
L-Type Calcium Channel: Predicting Pathogenic/Likely Pathogenic Status for Variants of Uncertain Clinical Significance


Abstract
:1. Introduction
2. Methods
2.1. Data Collection and Preprocessing
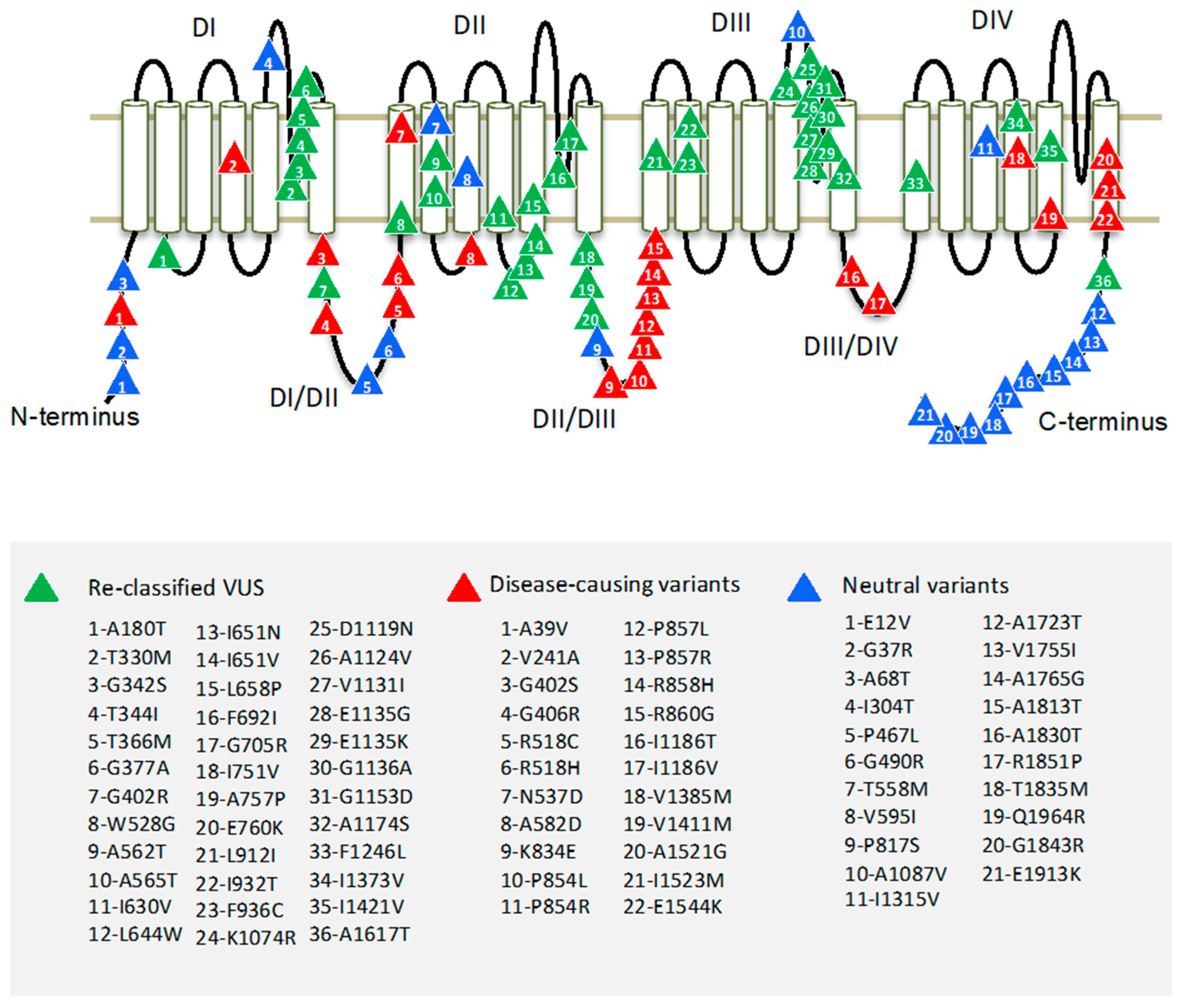
2.2. hCav1.2 Topology
2.3. Multiple Sequence Alignment and Paralogue Annotation
2.4. Annotation of Missense Variants
3. Results
3.1. Composing a Broad Dataset of Missense Variants for Channel hCav1.2 and Its Paralogues
3.2. Distribution of Missense Variants in Topological Regions of hCav1.2
3.3. Amino Acid Substitutions in P/LP and Benign Variants
3.4. Paralogue Annotation of Variants Identified in hCav1.2
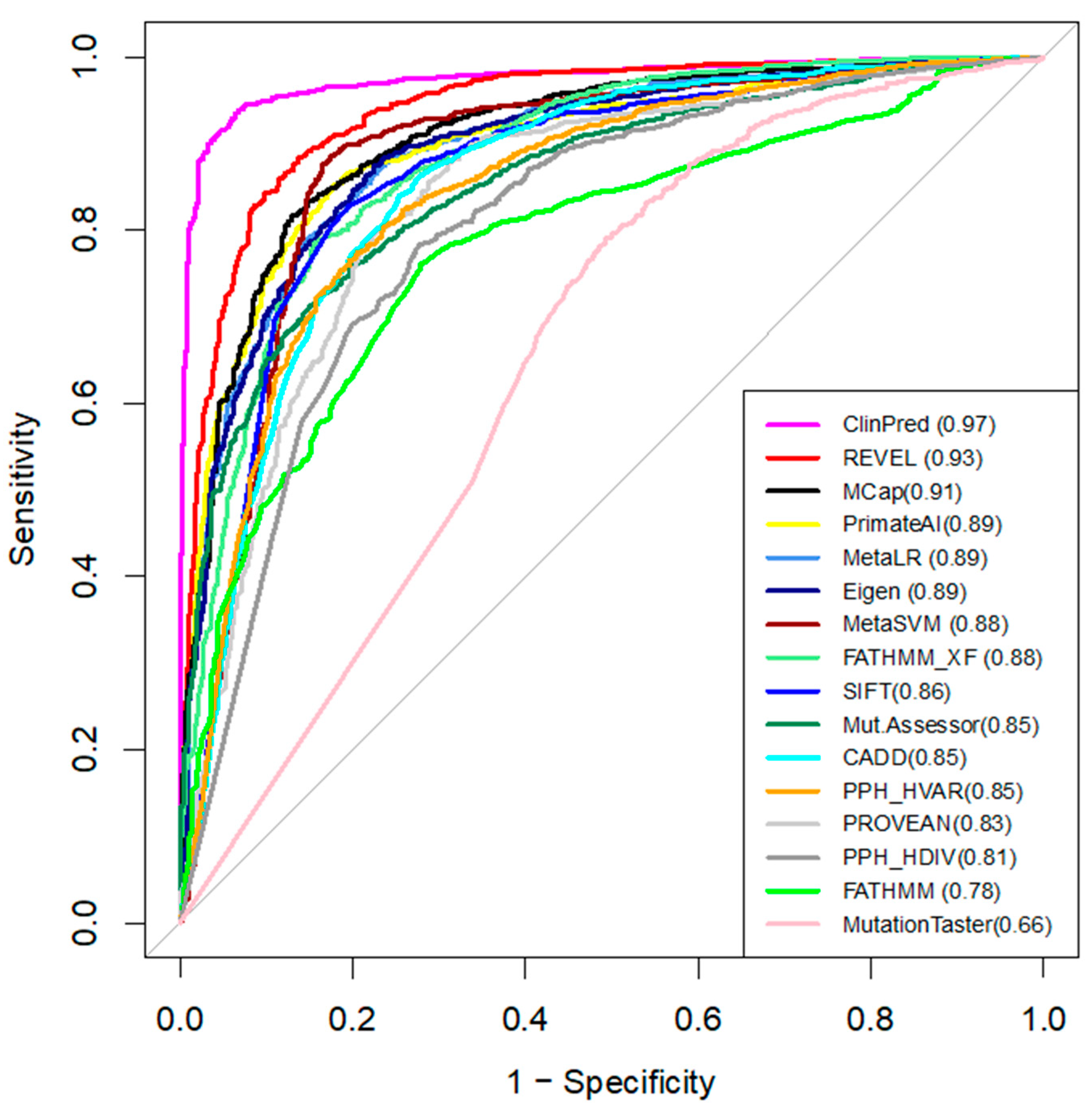
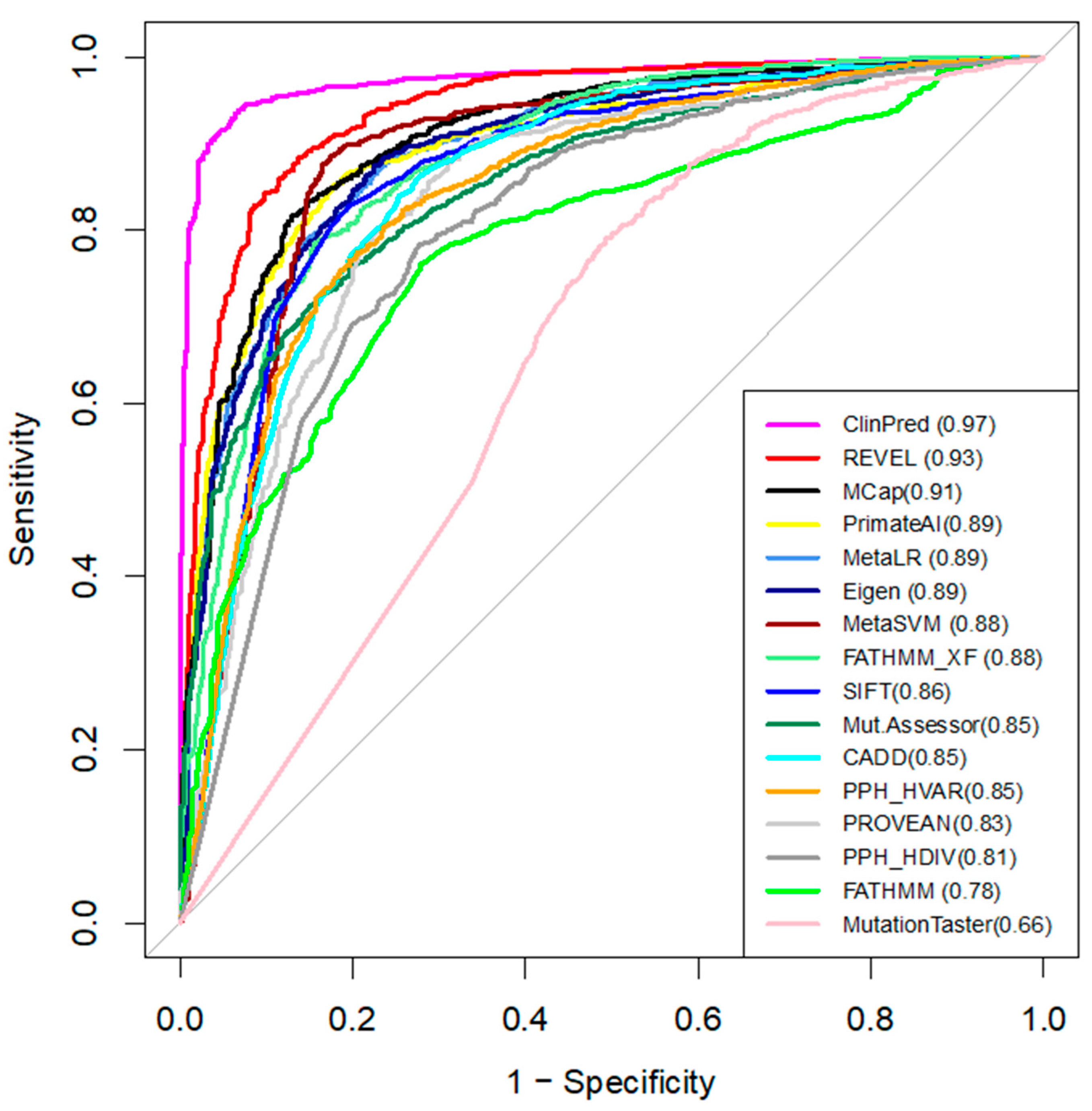
3.5. Comparing Performance of the Computational Tools
3.6. Reclassifying VUS Variants of hCav1.2 with ClinPred and Paralogue Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Benitah, J.-P.; Alvarez, J.L.; Gómez, A.M. L Type Ca2+ current in ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2010, 48, 26–36. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.; Lassmann, T. A phenotype centric benchmark of variant prioritisation tools. NPJ Genom. Med. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Tarnovskaya, S.I.; Korkosh, V.S.; Zhorov, B.S.; Frishman, D. Predicting novel disease mutations in the cardiac sodium channel. Biochem. Biophys. Res. Commun. 2020, 521, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yu, G. New insights into the pathogenicity of non-synonymous variants through multi-level analysis. