

Isoliquiritigenin Protects Neuronal Cells against Glutamate Excitotoxicity

 , , , ,
, , , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cortical Neuroglial Cell Culture Preparation
2.2. Evaluation of Neuronal Viability
2.3. Measuring of [Ca2+]i and ΔΨm in Cortical Neurons
2.4. Measurements of the Neuroglial Culture Oxygen Consumption Rate
2.5. Statistical Analysis
3. Results
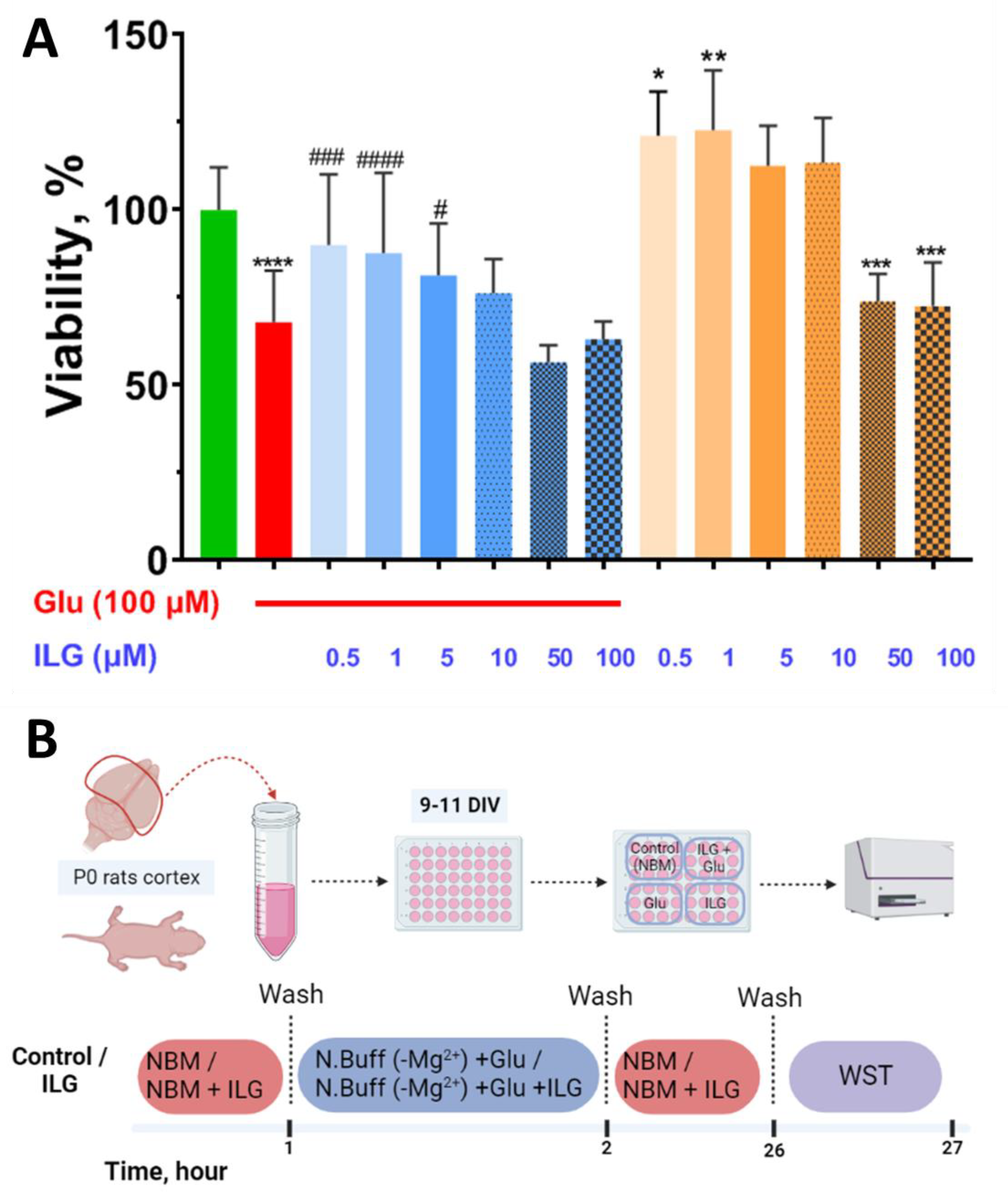
3.1. Measuring of Neuronal Viability by Biochemical WST-Test
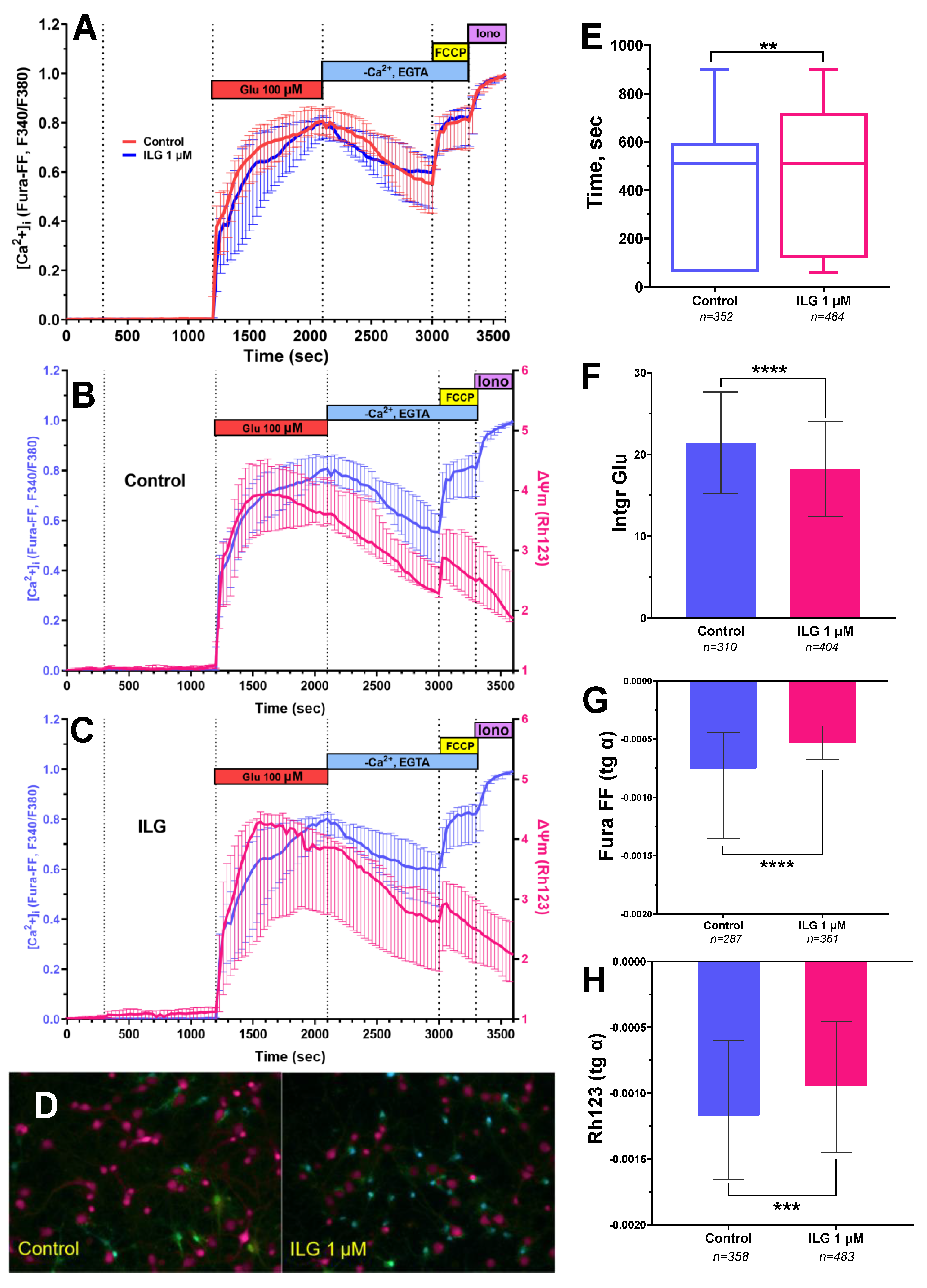
3.2. Measuring of [Ca2+]i and ΔΨm in Cortical Neurons
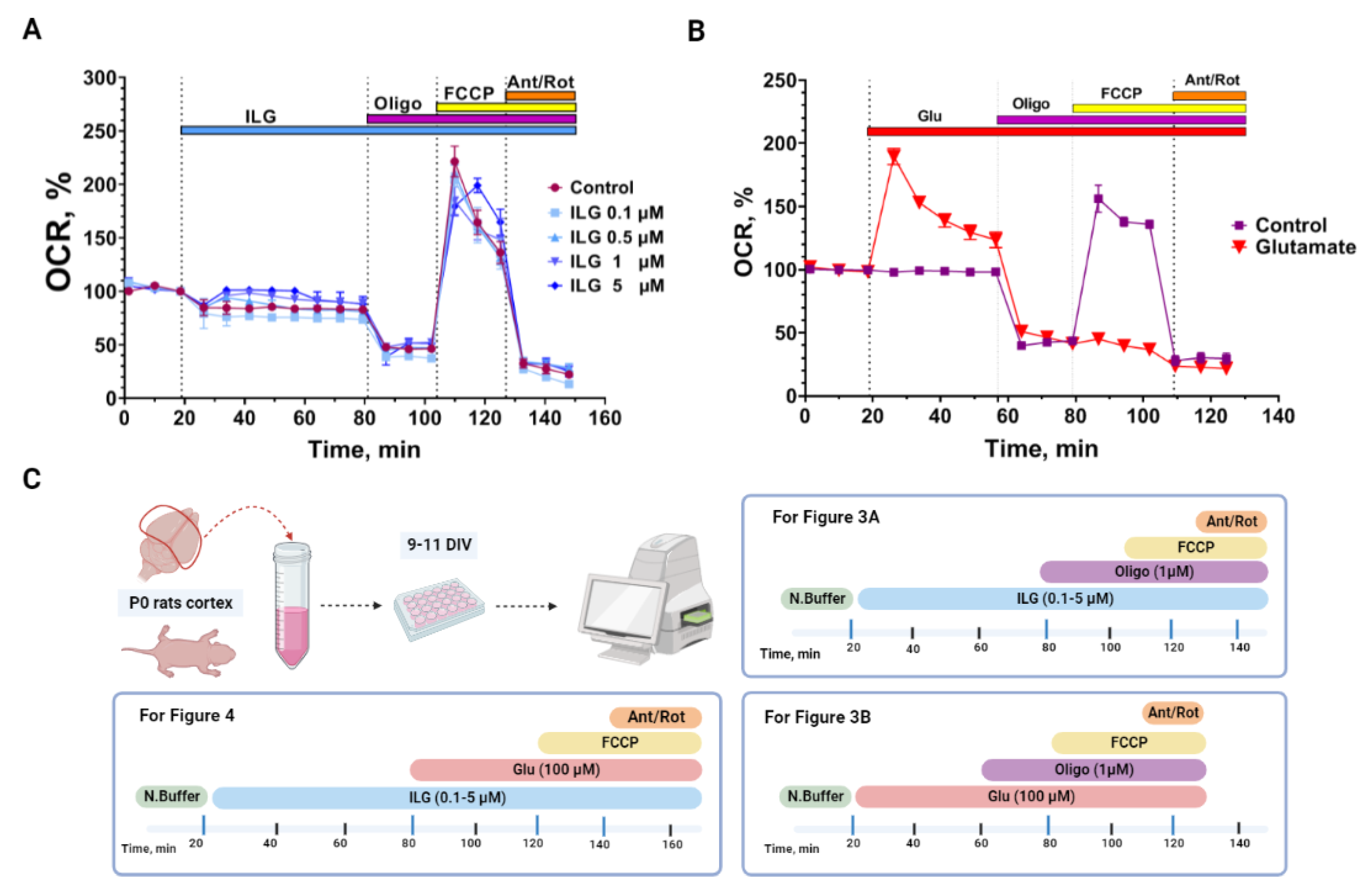
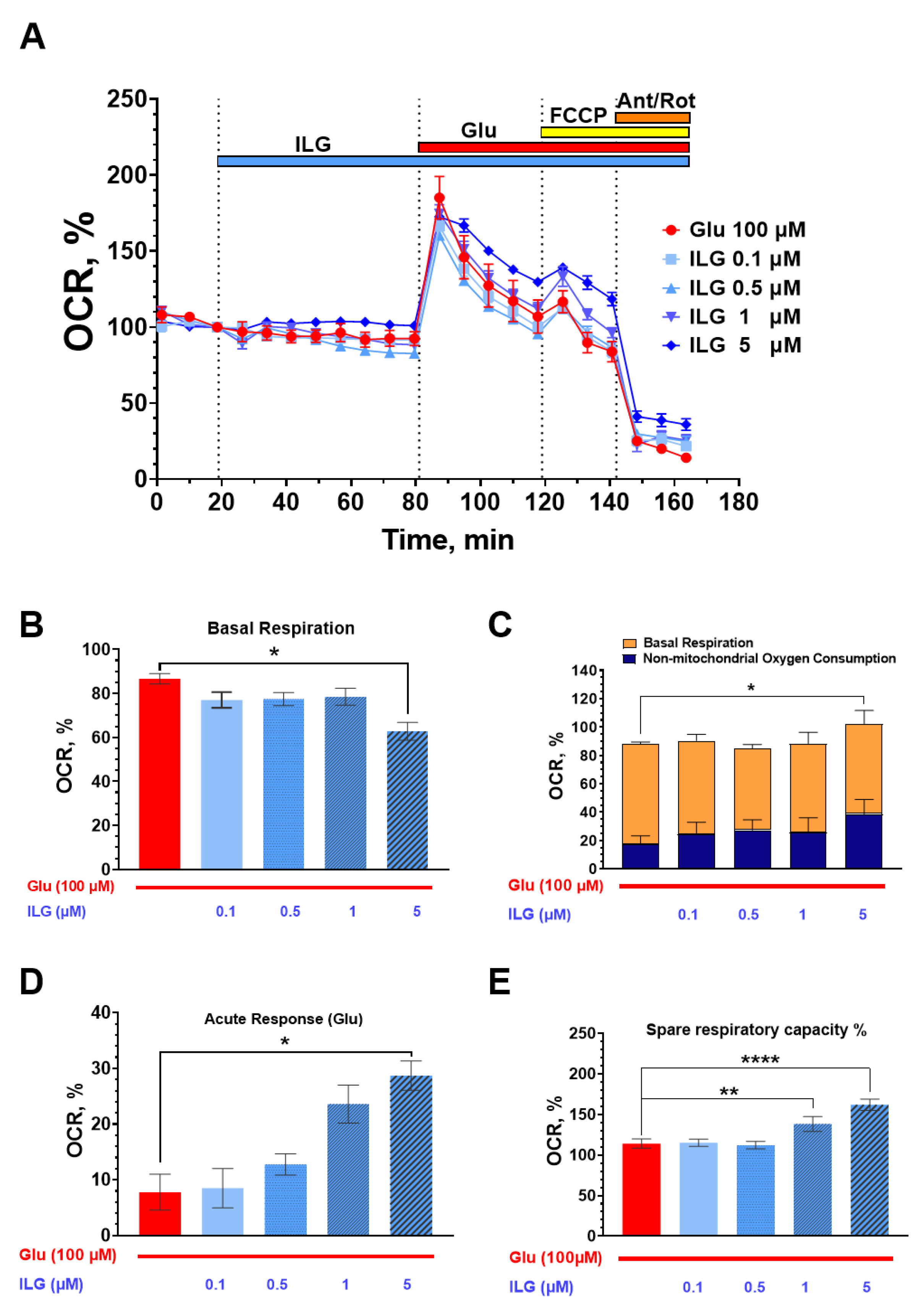
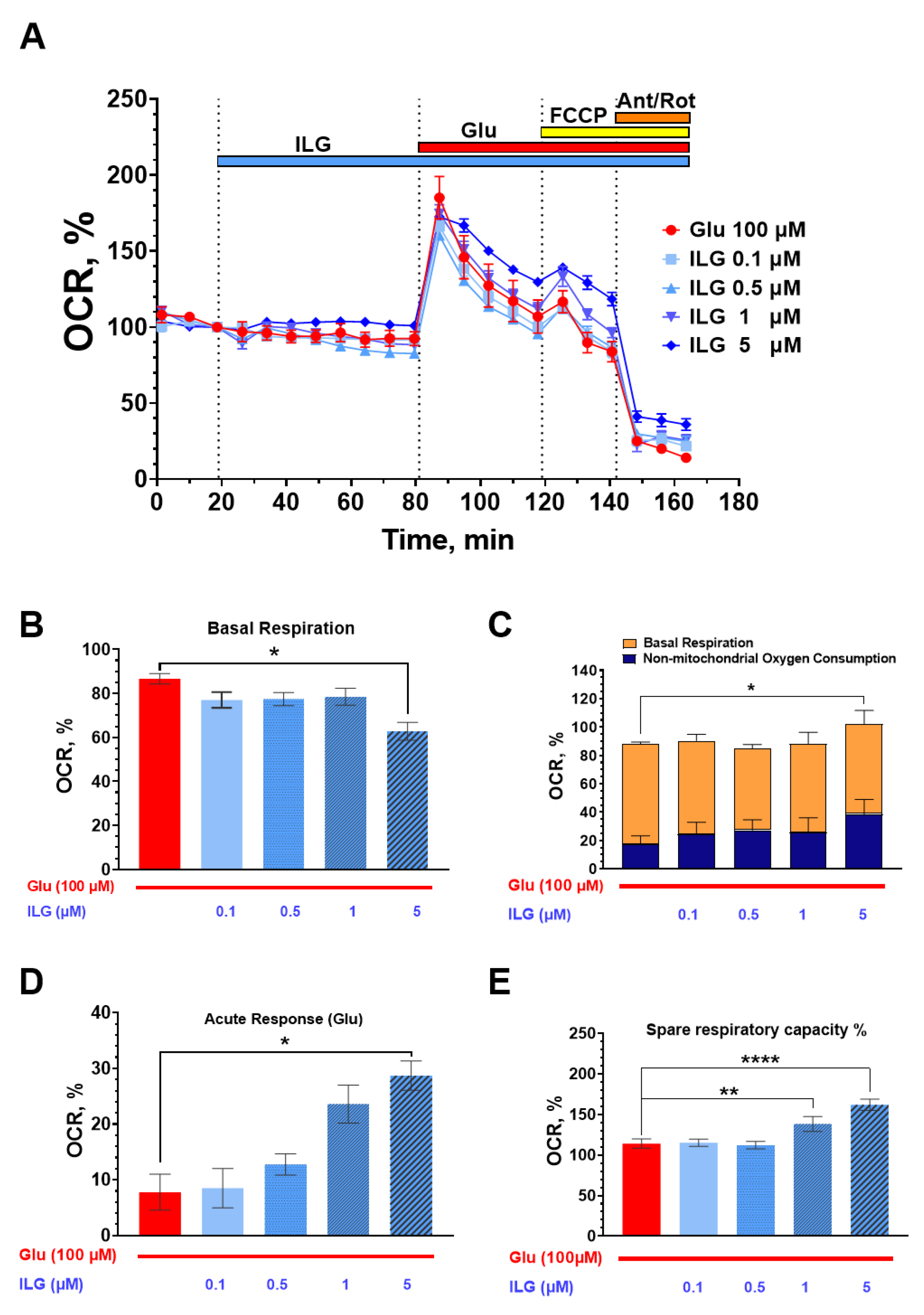
3.3. Measurements of the Neuroglial Culture Oxygen Consumption Rate (OCR)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lipton, S.A. Failures and Successes of NMDA Receptor Antagonists: Molecular Basis for the Use of Open-Channel Blockers like Memantine in the Treatment of Acute and Chronic Neurologic Insults. NeuroRx 2004, 1, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, S.M.; Connolly, C.N. Dendritic and Mitochondrial Changes during Glutamate Excitotoxicity. Neuropharmacology 2007, 53, 891–898. [Google Scholar] [CrossRef]
- Morland, C.; Nordengen, K. N-Acetyl-Aspartyl-Glutamate in Brain Health and Disease. Int. J. Mol. Sci. 2022, 23, 1268. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Arabaci, O.; Acari, A.; Ciftci, P.; Gozuacik, D. Glutamate Scavenging as a Neuroreparative Strategy in Ischemic Stroke. Front. Pharmacol. 2022, 13, 866738. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, Calcium and Mitochondria: A Triad in Synaptic Neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef]
- Bakaeva, Z.; Goncharov, M.; Krasilnikova, I.; Zgodova, A.; Frolov, D.; Grebenik, E.; Timashev, P.; Pinelis, V.; Surin, A. Acute and Delayed Effects of Mechanical Injury on Calcium Homeostasis and Mitochondrial Potential of Primary Neuroglial Cell Culture: Potential Causal Contributions to Post-Traumatic Syndrome. Int. J. Mol. Sci. 2022, 23, 3858. [Google Scholar] [CrossRef]
