
Design of Membrane Active Peptides Considering Multi-Objective Optimization for Biomedical Application


Abstract
:1. Introduction
2. Cell Penetrating Peptides
3. Uptake Mechanisms of Cell Penetrating Peptides
4. Antimicrobial Peptides
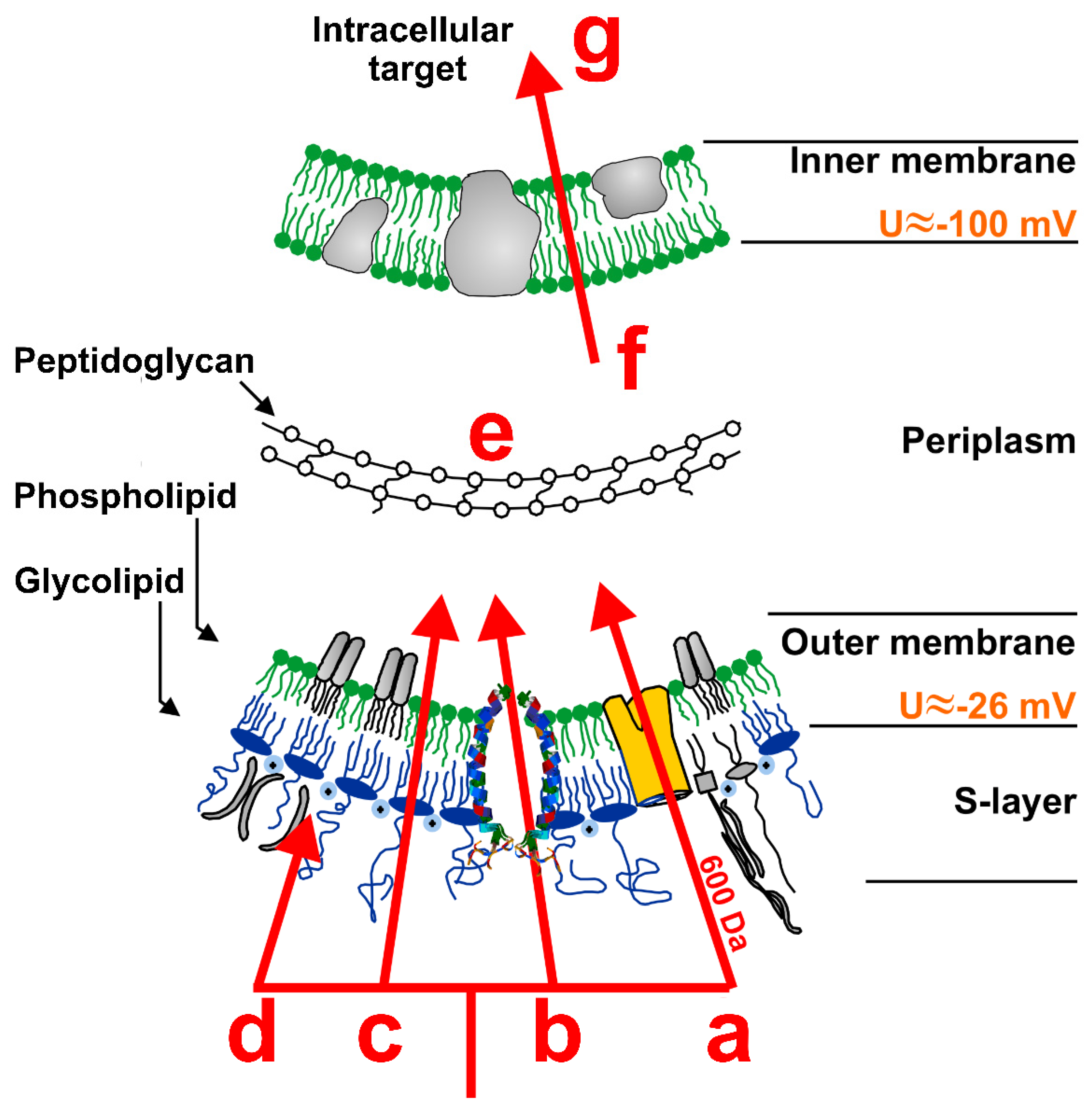
5. Membrane Interaction of Antimicrobial Peptides
6. Optimization of Membrane Active Peptides
6.1. Computational Methods for Optimization of Membrane Active Peptides
6.1.1. Machine Learning (ML) and Empirical Methods
6.1.2. Stochastic Methods
6.1.3. Data Driven Algorithms vs. Stochastic Methods
6.2. Combinatorial Synthesis Methods to Optimize Membrane Active Peptides
6.3. Assay Systems for Characterization of Membane Active Peptides
6.3.1. Membrane Activity Assays
6.3.2. Assays Quantifying other Optimization Criteria
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Hediger, M.A.; Romero, M.F.; Peng, J.-B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers. Arch.—Eur. J. Physiol. 2003, 447, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, G.; Alsenz, J.; van de Waterbeemd, H.; Folkers, G. Estimation of permeability by passive diffusion through Caco-2 cell monolayers using the drugs’ lipophilicity and molecular weight. Eur. J. Pharm. Sci. 1998, 6, 313–319. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. In Site-Specific Protein Labeling: Methods and Protocols; Gautier, A., Hinner, M.J., Eds.; Springer: New York, NY, USA, 2015; Volume 1266, pp. 29–53. [Google Scholar]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.; Rietschel, E.T. Invited review: Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef]
- Wiedemann, I.; Böttiger, T.; Bonelli, R.R.; Wiese, A.; Hagge, S.O.; Gutsmann, T.; Seydel, U.; Deegan, L.; Hill, C.; Ross, R.; et al. The mode of action of the lantibiotic lacticin 3147—A complex mechanism involving specific interaction of two peptides and the cell wall precursor lipid II. Mol. Microbiol. 2006, 61, 285–296. [Google Scholar] [CrossRef]