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niroula, A.; Vihinen, M. How good are pathogenicity predictors in detecting benign variants? PLoS Comput. Biol. 2019, 15, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Miosge, L.A.; Field, M.A.; Sontani, Y.; Cho, V.; Johnson, S.; Palkova, A.; Balakishnan, B.; Liang, R.; Zhang, Y.; Lyon, S.; et al. Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E5189–E5198. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jhong, J.-H.; Lee, J.; Koo, J.-Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017, 10, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaucha, J.; Heinzinger, M.; Tarnovskaya, S.; Rost, B.; Frishman, D. Family-specific analysis of variant pathogenicity prediction tools. NAR Genom. Bioinform. 2020, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ware, J.S.; Walsh, R.; Cunningham, F.; Birney, E.; Cook, S.A. Paralogous annotation of disease-causing variants in long qt syndrome genes. Hum. Mutat. 2012, 33, 1188–1191. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [Google Scholar] [CrossRef]
- Chen, Y.; Cunningham, F.; Rios, D.; McLaren, W.M.; Smith, J.; Pritchard, B.; Spudich, G.M.; Brent, S.; Kulesha, E.; Marin-Garcia, P.; et al. Ensembl variation resources. BMC Genom. 2010, 11, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- Kaltman, J.R.; Evans, F.; Fu, Y.-P. Re-evaluating pathogenicity of variants associated with the long QT syndrome. J. Cardiovasc. Electrophysiol. 2018, 29, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Peters, N.S.; Cook, S.A.; Ware, J.S. Paralogue annotation identifies novel pathogenic variants in patients with brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J. Med. Genet. 