- Ogino, Y.; Bernas, T.; Greer, J.E.; Povlishock, J.T. Axonal Injury Following Mild Traumatic Brain Injury Is Exacerbated by Repetitive Insult and Is Linked to the Delayed Attenuation of NeuN Expression without Concomitant Neuronal Death in the Mouse. Brain Pathol. 2022, 32, e13034. [Google Scholar] [CrossRef] [PubMed]
- Khodorov, B. Glutamate-Induced Deregulation of Calcium Homeostasis and Mitochondrial Dysfunction in Mammalian Central Neurones. Prog. Biophys. Mol. Biol. 2004, 86, 279–351. [Google Scholar] [CrossRef] [PubMed]
- Kiedrowski, L. N-Methyl-D-Aspartate Excitotoxicity: Relationships among Plasma Membrane Potential, Na+/Ca2+ Exchange, Mitochondrial Ca2+ Overload, and Cytoplasmic Concentrations of Ca2+ H+ and K+. Mol. Pharmacol. 1999, 56, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Nakamizo, T.; Inoue, R.; Sawada, H.; Kihara, T.; Honda, K.; Akaike, A.; Shimohama, S. N-Methyl-D-Aspartate Receptor-Mediated Mitochondrial Ca2+ Overload in Acute Excitotoxic Motor Neuron Death: A Mechanism Distinct from Chronic Neurotoxicity after Ca2+ Influx. J. Neurosci. Res. 2001, 63, 377–387. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria, Calcium-Dependent Neuronal Death and Neurodegenerative Disease. Pflugers Arch. Eur. J. Physiol. 2012, 464, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.M.; Lu, X.F.; Bhavnani, B.R. Equine Estrogens Differentially Inhibit DNA Fragmentation Induced by Glutamate in Neuronal Cells by Modulation of Regulatory Proteins Involved in Programmed Cell Death. BMC Neurosci. 2003, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.J.; Min, J.S.; Ku, H.Y.; Choi, H.S.; Park, M.K.; Kim, M.K.; Song, K.S.; Lee, D.S. Isoliquiritigenin Isolated from Glycyrrhiza Uralensis Protects Neuronal Cells against Glutamate-Induced Mitochondrial Dysfunction. Biochem. Biophys. Res. Commun. 