
Arterial Blood Pressure, Neuronal Excitability, Mineral Metabolism and Cell Volume Regulation Mechanisms Revealed by Xenopus laevis oocytes


{kind=link}
{kind=link}
Abstract
:1. Introduction
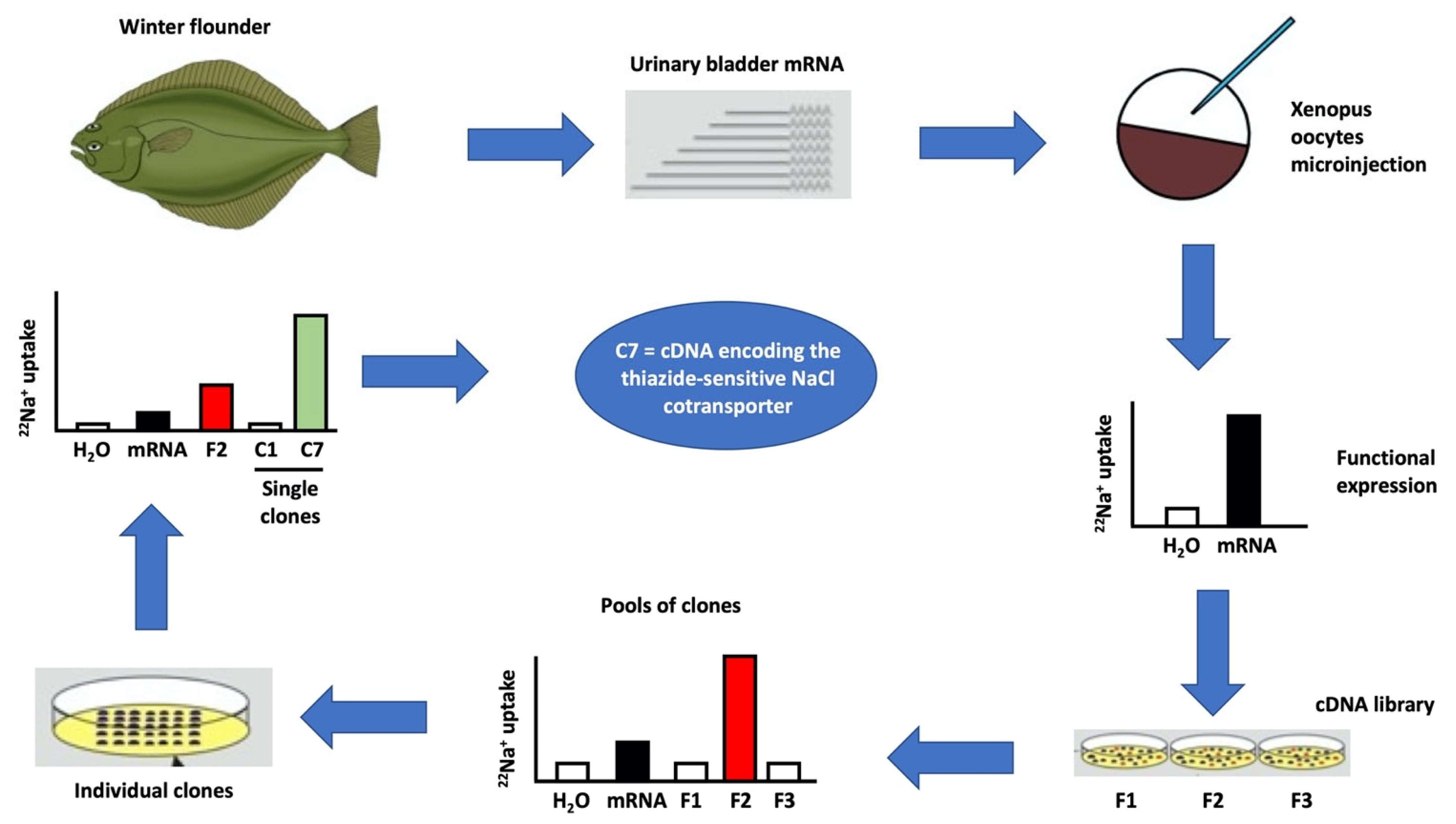
2. Xenopus laevis Oocytes Were Crucial for the Molecular Identification and Characterization of the CCCs
3. The CCCs Are Involved in Neuronal Excitability, Cell Volume and Blood Pressure Regulation
3.1. Neuronal Excitability
3.2. Cell Volume Regulation
3.3. Arterial Blood Pressure
4. The Birth of the Calcium-Sensing Field in Mineral Metabolism Was Possible Thanks to Xenopus laevis Oocytes
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gamba, G. Molecular physiology and pathophysiology of the electroneutral cation-chloride cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef]
- Mount, D.B.; Delpire, E.; Gamba, G.; Hall, A.E.; Poch, E.; Hoover, R.S., Jr.; Hebert, S.C. The electroneutral cation-chloride cotrans-porters. J. Exp. Biol. 1998, 201, 2091–2102. [Google Scholar] [CrossRef]
- Russell, J.M. Sodium-Potassium-Chloride Cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef] [PubMed]
- Hoorn, E.J.; Ellison, D.H. Diuretic Resistance. Am. J. Kidney Dis. 2017, 69, 136–142. [Google Scholar] [CrossRef]
- Bazua-Valenti, S.; Castaneda-Bueno, M.; Gamba, G. Physiological role of SLC12 family members in the kidney. Am. J. Physiol. Ren. Physiol. 2016, 311, F131–F144. [Google Scholar] [CrossRef]
- Bazzini, C.; Vezzoli, V.; Sironi, C.; Dossena, S.; Ravasio, A.; Debiasi, S.; Garavaglia, M.; Rodighiero, S.; Meyer, G.; Fascio, U.; et al. Thiazide-sensitive NaCl cotransporter in the intestine: Possible role of HCTZ in the intestinal Ca2+ uptake. J. Biol. Chem. 2005, 280, 19902–19910. [Google Scholar] [CrossRef]
- Dvorak, M.M.; De Joussineau, C.; Carter, D.H.; Pisitkun, T.; Knepper, M.A.; Gamba, G.; Kemp, P.J.; Riccardi, D. Thiazide Diuretics Directly Induce Osteoblast Differentiation and Mineralized Nodule Formation by Interacting with a Sodium Chloride Co-Transporter in Bone. J. Am. Soc. Nephrol. 2007, 18, 2509–2516. [Google Scholar] [CrossRef]
- Kelly, L.; Almutairi, M.M.; Kursan, S.; Pacheco, R.; Dias-Junior, E.; Castrop, H.; Di Fulvio, M. Impaired glucose tolerance, glucagon, and insulin responses in mice lacking the loop diuretic-sensitive Nkcc2a transporter. Am. J. Physiol. Cell Physiol. 2019, 317, C843–C856. [Google Scholar] [CrossRef]
- Mao, S.; Garzon-Muvdi, T.; Di Fulvio, M.; Chen, Y.; Delpire, E.; Alvarez, F.J.; Alvarez-Leefmans, F.J. Molecular and functional expression of cation-chloride cotransporters in dorsal root ganglion neurons during postnatal maturation. J. Neurophysiol. 2012, 108, 834–852. [Google Scholar] [CrossRef]
- Moreno, E.; Plata, C.; Rodriguez-Gama, A.; Argaiz, E.R.; Vazquez, N.; Leyva-Rios, K.; Islas, L.; Cutler, C.; Pacheco-Alvarez, D.; Mercado, A.; et al. The European Eel NCCbeta Gene Encodes a Thiazide-resistant Na-Cl Cotransporter. J. Biol. Chem. 2016, 291, 22472–22481. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol. Asp. Med. 2013, 34, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Lauf, P.K.; McManus, T.J.; Haas, M.; Forbush, I.B.; Duhm, J.; Flatman, P.W.; Saier, M.H.; Russell, J.M. Physiology and biophysics of chloride and cation cotransport across cell membranes. Fed. Proc. 1987, 46, 2377–2394. [Google Scholar] [PubMed]
