.; Zieger, H.L.; Bartol, T.M.; Arttamangkul, S.; Sejnowski, T.J.; Hosy, E.; von Zastrow, M. A Discrete Presynaptic Vesicle Cycle for Neuromodulator Receptors. Neuron 2020, 105, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, O.; Torres, R.; Ferguson, R.S.; Josephs, T.; Rosenthal, S.J. Single Quantum Dot Tracking Unravels Agonist Effects on the Dopamine Receptor Dynamics. Biochemistry 2021, 60, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Felce, J.H.; Davis, S.J.; Klenerman, D. Single-Molecule Analysis of G Protein-Coupled Receptor Stoichiometry: Approaches and Limitations. Trends Pharmacol. Sci. 2018, 39, 96–108. [Google Scholar] [CrossRef]
- Asher, W.B.; Mathiasen, S.; Holsey, M.D.; Grinnell, S.G.; Lambert, N.A.; Javitch, J.A. Extreme Vetting of Dopamine Receptor Oligomerization. In G-Protein-Coupled Receptor Dimers; Herrick-Davis, K., Milligan, G., Di Giovanni, G., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 99–127. [Google Scholar]
- Calebiro, D.; Koszegi, Z. The subcellular dynamics of GPCR signaling. Mol. Cell. Endocrinol. 2019, 483, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, I.D.; Kovtun, O.; Crescentini, T.M.; Rosenthal, S.J. Biotinylated-spiperone ligands for quantum dot labeling of the dopamine D2 receptor in live cell cultures. Bioorg. Med. Chem. Lett. 2019, 29, 959–964. [Google Scholar] [CrossRef]
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182. [Google Scholar] [CrossRef] [Green Version]
- Sibley, D.R.; Monsma, F.J. Molecular biology of dopamine receptors. Trends Pharmacol. Sci. 1992, 13, 61–69. [Google Scholar] [CrossRef]
- Wang, M.; Pei, L.; Fletcher, P.J.; Kapur, S.; Seeman, P.; Liu, F. Schizophrenia, amphetamine-induced sensitized state and acute amphetamine exposure all show a common alteration: Increased dopamine D2 receptor dimerization. Mol. Brain 2010, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jönsson, E.G.; Sillén, A.; Vares, M.; Ekholm, B.; Terenius, L.; Sedvall, G.C. Dopamine D2 receptor gene Ser311Cys variant and schizophrenia: Association study and meta-analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2003, 119B, 28–34. [Google Scholar] [CrossRef]
- Tataranni, P.A.; Baier, L.; Jenkinson, C.; Harper, I.; Del Parigi, A.; Bogardus, C. A Ser311Cys Mutation in the Human Dopamine Receptor D2 Gene Is Associated With Reduced Energy Expenditure. Diabetes 2001, 50, 901. [Google Scholar] [CrossRef] [Green Version]
- Cravchik, A.; Sibley, D.R.; Gejman, P.V. Functional Analysis of the Human D2 Dopamine Receptor Missense Variants. J. Biol. Chem. 1996, 271, 26013–26017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and Functional Basis of Dopamine Receptors and Their Role in Neurogenesis: Possible Implication for Parkinson’s disease. J. Exp. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Faron-Górecka, A.; Kuśmider, M.; Solich, J.; Górecki, A.; Dziedzicka-Wasylewska, M. Chapter Nine - Genetic variants in dopamine receptors influence on heterodimerization in the context of antipsychotic drug action. In Progress in Molecular Biology and Translational Science; Giraldo, J., Ciruela, F., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 169, pp. 279–296. [Google Scholar]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Masri, B.; Salahpour, A.; Didriksen, M.; Ghisi, V.; Beaulieu, J.