2012, 421, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of Reactive Oxygen Species Generation in Cell Signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus Extrasynaptic NMDA Receptor Signalling: Implications for Neurodegenerative Disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.-H.; Hazell, A.S. Excitotoxic Mechanisms and the Role of Astrocytic Glutamate Transporters in Traumatic Brain Injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, C.R.; McGuire, J.L.; Niedzielko, T.L.; Depasquale, E.A.K.; Meller, J.; Floyd, C.L.; McCullumsmith, R.E. Traumatic Brain Injury Induces Alterations in Cortical Glutamate Uptake without a Reduction in Glutamate Transporter-1 Protein Expression. J. Neurotrauma 2017, 34, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrov, M.Y.; Lizhin, A.A.; Andrianova, E.L.; Gretskaya, N.M.; Frumkina, L.E.; Khaspekov, L.G.; Bezuglov, V.V. Antioxidant and Neuroprotective Properties of N-Arachidonoyldopamine. Neurosci. Lett. 2008, 431, 6–11. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Brand, M.D.; Gerencser, A.A. Mitochondrial Bioenergetics and Neuronal Survival Modelled in Primary Neuronal Culture and Isolated Nerve Terminals. J. Bioenerg. Biomembr. 2015, 47, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Bakaeva, Z.V.; Surin, A.M.; Lizunova, N.V.; Zgodova, A.E.; Krasilnikova, I.A.; Fisenko, A.P.; Frolov, D.A.; Andreeva, L.A.; Myasoedov, N.F.; Pinelis, V.G. Neuroprotective Potential of Peptides HFRWPGP (ACTH6–9PGP), KKRRPGP, and PyrRP in Cultured Cortical Neurons at Glutamate Excitotoxicity. Dokl. Biochem. Biophys. 2020, 491, 62–66. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Sharipov, R.R.; Boyarkin, D.P.; Belosludtseva, N.V.; Dubinin, M.V.; Krasilnikova, I.A.; Bakaeva, Z.V.; Zgodova, A.E.; Pinelis, V.G.; Surin, A.M. The Effect of DS16570511, a New Inhibitor of Mitochondrial Calcium Uniporter, on Calcium Homeostasis, Metabolism, and Functional State of Cultured Cortical Neurons and Isolated Brain Mitochondria. Biochim. Biophys. Acta-Gen. Subj. 2021, 1865, 129847. [Google Scholar] [CrossRef]