- Gamba, G.; Saltzberg, S.N.; Lombardi, M.; Miyanoshita, A.; Lytton, J.; Hediger, M.A.; Brenner, B.M.; Hebert, S.C. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc. Natl. Acad. Sci. USA 1993, 90, 2749–2753. [Google Scholar] [CrossRef]
- Renfro, J.L. Interdependence of active Na+ and Cl− transport by the isolated urinary bladder of the teleost, Pseudopleuronectes americanus. J. Exp. Zoo 1977, 199, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.B.; Lee, I.; D’Amico, M. Sodium Chloride absorption by the urinary bladder of the winter flounder. A thiazide-sensitive, electrically neutral transport system. J. Clin. Investig. 1984, 74, 7–16. [Google Scholar] [CrossRef]
- Gamba, G.; Miyanoshita, A.; Lombardi, M.; Lytton, J.; Lee, W.S.; Hediger, M.A.; Hebert, S.C. Molecular cloning, primary structure and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J. Biol. Chem. 1994, 269, 17713–17722. [Google Scholar] [CrossRef]
- Delpire, E.; Rauchman, M.I.; Beier, D.R.; Hebert, S.C.; Gullans, S.R. Molecular cloning and chromosome localization of a putative basolateral Na+-K+-2Cl—Cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J. Biol. Chem. 1994, 269, 25677–25683. [Google Scholar] [CrossRef]
- Moreno, E.; de Los Heros, P.; Plata, C.; Cutler, C.; Vega-Mateos, A.; Vazquez, N.; Gamba, G. Structure-function relationships in the renal NaCl cotransporter (NCC). Curr. Top Membr. 2019, 83, 177–204. [Google Scholar]
- Macnamara, E.F.; Koehler, A.E.; D’Souza, P.; Estwick, T.; Lee, P.; Vezina, G.; Undiagnosed Diseases, N.; Fauni, H.; Braddock, S.R.; Torti, E.; et al. Kilquist syndrome: A novel syndromic hearing loss disorder caused by homozygous deletion of SLC12A2. Hum. Mutat. 2019, 40, 532–538. [Google Scholar] [CrossRef]
- Delpire, E.; Mount, D.B. Human and murine phenotypes associated with defects in cation-chloride cotransport. Annu. Rev. Physiol. 2002, 64, 803–843. [Google Scholar] [CrossRef]
- Gillen, C.M.; Brill, S.; Payne, J.A.; Forbush, I.B. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat and human. A new member of the cation-chloride cotransporter family. J. Biol. Chem. 1996, 271, 16237–16244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.A.; Stevenson, T.J.; Donaldson, L.F. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neu-ronal-specific isoform. J. Biol. Chem. 1996, 271, 16245–16252. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Mercado, A.; Vazquez, N.; Xie, Q.; Desai, R.; George, A.L.; Gamba, G.; Mount, D.B. Molecular, functional, and genomic characterization of human KCC2, the neuronal K-Cl cotransporter. Brain Res. Mol. Brain Res. 2002, 103, 91–105. [Google Scholar] [CrossRef]