2014, 51, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Zvelebil, M.J.; Barton, G.J.; Taylor, W.R.; Sternberg, M.J.E. Prediction of protein secondary structure and active sites using the alignment of homologous sequences. J. Mol. Biol. 1987, 195, 957–961. [Google Scholar] [CrossRef]
- Manning, J.R.; Jefferson, E.R.; Barton, G.J. The Contrasting Properties of Conservation and Correlated Phylogeny in Protein Functional Residue Prediction. BMC Bioinforma. 2008, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, C.; Mou, C.; Dong, Y.; Tu, Y. DbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020, 12, 1–8. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. PROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Niroula, A.; Vihinen, M. Variation interpretation predictors: Principles, types, performance, and choice. Hum. Mutat. 2016, 37, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Alirezaie, N.; Kernohan, K.D.; Hartley, T.; Majewski, J.; Hocking, T.D. ClinPred: Prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. Am. J. Hum. Genet. 2018, 103, 474–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Gene | Channel | Benign | P/LP | VUS |
|---|---|---|---|---|
| CACNA1A | hCav2.1 | 5 | 60 | 346 |
| CACNA1B | hCav2.2 | 48 | 0 | 10 |
| CACNA1C | hCav1.2 | 21 | 22 | 309 |
| CACNA1D | hCav1.3 | 4 | 7 | 46 |
| CACNA1E | hCav2.3 | 36 | 17 | 18 |
| CACNA1F | hCav1.4 | 39 | 28 | 39 |
| CACNA1G | hCav3.1 | 42 | 5 | 45 |
| CACNA1H | hCav3.2 | 116 | 21 | 454 |
| CACNA1I | hCav3.3 | 61 | 0 | 1 |