- Hamsalakshmi; Alex, A.M.; Arehally Marappa, M.; Joghee, S.; Chidambaram, S.B. Therapeutic Benefits of Flavonoids against Neuroinflammation: A Systematic Review. Inflammopharmacology 2022, 30, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, C.D.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef]
- Wang, X.; Qi, Y.; Zheng, H. Dietary Polyphenol, Gut Microbiota, and Health Benefits. Antioxidants 2022, 11, 1212. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.W.; Kim, S.G. AMPK-Mediated GSK3β Inhibition by Isoliquiritigenin Contributes to Protecting Mitochondria against Iron-Catalyzed Oxidative Stress. Biochem. Pharmacol. 2010, 79, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Manach, C.; Morand, C.; Rémésy, C.; Jiménez, L. Dietary Polyphenols and the Prevention of Diseases. Crit. Rev. Food Sci. Nutr. 2005, 45, 287–306. [Google Scholar] [CrossRef]
- Pavlova, S.I.; Albegova, D.Z.; Vorob, Y.S.; Laptev, O.S.; Kozlov, I.G. Molecular-Biological Problems of Drug Design and Mechanism of Drug Action Flavonoids as Potential Immunosuppressants Affecting Intracellular Signaling Pathways ( A Review ). Pharm. Chem. J. 2016, 49, 645–652. [Google Scholar] [CrossRef]
- Yang, E.J.; Kim, M.; Woo, J.E.; Lee, T.; Jung, J.W.; Song, K.S. The Comparison of Neuroprotective Effects of Isoliquiritigenin and Its Phase I Metabolites against Glutamate-Induced HT22 Cell Death. Bioorganic Med. Chem. Lett. 2016, 26, 5639–5643. [Google Scholar] [CrossRef]
- Lee, D.G.; Min, J.S.; Lee, H.S.; Lee, D.S. Isoliquiritigenin Attenuates Glutamate-Induced Mitochondrial Fission via Calcineurin-Mediated Drp1 Dephosphorylation in HT22 Hippocampal Neuron Cells. Neurotoxicology 2018, 68, 133–141. [Google Scholar] [CrossRef]
- Selvaraj, B.; Won, D.; Huh, G.; Lee, H.; Kang, K. Bioorganic & Medicinal Chemistry Letters Synthesis and Biological Evaluation of Isoliquiritigenin Derivatives as a Neuroprotective Agent against Glutamate Mediated Neurotoxicity in HT22 Cells. Bioorg. Med. Chem. Lett. 2020, 30, 127058. [Google Scholar] [CrossRef] [PubMed]