-M.; Gainetdinov, R.R.; Caron, M.G. Antagonism of dopamine D2 receptor/β-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. USA 2008, 105, 13656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayport, S.; Sulzer, D. Visualization of Antipsychotic Drug Binding to Living Mesolimbic Neurons Reveals D2 Receptor, Acidotropic, and Lipophilic Components. J. Neurochem. 1995, 65, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Monsma, F.J.; McVittie, L.D.; Gerfen, C.R.; Mahan, L.C.; Sibley, D.R. Multiple D2 dopamine receptors produced by alternative RNA splicing. Nature 1989, 342, 926–929. [Google Scholar] [CrossRef]
- Usiello, A.; Baik, J.-H.; Rougé-Pont, F.; Picetti, R.; Dierich, A.; LeMeur, M.; Piazza, P.V.; Borrelli, E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature 2000, 408, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Kasai, R.S.; Ito, S.V.; Awane, R.M.; Fujiwara, T.K.; Kusumi, A. The Class-A GPCR Dopamine D2 Receptor Forms Transient Dimers Stabilized by Agonists: Detection by Single-Molecule Tracking. Cell Biochem. Biophys. 2018, 76, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Sung, Y.-M.; Wilkins, A.D.; Rodriguez, G.J.; Wensel, T.G.; Lichtarge, O. Intramolecular allosteric communication in dopamine D2 receptor revealed by evolutionary amino acid covariation. Proc. Natl. Acad. Sci. USA 2016, 113, 3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichter, K.M.; Flajolet, M.; Greengard, P.; Vu, T.Q. Kinetics of G-protein–coupled receptor endosomal trafficking pathways revealed by single quantum dots. Proc. Natl. Acad. Sci. USA 2010, 107, 18658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabor, A.; Weisenburger, S.; Banerjee, A.; Purkayastha, N.; Kaindl, J.M.; Hübner, H.; Wei, L.; Grömer, T.W.; Kornhuber, J.; Tschammer, N.; et al. Visualization and ligand-induced modulation of dopamine receptor dimerization at the single molecule level. Sci. Rep. 2016, 6, 33233. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, O.; Tomlinson, I.D.; Bailey, D.M.; Thal, L.B.; Ross, E.J.; Harris, L.; Frankland, M.P.; Ferguson, R.S.; Glaser, Z.; Greer, J.; et al. Single quantum dot tracking illuminates neuroscience at the nanoscale. Chem. Phys. Lett. 2018, 706, 741–752. [Google Scholar] [CrossRef]
- Rosenthal, S.J.; Chang, J.C.; Kovtun, O.; McBride, J.R.; Tomlinson, I.D. Biocompatible Quantum Dots for Biological Applications. Chem. Biol. 2011, 18, 10–24. [Google Scholar] [CrossRef] [Green Version]
- Cravchik, A.; Sibley, D.R.; Gejman, P.V. Analysis of neuroleptic binding affinities and potencies for the different human D2 dopamine receptor missense variants. Pharm. Genom. 1999, 9, 17–23. [Google Scholar] [CrossRef]
- Low-Nam, S.T.; Lidke, K.A.; Cutler, P.J.; Roovers, R.C.; van Bergen en Henegouwen, P.M.P.; Wilson, B.S.; Lidke, D.S. ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat. Struct. Mol. Biol. 2011, 18, 1244–1249. [Google Scholar] [CrossRef] [Green Version]
- Kusumi, A.; Tsunoyama, T.A.; Hirosawa, K.M.; Kasai, R.S.; Fujiwara, T.K. Tracking single molecules at work in living cells. Nat. Chem. Biol. 2014, 10, 524–532. [Google Scholar] [CrossRef]
- Levet, F.; Julien, G.; Galland, R.; Butler, C.; Beghin, A.; Chazeau, A.; Hoess, P.; Ries, J.; Giannone, G.; Sibarita, J.-B. A tessellation-based colocalization analysis approach for single-molecule localization microscopy. Nat. Commun. 2019, 10, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levet, F.; Hosy, E.; Kechkar, A.; Butler, C.; Beghin, A.; Choquet, D.; Sibarita, J.-B. SR-Tesseler: A method to segment and quantify localization-based super-resolution microscopy data. Nat. Methods 2015, 12, 1065–1071. [Google Scholar] [CrossRef]