| CACNA1S | hCav1.1 | 57 | 11 | 290 |
| NALCN | hNavi2.1 | 18 | 31 | 17 |
| SCN1A | hNav1.1 | 18 | 605 | 482 |
| SCN2A | hNav1.2 | 15 | 166 | 301 |
| SCN3A | hNav1.3 | 16 | 8 | 202 |
| SCN4A | hNav1.4 | 47 | 78 | 312 |
| SCN5A | hNav1.5 | 43 | 350 | 705 |
| SCN7A | hNav2.1 | 65 | 0 | 0 |
| SCN8A | hNav1.6 | 6 | 93 | 249 |
| SCN9A | hNav1.7 | 11 | 32 | 505 |
| SCN10A | hNav1.8 | 66 | 3 | 291 |
| SCN11A | hNav1.9 | 29 | 12 | 230 |
| Tool | Custom Threshold | Sensitivity | Specificity | Accuracy |
|---|---|---|---|---|
| ClinPred | >0.66 | 0.95 | 0.94 | 0.95 |
| REVEL | >0.73 | 0.89 | 0.87 | 0.88 |
| MetaLR | >0.91 | 0.86 | 0.84 | 0.85 |
| Eigen | >0.55 | 0.81 | 0.87 | 0.84 |
| MCap | >0.59 | 0.82 | 0.87 | 0.84 |
| PrimateAI | >0.69 | 0.88 | 0.79 | 0.84 |
| MetaSVM | >0.92 | 0.88 | 0.85 | 0.87 |
| FATHMM_XF | >0.85 | 0.82 | 0.84 | 0.83 |
| SIFT | <0.0025 | 0.83 | 0.83 | 0.83 |
| PROVEAN | <−3.42 | 0.80 | 0.80 | 0.80 |
| CADD | >24 | 0.88 | 0.72 | 0.80 |
| MutationAssessor | >2.51 | 0.77 | 0.81 | 0.79 |
| Polyphen HVAR | >0.6 | 0.80 | 0.70 | 0.75 |
| FATHMM | <−4.305 | 0.76 | 0.73 | 0.74 |
| Polyphen HDIV | >0.96 | 0.74 | 0.71 | 0.73 |
| MutationTaster | >0.99 | 0.94 | 0.31 | 0.62 |
| Type of Data | WT Residue | Mutant Residue | Preferred Substitutions |
|---|---|---|---|
| All variants (n = 2312) | R, A, V, L, G | V, T, S, R, I | R > Q, L > P, R > H, A > T, R > C |
| P/LP variants (n = 1549) | R, L, V, G, A | M, R, C, D, H | L > P, G > R, R > Q, R > H, R > C |
| Benign variants (n = 763) | R, A, P, V, G | V, S, T, A, R | A > T, R > Q, P > L, R > H, V > I |