- Denzer, I.; Münch, G.; Pischetsrieder, M.; Friedland, K. S-Allyl-l-Cysteine and Isoliquiritigenin Improve Mitochondrial Function in Cellular Models of Oxidative and Nitrosative Stress. Food Chem. 2016, 194, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Z.; Ikarashi, Y.; Kase, Y. Isoliquiritigenin Is a Novel NMDA Receptor Antagonist in Kampo Medicine Yokukansan. Cell. Mol. Neurobiol. 2011, 31, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Präbst, K.; Engelhardt, H.; Ringgeler, S.; Hübner, H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. Methods Mol. Biol. 2017, 1601, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bakaeva, Z.; Lizunova, N.; Tarzhanov, I.; Boyarkin, D.; Petrichuk, S.; Pinelis, V.; Fisenko, A.; Tuzikov, A.; Sharipov, R.; Surin, A. Lipopolysaccharide From E. Coli Increases Glutamate-Induced Disturbances of Calcium Homeostasis, the Functional State of Mitochondria, and the Death of Cultured Cortical Neurons. Front. Mol. Neurosci. 2022, 14, 811171. [Google Scholar] [CrossRef]
- Gerencser, A.A.; Neilson, A.; Choi, S.W.; Edman, U.; Yadava, N.; Oh, R.J.; Ferrick, D.A.; Nicholls, D.G.; Brand, M.D. Quantitative Microplate-Based Respirometry with Correction for Oxygen Diffusion. Anal. Chem. 2009, 81, 6868–6878. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G.; Budd, S.L. Mitochondria and Neuronal Survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodorov, B.I.; Storozhevykh, T.P.; Surin, A.M.; Yuryavichyus, A.I.; Sorokina, E.G.; Borodin, A.V.; Vinskaya, N.P.; Khaspekov, L.G.; Pinelis, V.G. The Leading Role of Mitochondrial Depolarization in the Mechanism of Glutamate-Induced Disruptions in Ca2+ Homeostasis. Neurosci. Behav. Physiol. 2002, 32, 541–547. [Google Scholar] [CrossRef]
- Nicholls, D.G. Spare Respiratory Capacity, Oxidative Stress and Excitotoxicity. Biochem. Soc. Trans. 2009, 37, 1385–1388. [Google Scholar] [CrossRef]