- Gormal, R.S.; Padmanabhan, P.; Kasula, R.; Bademosi, A.T.; Coakley, S.; Giacomotto, J.; Blum, A.; Joensuu, M.; Wallis, T.P.; Lo, H.P.; et al. Modular transient nanoclustering of activated β2-adrenergic receptors revealed by single-molecule tracking of conformation-specific nanobodies. Proc. Natl. Acad. Sci. USA 2020, 117, 30476. [Google Scholar] [CrossRef] [PubMed]
- Thal, L.B.; Kovtun, O.; Rosenthal, S.J. Labeling Neuronal Proteins with Quantum Dots for Single-Molecule Imaging. In Quantum Dots: Applications in Biology; Fontes, A., Santos, B.S., Eds.; Springer: New York, NY, USA, 2020; pp. 169–177. [Google Scholar]
- Kovtun, O.; Thal, L.B.; Josephs, T.; Rosenthal, S.J. Quantitative Analysis of Single Quantum Dot Trajectories. In Quantum Dots: Applications in Biology; Fontes, A., Santos, B.S., Eds.; Springer: New York, NY, USA, 2020; pp. 109–123. [Google Scholar]
- Crocker, J.C.; Grier, D.G. Methods of Digital Video Microscopy for Colloidal Studies. J. Colloid Interface Sci. 1996, 179, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Jaqaman, K.; Loerke, D.; Mettlen, M.; Kuwata, H.; Grinstein, S.; Schmid, S.L.; Danuser, G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 2008, 5, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Tinevez, J.-Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Michalet, X.; Berglund, A.J. Optimal diffusion coefficient estimation in single-particle tracking. Phys. Rev. E 2012, 85, 061916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berglund, A.J. Statistics of camera-based single-particle tracking. Phys. Rev. E 2010, 82, 011917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, M.J. A Biological Interpretation of Transient Anomalous Subdiffusion. I. Qualitative Model. Biophys. J. 2007, 92, 1178–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahan, M.; Lévi, S.; Luccardini, C.; Rostaing, P.; Riveau, B.; Triller, A. Diffusion Dynamics of Glycine Receptors Revealed by Single-Quantum Dot Tracking. Science 2003, 302, 442. [Google Scholar] [CrossRef]
- Kovtun, O.; Tomlinson, I.D.; Ferguson, R.S.; Rosenthal, S.J. Quantum dots reveal heterogeneous membrane diffusivity and dynamic surface density polarization of dopamine transporter. PLoS ONE 2019, 14, e0225339. [Google Scholar] [CrossRef]
- Ovesný, M.; Křížek, P.; Borkovec, J.; Švindrych, Z.; Hagen, G.M. ThunderSTORM: A comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 2014, 30, 2389–2390. [Google Scholar] [CrossRef]
- Haggie, P.M.; Kim, J.K.; Lukacs, G.L.; Verkman, A.S. Tracking of Quantum Dot-labeled CFTR Shows Near Immobilization by C-Terminal PDZ Interactions. Mol. Biol. Cell 2006, 17, 4937–4945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinkamp Mara, P.; Low-Nam Shalini, T.; Yang, S.; Lidke Keith, A.; Lidke Diane, S.; Wilson Bridget, S. erbB3 Is an Active Tyrosine Kinase Capable of Homo- and Heterointeractions. Mol. Cell. Biol. 2014, 34, 965–977. [Google Scholar] [CrossRef] [Green Version]
- Tabor, A.; Möller, D.; Hübner, H.; Kornhuber, J.; Gmeiner, P. Visualization of ligand-induced dopamine D2S and D2L receptor internalization by TIRF microscopy. Sci. Rep. 2017, 7, 10894. [Google Scholar] [CrossRef] [PubMed]