- Mount, D.B.; Mercado, A.; Song, L.; Xu, J.; George, A.L., Jr.; Delpire, E.; Gamba, G. Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J. Biol. Chem. 1999, 274, 16355–16362. [Google Scholar] [CrossRef]
- Race, J.E.; Makhlouf, F.N.; Logue, P.J.; Wilson, F.H.; Dunham, P.B.; Holtzman, E.J. Molecular cloning and functional charac-terization of KCC3, a new K-Cl cotransporter. Am. J. Physiol. 1999, 277, C1210–C1219. [Google Scholar] [CrossRef]
- Lytle, C.; Forbush, I.B. The Na-K-Cl cotransport protein of shark rectal gland. II Regulation by direct phosphorylation. J. Biol. Chem. 1992, 267, 25438–25443. [Google Scholar] [CrossRef]
- Pacheco-Alvarez, D.; San Cristobal, P.; Meade, P.; Moreno, E.; Vazquez, N.; Munoz, E.; Diaz, A.; Juarez, M.E.; Gimenez, I.; Gamba, G. The Na-Cl cotransporter is activated and phosphorylated at the amino terminal domain upon intracellular chloride depletion. J. Biol. Chem. 2006, 281, 28755–28763. [Google Scholar] [CrossRef]
- Ponce-Coria, J.; San Cristobal, P.; Kahle, K.T.; Vazquez, N.; Pacheco-Alvarez, D.; De Los Heros, P.; Juarez, P.; Munoz, E.; Michel, G.; Bobadilla, N.A.; et al. Regulation of NKCC2 by a chloride-sensing mechanism in-volving the WNK3 and SPAK kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 8458–8463. [Google Scholar] [CrossRef]
- Alessi, D.R.; Zhang, J.; Khanna, A.; Hochdorfer, T.; Shang, Y.; Kahle, K.T. The WNK-SPAK/OSR1 pathway: Master regulator of cation-chloride cotransporters. Sci. Signal. 2014, 7, re3. [Google Scholar] [CrossRef]
- De Los Heros, P.; Kahle, K.T.; Rinehart, J.; Bobadilla, N.A.; Vazquez, N.; San Cristobal, P.; Mount, D.B.; Lifton, R.P.; Hebert, S.C.; Gamba, G. WNK3 bypasses the tonicity requirement for K-Cl cotransporter activation via a phosphatase-dependent pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 1976–1981. [Google Scholar] [CrossRef]
- Rinehart, J.; Maksimova, Y.D.; Tanis, J.E.; Stone, K.L.; Hodson, C.A.; Zhang, J.; Risinger, M.; Pan, W.; Wu, D.; Colangelo, C.M.; et al. Sites of regulated phosphorylation that control K-Cl co-transporter activity. Cell 2009, 138, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piechotta, K.; Lu, J.; Delpire, E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related pro-line-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J. Biol. Chem. 2002, 277, 50812–50819. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.B.; Delpire, E. Molecular Physiology of SPAK and OSR1: Two Ste20-Related Protein Kinases Regulating Ion Transport. Physiol. Rev. 2012, 92, 1577–1617. [Google Scholar] [CrossRef] [PubMed]
- Vitari, A.C.; Deak, M.; Morrice, N.A.; Alessi, D.R. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hyper-tension syndrome, phosphorylate and active SPAK and OSR1 protein kinases. Biochem. J. 2005, 391, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Verissimo, F.; Jordan, P. WNK kinases, a novel protein kinase subfamily in multi-cellular organisms. Oncogene 2001, 20, 5562–5569. [Google Scholar] [CrossRef] [PubMed]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef]