- Brustovetsky, T.; Li, V.; Brustovetsky, N. Stimulation of Glutamate Receptors in Cultured Hippocampal Neurons Causes Ca2+-Dependent Mitochondrial Contraction. Cell Calcium 2009, 46, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.A.; Rios, J.A.; Picón-Pagès, P.; Herrera-Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández-Fernández, J.M.; Muñoz, F.J. Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration. Biomolecules 2021, 11, 1012. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carajoli, E. Intracellular Calcium; Annual Reviews Inc.: San Mateo, CA, USA, 1987. [Google Scholar]
- Blaustein, M.P.; Lederer, W.J. Sodium/Calcium Exchange: Its Physiological Implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef] [PubMed]
- Ontiveros, M.; Rinaldi, D.; Marder, M.; Espelt, M.V.; Mangialavori, I.; Vigil, M.; Rossi, J.P.; Ferreira-Gomes, M. Natural Flavonoids Inhibit the Plasma Membrane Ca2+ -ATPase. Biochem. Pharmacol. 2019, 166, 1–11. [Google Scholar] [CrossRef]
- Shen, Y.; Tian, Y.; Shi, X.; Yang, J.; Ouyang, L.; Gao, J.; Lu, J. Exposure to High Glutamate Concentration Activates Aerobic Glycolysis but Inhibits ATP-Linked Respiration in Cultured Cortical Astrocytes. Cell Biochem. Funct. 2014, 32, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Shi, Z.F.; Xu, L.X.; Li, J.X.; Wu, M.; Wang, X.X.; Jia, M.; Dong, L.P.; Yang, S.H.; Yuan, F. Glutamate Impairs Mitochondria Aerobic Respiration Capacity and Enhances Glycolysis in Cultured Rat Astrocytes. Biomed. Environ. Sci. 2017, 30, 44–51. [Google Scholar] [CrossRef]
- Pinelis, V.; Krasilnikova, I.; Bakaeva, Z.; Surin, A.; Boyarkin, D.; Fisenko, A.; Krasilnikova, O.; Pomytkin, I. Insulin Diminishes Superoxide Increase in Cytosol and Mitochondria of Cultured Cortical Neurons Treated with Toxic Glutamate. Int. J. Mol. Sci. 2022, 23, 12593. [Google Scholar] [CrossRef]
- Brennan-Minnella, A.M.; Shen, Y.; Swanson, R.A. Phosphoinositide 3-Kinase Couples NMDA Receptors to Superoxide Release in Excitotoxic Neuronal Death. Cell Death Dis. 2013, 4, e580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, A.M.; Won Suh, S.; Joon Won, S.