- Chenouard, N.; Smal, I.; de Chaumont, F.; Maška, M.; Sbalzarini, I.F.; Gong, Y.; Cardinale, J.; Carthel, C.; Coraluppi, S.; Winter, M.; et al. Objective comparison of particle tracking methods. Nat. Methods 2014, 11, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovtun, O.; Rosenthal, S.J. Ensemble and Single Quantum Dot Fluorescence Methods in Neurotransmitter Transporter Research. In Neurotransmitter Transporters: Investigative Methods; Bönisch, H., Sitte, H.H., Eds.; Springer: New York, NY, USA, 2016; pp. 129–141. [Google Scholar]
- Thal, L.B.; Mann, V.R.; Sprinzen, D.; McBride, J.R.; Reid, K.R.; Tomlinson, I.D.; McMahon, D.G.; Cohen, B.E.; Rosenthal, S.J. Ligand-conjugated quantum dots for fast sub-diffraction protein tracking in acute brain slices. Biomater. Sci. 2020, 8, 837–845. [Google Scholar] [CrossRef]
- Vega, A.R.; Freeman, S.A.; Grinstein, S.; Jaqaman, K. Multistep Track Segmentation and Motion Classification for Transient Mobility Analysis. Biophys. J. 2018, 114, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.H.; Marques, T.R.; Jauhar, S.; Nour, M.M.; Goodwin, G.M.; Young, A.H.; Howes, O.D. The dopamine hypothesis of bipolar affective disorder: The state of the art and implications for treatment. Mol. Psychiatry 2017, 22, 666–679. [Google Scholar] [CrossRef]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524. [Google Scholar] [CrossRef]
- Shioda, N. Dopamine D2L receptor-interacting proteins regulate dopaminergic signaling. J. Pharmacol. Sci. 2017, 135, 51–54. [Google Scholar] [CrossRef]
- Kos, M.Z.; Duan, J.; Sanders, A.R.; Blondell, L.; Drigalenko, E.I.; Carless, M.A.; Gejman, P.V.; Göring, H.H.H.; Gejman, P.V.; Sanders, A.R.; et al. Dopamine perturbation of gene co-expression networks reveals differential response in schizophrenia for translational machinery. Transl. Psychiatry 2018, 8, 278. [Google Scholar] [CrossRef]
- Lee, F.J.S.; Pei, L.; Moszczynska, A.; Vukusic, B.; Fletcher, P.J.; Liu, F. Dopamine transporter cell surface localization facilitated by a direct interaction with the dopamine D2 receptor. EMBO J. 2007, 26, 2127–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orfield, N.J.; McBride, J.R.; Keene, J.D.; Davis, L.M.; Rosenthal, S.J. Correlation of Atomic Structure and Photoluminescence of the Same Quantum Dot: Pinpointing Surface and Internal Defects That Inhibit Photoluminescence. ACS Nano 2015, 9, 831–839. [Google Scholar] [CrossRef]
- Yanagawa, M.; Sako, Y. Workflows of the Single-Molecule Imaging Analysis in Living Cells: Tutorial Guidance to the Measurement of the Drug Effects on a GPCR. In Live Cell Imaging: Methods and Protocols; Kim, S.-B., Ed.; Springer: New York, NY, USA, 2021; pp. 391–441. [Google Scholar]
- Lycas, M.D.; Ejdrup, A.L.; Sørensen, A.T.; Haahr, N.O.; Jørgensen, S.H.; Guthrie, D.A.; Støier, J.F.; Werner, C.; Newman, A.H.; Sauer, M.; et al. Nanoscopic dopamine transporter distribution and conformation are inversely regulated by excitatory drive and D2-autoreceptor activity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar] [CrossRef] [Green Version]
- Hammond, G.R.V.; Sim, Y.; Lagnado, L.; Irvine, R.F. Reversible binding and rapid diffusion of proteins in complex with inositol lipids serves to coordinate free movement with spatial information. J. Cell Biol. 2009, 184, 297–308. [Google Scholar] [CrossRef] [Green Version]