| # | Variant | Location | Paralogue |
|---|---|---|---|
| 1 | A180T | DI-S2/S3 | SCN5A-A178G, SCN1A-A175V, SCN1A-A175T |
| 2 | T330M | DI-S5/S6 | SCN1A-Y349C |
| 3 | G342S | DI-S5/S6 | SCN5A-G351V, SCN5A-G351D |
| 4 | T344I | DI-S5/S6 | SCN1A-T363R, SCN1A-T363P, SCN5A-T353I |
| 5 | T366M | DI-S5/S6 | SCN1A-E385Q |
| 6 | G377A | DI-S5/S6 | SCN5A-G386R, SCN5A-G386E |
| 7 | G402R | DI-DII | CACNA1E-G348R, SCN1A-A420V, CACNA1D-G403D, CACNA1F-G369D |
| 8 | W528G | DII-S1 | SCN1A-L772P |
| 9 | A562T | DII-S2 | CACNA1H-V831M |
| 10 | A565T | DII-S2 | SCN5A-G758E |
| 11 | I630V | DII-S4 | SCN1A-L869S, SCN1A-L869F |
| 12 | L644W | DII-S4/S5 | SCN9A-I859T, SCN3A-I875T, SCN4A-I693T, SCN8A-I868T |
| 13 | I651N | DII-S4/S5 | SCN5A-L839P, CACNA1E-I603L, SCN8A-L875Q, CACNA1A-I614M, SCN1A-L890P |
| 14 | I651V | DII-S4/S5 | SCN5A-L839P, CACNA1E-I603L, SCN8A-L875Q, CACNA1A-I614M, SCN1A-L890P |
| 15 | L658P | DII-S5 | NALCN-T513N, SCN1A-L897S, SCN1A-L897F |
| 16 | F692I | DII-S5/S6 | SCN2A-F928C |
| 17 | G705R | DII-S5/S6 | SCN1A-G950E, SCN1A-G950R |
| 18 | I751V | DII-DIII | CACNA1D-I750F, CACNA1A-I712V, CACNA1E-I701V, CACNA1F-I756T, CACNA1D-I750M |
| 19 | A757P | DII-DIII | SCN2A-S987I |
| 20 | E760K | DII-DIII | SCN1A-D998G |
| 21 | L912I | DIII-S1 | SCN1A-L1230F |
| 22 | I932T | DIII-S2 | SCN1A-M1251R, SCN5A-L1238P |
| 23 | F936C | DIII-S2 | SCN1A-A1255D, SCN1A-A1255P |
| 24 | K1074R | DIII-S5/S6 | SCN5A-K1359N |
| 25 | D1119N | DIII-S5/S6 | SCN1A-D1416G |
| 26 | A1124V | DIII-S5/S6 | SCN1A-G1421R, SCN1A-G1421E, SCN5A-G1408R |
| 27 | V1131I | DIII-S5/S6 | CACNA1A-V1456L, SCN1A-V1428A, SCN1A-V1428F |