- Pacheco-Alvarez, D.; Carrillo-Perez, D.L.; Mercado, A.; Leyva-Rios, K.; Moreno, E.; Hernandez-Mercado, E.; Castaneda-Bueno, M.; Vazquez, N.; Gamba, G. WNK3 and WNK4 exhibit opposite sensitivity with respect to cell volume and intracellular chloride concentration. Am. J. Physiol. Cell Physiol. 2020, 319, C371–C380. [Google Scholar] [CrossRef]
- Bazua-Valenti, S.; Chavez-Canales, M.; Rojas-Vega, L.; Gonzalez-Rodriguez, X.; Vazquez, N.; Rodriguez-Gama, A.; Argaiz, E.R.; Melo, Z.; Plata, C.; Ellison, D.H.; et al. The Effect of WNK4 on the Na+-Cl− Cotransporter Is Modulated by Intracellular Chloride. J. Am. Soc. Nephrol. 2015, 26, 1781–1786. [Google Scholar] [CrossRef]
- Chavez-Canales, M.; Zhang, C.; Soukaseum, C.; Moreno, E.; Pacheco-Alvarez, D.; Vidal-Petiot, E.; Castaneda-Bueno, M.; Vazquez, N.; Rojas-Vega, L.; Meermeier, N.P.; et al. WNK-SPAK-NCC Cascade Revisited: WNK1 Stimulates the Activity of the Na-Cl Cotransporter via SPAK, an Effect Antagonized by WNK4. Hypertension 2014, 64, 1047–1053. [Google Scholar] [CrossRef]
- Rinehart, J.; Vazquez, N.; Kahle, K.T.; Hodson, C.A.; Ring, A.M.; Gulcicek, E.E.; Louvi, A.; Bobadilla, N.A.; Gamba, G.; Lifton, R.P. WNK2 is a novel regulator of essential neuronal cation-chloride cotransporters. J. Biol. Chem. 2011, 286, 30171–30180. [Google Scholar] [CrossRef]
- Pacheco-Alvarez, D.; Gamba, G. WNK3 is a Putative Chloride-Sensing Kinase. Cell. Physiol. Biochem. 2011, 28, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Pacheco-Alvarez, D.; Gagnon, K.B.; Vazquez, N.; Ponce-Coria, J.; Moreno, E.; Delpire, E.; Gamba, G. WNK4 Kinase is a Negative Regulator of K+-Cl-Cotransporters. Am. J. Physiol. Ren. Physiol. 2007, 292, F1197–F1207. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.; Kahle, K.T.; De Los Heros, P.; Vazquez, N.; Meade, P.; Wilson, F.H.; Hebert, S.C.; Gimenez, I.; Gamba, G.; Lifton, R.P. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl− cotransporters required for normal blood pressure ho-meostasis. Proc. Natl. Acad. Sci. USA 2005, 102, 16777–16782. [Google Scholar] [CrossRef]
- Kahle, K.T.; Rinehart, J.; De Los Heros, P.; Louvi, A.; Meade, P.; Vazquez, N.; Hebert, S.C.; Gamba, G.; Gimenez, I.; Lifton, R.P. WNK3 modulates transport of Cl− in and out of cells: Implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. USA 2005, 102, 16783–16788. [Google Scholar] [CrossRef] [PubMed]
- Piala, A.T.; Moon, T.M.; Akella, R.; He, H.; Cobb, M.H.; Goldsmith, E.J. Chloride Sensing by WNK1 Involves Inhibition of Au-tophosphorylation. Sci. Signal. 2014, 7, ra41. [Google Scholar] [CrossRef]
- Terker, A.S.; Zhang, C.; Erspamer, K.J.; Gamba, G.; Yang, C.L.; Ellison, D.H. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016, 89, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Murillo-de-Ozores, A.R.; Chavez-Canales, M.; de Los Heros, P.; Gamba, G.; Castaneda-Bueno, M. Physiological Processes Modu-lated by the Chloride-Sensitive WNK-SPAK/OSR1 Kinase Signaling Pathway and the Cation-Coupled Chloride Cotransporters. Front. Physiol. 2020, 11, 585907. [Google Scholar] [CrossRef]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K + /Cl—Co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef]