- McKinney, S.A.; Murphy, C.S.; Hazelwood, K.L.; Davidson, M.W.; Looger, L.L. A bright and photostable photoconvertible fluorescent protein. Nat. Methods 2009, 6, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Nienhaus, K.; Nienhaus, G.U.; Wiedenmann, J.; Nar, H. Structural basis for photo-induced protein cleavage and green-to-red conversion of fluorescent protein EosFP. Proc. Natl. Acad. Sci. USA 2005, 102, 9156. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Tehrani, K.F.; Kner, P. Multicolor 3D Super-resolution Imaging by Quantum Dot Stochastic Optical Reconstruction Microscopy. ACS Nano 2015, 9, 2917–2925. [Google Scholar] [CrossRef]
- Appelhans, T.; Richter, C.P.; Wilkens, V.; Hess, S.T.; Piehler, J.; Busch, K.B. Nanoscale Organization of Mitochondrial Microcompartments Revealed by Combining Tracking and Localization Microscopy. Nano Lett. 2012, 12, 610–616. [Google Scholar] [CrossRef]
- Platzer, R.; Rossboth, B.K.; Schneider, M.C.; Sevcsik, E.; Baumgart, F.; Stockinger, H.; Schütz, G.J.; Huppa, J.B.; Brameshuber, M. Unscrambling fluorophore blinking for comprehensive cluster detection via photoactivated localization microscopy. Nat. Commun. 2020, 11, 4993. [Google Scholar] [CrossRef] [PubMed]
- van den Bogaart, G.; Meyenberg, K.; Risselada, H.J.; Amin, H.; Willig, K.I.; Hubrich, B.E.; Dier, M.; Hell, S.W.; Grubmüller, H.; Diederichsen, U.; et al. Membrane protein sequestering by ionic protein–lipid interactions. Nature 2011, 479, 552–555. [Google Scholar] [CrossRef]
- Wang, J.; Richards, D.A. Segregation of PIP2 and PIP3 into distinct nanoscale regions within the plasma membrane. Biol. Open 2012, 1, 857–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C.; Zhang, Y.; Xu, P.; Xu, T.; Lou, X. Nanoscale Landscape of Phosphoinositides Revealed by Specific Pleckstrin Homology (PH) Domains Using Single-molecule Superresolution Imaging in the Plasma Membrane. J. Biol. Chem. 2015, 290, 26978–26993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miklosi, A.G.; Del Favero, G.; Bulat, T.; Höger, H.; Shigemoto, R.; Marko, D.; Lubec, G. Super-resolution Microscopical Localization of Dopamine Receptors 1 and 2 in Rat Hippocampal Synaptosomes. Mol. Neurobiol. 2018, 55, 4857–4869. [Google Scholar] [CrossRef]
- Rosenthal, S.J. Nanotechnology in Neuroscience Reveals Membrane Mobility Matters. ACS Chem. Neurosci. 2019, 10, 30–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovtun, O.; Torres, R.; Bellocchio, L.G.; Rosenthal, S.J. Membrane Nanoscopic Organization of D2L Dopamine Receptor Probed by Quantum Dot Tracking. Membranes 2021, 11, 578. https://doi.org/10.3390/membranes11080578
Kovtun O, Torres R, Bellocchio LG, Rosenthal SJ. Membrane Nanoscopic Organization of D2L Dopamine Receptor Probed by Quantum Dot Tracking. Membranes. 2021; 11(8):578. https://doi.org/10.3390/membranes11080578
Chicago/Turabian StyleKovtun, Oleg, Ruben Torres, Laurel G. Bellocchio, and Sandra Jean Rosenthal. 2021. "Membrane Nanoscopic Organization of D2L Dopamine Receptor Probed by Quantum Dot Tracking" Membranes 11, no. 8: 578. https://doi.org/10.3390/membranes11080578
APA StyleKovtun, O., Torres, R., Bellocchio, L. G., & Rosenthal, S. J. (2021). Membrane Nanoscopic Organization of D2L Dopamine Receptor Probed by Quantum Dot Tracking. Membranes, 11(8), 578. https://doi.org/10.3390/membranes11080578