| 28 | E1135G | DIII-S5/S6 | SCN1A-K1432I, SCN5A-K1419E, SCN2A-K1422E |
| 29 | E1135K | DIII-S5/S6 | SCN1A-K1432I, SCN5A-K1419E, SCN2A-K1422E |
| 30 | G1136A | DIII-S5/S6 | SCN1A-G1433V, SCN5A-G1420R, SCN5A-G1420V, SCN1A-G1433R, SCN1A-G1433E |
| 31 | G1153D | DIII-S5/S6 | SCN1A-Q1450K, SCN1A-Q1450R |
| 32 | A1174S | DIII-S6 | SCN5A-S1458Y, SCN1A-S1471F |
| 33 | F1246L | DIV-S1 | SCN4A-M1360V, SCN2A-M1538I |
| 34 | I1373V | DIV-S4 | SCN1A-P1632S |
| 35 | I1421V | DIV-S5 | SCN1A-I1683F, SCN4A-I1495F, SCN1A-I1683T |
| 36 | A1617T | C-term | SCN5A-V1861I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarnovskaya, S.I.; Kostareva, A.A.; Zhorov, B.S. L-Type Calcium Channel: Predicting Pathogenic/Likely Pathogenic Status for Variants of Uncertain Clinical Significance. Membranes 2021, 11, 599. https://doi.org/10.3390/membranes11080599
Tarnovskaya SI, Kostareva AA, Zhorov BS. L-Type Calcium Channel: Predicting Pathogenic/Likely Pathogenic Status for Variants of Uncertain Clinical Significance. Membranes. 2021; 11(8):599. https://doi.org/10.3390/membranes11080599
Chicago/Turabian StyleTarnovskaya, Svetlana I., Anna A. Kostareva, and Boris S. Zhorov. 2021. "L-Type Calcium Channel: Predicting Pathogenic/Likely Pathogenic Status for Variants of Uncertain Clinical Significance" Membranes 11, no. 8: 599. https://doi.org/10.3390/membranes11080599
APA StyleTarnovskaya, S. I., Kostareva, A. A., & Zhorov, B. S. (2021). L-Type Calcium Channel: Predicting Pathogenic/Likely Pathogenic Status for Variants of Uncertain Clinical Significance. Membranes, 11(8), 599. https://doi.org/10.3390/membranes11080599