- Woo, N.S.; Lu, J.; England, R.; McClellan, R.; Dufour, S.; Mount, D.B.; Deutch, A.Y.; Lovinger, D.M.; Delpire, E. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus 2002, 12, 258–268. [Google Scholar] [CrossRef]
- Stodberg, T.; McTague, A.; Ruiz, A.J.; Hirata, H.; Zhen, J.; Long, P.; Farabella, I.; Meyer, E.; Kawahara, A.; Vassallo, G.; et al. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 2015, 6, 8038. [Google Scholar] [CrossRef]
- Fukuda, A.; Watanabe, M. Pathogenic potential of human SLC12A5 variants causing KCC2 dysfunction. Brain Res. 2019, 1710, 1–7. [Google Scholar] [CrossRef]
- Kim, Y.B.; Colwell, C.S.; Kim, Y.I. Long-term ionic plasticity of GABAergic signalling in the hypothalamus. J. Neuroend. 2019, 31, e12753. [Google Scholar] [CrossRef] [PubMed]
- Howard, H.C.; Mount, D.B.; Rochefort, D.; Byun, N.; Dupre, N.; Lu, J.; Fan, X.; Song, L.; Riviere, J.B.; Prevost, C.; et al. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat. Genet. 2002, 32, 384–392. [Google Scholar] [CrossRef]
- De Los Heros, P.; Pacheco-Alvarez, D.; Gamba, G. Role of WNK Kinases in the Modulation of Cell Volume. Curr. Top. Membr. 2018, 81, 207–235. [Google Scholar]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Cruz-Rangel, S.; Gamba, G.; Ramos-Mandujano, G.; Pasantes-Morales, H. Influence of WNK3 on intracellular chloride concentra-tion and volume regulation in HEK293 cells. Pflug. Arch. 2012, 464, 317–330. [Google Scholar] [CrossRef]
- Bazua-Valenti, S.; Rojas-Vega, L.; Castaneda-Bueno, M.; Barrera-Chimal, J.; Bautista, R.; Cervantes-Perez, L.G.; Vazquez, N.; Plata, C.; Murillo-de-Ozores, A.R.; Gonzalez-Mariscal, L.; et al. The Calcium-Sensing Receptor Increases Activity of the Renal NCC through the WNK4-SPAK Pathway. J. Am. Soc. Nephrol. 2018, 29, 1838–1848. [Google Scholar] [CrossRef]
- Kunchaparty, S.; Palcso, M.; Berkman, J.; Velazquez, H.; Desir, G.V.; Bernstein, P.; Reilly, R.F.; Ellison, D.H. Defective processing and expression of thiazide-sensitive Na-Cl cotransporter as a cause of Gitelman’s syndrome. Am. J. Physiol. 1999, 277, F643–F649. [Google Scholar] [CrossRef]
- Sabath, E.; Meade, P.; Berkman, J.; De Los Heros, P.; Moreno, E.; Bobadilla, N.A.; Vazquez, N.; Ellison, D.H.; Gamba, G. Patho-physiology of Functional Mutations of the Thiazide-sensitive Na-Cl Cotransporter in Gitelman Disease. Am. J. Physiol. Ren. Physiol. 2004, 287, F195–F203. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Rafiqi, F.H.; Karlsson, H.K.; Moleleki, N.; Vandewalle, A.; Campbell, D.G.; Morrice, N.A.; Alessi, D.R. Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J. Cell Sci. 2008, 121, 675–684. [Google Scholar] [CrossRef]
- Hadchouel, J.; Ellison, D.H.; Gamba, G. Regulation of Renal Electrolyte Transport by WNK and SPAK-OSR1 Kinases. Annu. Rev. Physiol. 2016, 78, 367–389. [Google Scholar] [CrossRef]