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH Oxidase Is the Primary Source of Superoxide Induced by NMDA Receptor Activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girouard, H.; Wang, G.; Gallo, E.F.; Anrather, J.; Zhou, P.; Pickel, V.M.; Iadecola, C. NMDA Receptor Activation Increases Free Radical Production through Nitric Oxide and NOX2. J. Neurosci. 2009, 29, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Sang, W.S.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic Neuronal Death Is Triggered by Glucose Reperfusion and Activation of Neuronal NADPH Oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef]
- Dikalov, S. Cross Talk between Mitochondria and NADPH Oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Horton, R.M.; Bader, D.A.; Liu, F.; Sun, Q.; Kinney, P.L. Long-Term Projections of Temperature-Related Mortality Risks for Ischemic Stroke, Hemorrhagic Stroke, and Acute Ischemic Heart Disease under Changing Climate in Beijing, China. Environ. Int. 2018, 112, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lv, X.; Yang, H.; Peng, L.; Ci, X. Isoliquiritigenin Exerts Antioxidative and Anti-Inflammatory Effects: Via Activating the KEAP-1/Nrf2 Pathway and Inhibiting the NF-ΚB and NLRP3 Pathways in Carrageenan-Induced Pleurisy. Food Funct. 2020, 11, 2522–2534. [Google Scholar] [CrossRef]
- Matthews, G.A.; Nieh, E.H.; Vander Weele, C.M.; Halbert, S.A.; Pradhan, R.V.; Yosafat, A.S.; Glober, G.F.; Izadmehr, E.M.; Thomas, R.E.; Lacy, G.D.; et al. Dorsal Raphe Dopamine Neurons Represent the Experience of Social Isolation. Cell 2016, 164, 617–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prajapati, R.; Seong, S.H.; Park, S.E.; Paudel, P.; Jung, H.A.; Choi, J.S. Isoliquiritigenin, a Potent Human Monoamine Oxidase Inhibitor, Modulates Dopamine D1, D3, and Vasopressin V1A Receptors. Sci. Rep. 2021, 11, 23528. [Google Scholar] [CrossRef]
- Mancuso, C. Heme Oxygenase and Its Products in the Nervous System. Antioxid. Redox Signal. 2004, 6, 878–887. [Google Scholar] [CrossRef]