- Boettger, T.; Hubner, C.A.; Maier, H.; Rust, M.B.; Beck, F.X.; Jentsch, T.J. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature 2002, 416, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Noriega, L.G.; Melo, Z.; Rajaram, R.D.; Mercado, A.; Tovar, A.R.; Velazquez-Villegas, L.A.; Castaneda-Bueno, M.; Reyes-Lopez, Y.; Ryu, D.; Rojas-Vega, L.; et al. SIRT7 modulates the stability and activity of the renal K-Cl cotransporter KCC4 through deacetylation. EMBO Rep. 2021, 22, e50766. [Google Scholar] [CrossRef]
- Brown, E.M.; Gamba, G.; Riccardi, D.; Lombardi, M.; Butters, R.; Kifor, O.; Sun, A.; Hediger, M.A.; Lytton, J.; Hebert, S.C. Cloning and characterization of an extracellular Ca 2+ sensing receptor from bovine parathyroid. Nature 1993, 366, 575–580. [Google Scholar] [CrossRef]
- Brown, E.M.; MacLeod, R.J. Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 2001, 81, 239–297. [Google Scholar] [CrossRef]
- Brown, E.M.; Pollak, M.R.; Hebert, S.C. Sensing of extracellular Ca2+ by parathyroid and kidney cells: Cloning and characteri-zation of an extracellular Ca2+-sensing receptor. Am. J. Kidney Dis. 1995, 25, 506–513. [Google Scholar] [CrossRef]
- Brown, E.M. Extracellular Ca2+ sensing, regulation of parathyroid cell function, and role of Ca2+ and other ions as extracellular (first) messengers. Physiol. Rev. 1991, 71, 371–411. [Google Scholar] [CrossRef]
- Alfadda, T.I.; Saleh, A.M.; Houillier, P.; Geibel, J.P. Calcium-sensing receptor 20 years later. Am. J. Physiol. Cell Physiol. 2014, 307, C221–C231. [Google Scholar] [CrossRef]
- Hannan, F.M.; Kallay, E.; Chang, W.; Brandi, M.L.; Thakker, R.V. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 2018, 15, 33–51. [Google Scholar] [CrossRef]
- Block, G.A.; Martin, K.J.; de Francisco, A.L.; Turner, S.A.; Avram, M.M.; Suranyi, M.G.; Hercz, G.; Cunningham, J.; Abu-Alfa, A.K.; Messa, P.; et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N. Engl. J. Med. 2004, 350, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.F. Translational implications of the parathyroid calcium receptor. N. Engl. J. Med. 2004, 351, 324–326. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamba, G. Arterial Blood Pressure, Neuronal Excitability, Mineral Metabolism and Cell Volume Regulation Mechanisms Revealed by Xenopus laevis oocytes. Membranes 2022, 12, 911. https://doi.org/10.3390/membranes12100911
Gamba G. Arterial Blood Pressure, Neuronal Excitability, Mineral Metabolism and Cell Volume Regulation Mechanisms Revealed by Xenopus laevis oocytes. Membranes. 2022; 12(10):911. https://doi.org/10.3390/membranes12100911
Chicago/Turabian StyleGamba, Gerardo. 2022. "Arterial Blood Pressure, Neuronal Excitability, Mineral Metabolism and Cell Volume Regulation Mechanisms Revealed by Xenopus laevis oocytes" Membranes 12, no. 10: 911. https://doi.org/10.3390/membranes12100911
APA StyleGamba, G. (2022). Arterial Blood Pressure, Neuronal Excitability, Mineral Metabolism and Cell Volume Regulation Mechanisms Revealed by Xenopus laevis oocytes. Membranes, 12(10), 911. https://doi.org/10.3390/membranes12100911