- Sharp, F.R.; Zhan, X.; Liu, D. Heat Shock Proteins in the Brain: Role of Hsp70, Hsp 27, and HO-1 ( Hsp32) and Their Therapeutic Potential. Transl. Stroke Res. 2013, 4, 685–692. [Google Scholar] [CrossRef]
- Yao, D.; Shi, B.; Wang, S.; Bao, L.; Tan, M.; Shen, H.; Zhang, Z.; Pan, X.; Yang, Y.; Wu, Y.; et al. Isoliquiritigenin Ameliorates Ischemia-Induced Myocardial Injury via Modulating the Nrf2/HO-1 Pathway in Mice. Drug Des. Devel. Ther. 2022, 16, 1273–1287. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Tian, R.; Yang, Z.; Peng, Y.Y.; Lu, N. Quercetin Suppressed NADPH Oxidase-Derived Oxidative Stress via Heme Oxygenase-1 Induction in Macrophages. Arch. Biochem. Biophys. 2019, 671, 69–76. [Google Scholar] [CrossRef]
- Wang, H.U.I.; Xu, Y.O.U.S.; Wang, M.L.I.N.; Cheng, C.; Bian, R.U.I.; Yuan, H.A.O.; Wang, Y.I.; Guo, T.; Zhu, L.L.; Zhou, H. Protective Effect of Naringin against the LPS-Induced Apoptosis of PC12 Cells: Implications for the Treatment of Neurodegenerative Disorders. Int. J. Mol. Med. 2017, 39, 819–830. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zgodova, A.; Pavlova, S.; Nekrasova, A.; Boyarkin, D.; Pinelis, V.; Surin, A.; Bakaeva, Z. Isoliquiritigenin Protects Neuronal Cells against Glutamate Excitotoxicity. Membranes 2022, 12, 1052. https://doi.org/10.3390/membranes12111052
Zgodova A, Pavlova S, Nekrasova A, Boyarkin D, Pinelis V, Surin A, Bakaeva Z. Isoliquiritigenin Protects Neuronal Cells against Glutamate Excitotoxicity. Membranes. 2022; 12(11):1052. https://doi.org/10.3390/membranes12111052
Chicago/Turabian StyleZgodova, Arina, Svetlana Pavlova, Anastasia Nekrasova, Dmitriy Boyarkin, Vsevolod Pinelis, Alexander Surin, and Zanda Bakaeva. 2022. "Isoliquiritigenin Protects Neuronal Cells against Glutamate Excitotoxicity" Membranes 12, no. 11: 1052. https://doi.org/10.3390/membranes12111052
APA StyleZgodova, A., Pavlova, S., Nekrasova, A., Boyarkin, D., Pinelis, V., Surin, A., & Bakaeva, Z. (2022). Isoliquiritigenin Protects Neuronal Cells against Glutamate Excitotoxicity. Membranes, 12(11), 1052. https://doi.org/10.3390/membranes12111052