- Avci, F.G.; Akbulut, B.S.; Ozkirimli, E. Membrane Active Peptides and Their Biophysical Characterization. Biomolecules 2018, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, M.D.T.; Sothiselvam, S.; Lu, T.K.; De La Fuente-Nunez, C. Peptide Design Principles for Antimicrobial Applications. J. Mol. Biol. 2019, 431, 3547–3567. [Google Scholar] [CrossRef]
- Drexelius, M.; Reinhardt, A.; Grabeck, J.; Cronenberg, T.; Nitsche, F.; Huesgen, P.F.; Maier, B.; Neundorf, I. Multistep optimization of a cell-penetrating peptide towards its antimicrobial activity. Biochem. J. 2021, 478, 63–78. [Google Scholar] [CrossRef]
- Buccini, D.F.; Cardoso, M.H.; Franco, O.L. Antimicrobial Peptides and Cell-Penetrating Peptides for Treating Intracellular Bacterial Infections. Front. Cell. Infect. Microbiol. 2021, 10, 612931. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Virès, E.; Granier, C.; Prevot, P.; LeBleu, B. Structure-activity relationship study of the plasma membrane translocating potential of a short peptide from HIV-1 Tat protein. Int. J. Pept. Res. Ther. 1997, 4, 429–436. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, Ü. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Elmquist, A.; Lindgren, M.; Bartfai, T.; Langel, Ü. VE-Cadherin-Derived Cell-Penetrating Peptide, pVEC, with Carrier Functions. Exp. Cell Res. 2001, 269, 237–244. [Google Scholar] [CrossRef]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Hansen, M.; Kilk, K.; Langel, Ü. Predicting cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 572–579. [Google Scholar] [CrossRef]
- Elmquist, A.; Hansen, M.; Langel, Ü. Structure–activity relationship study of the cell-penetrating peptide pVEC. Biochim. Biophys. Acta—Biomembr. 2006, 1758, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Hällbrink, M.; Kilk, K.; Elmquist, A.; Lundberg, P.; Lindgren, M.; Jiang, Y.; Pooga, M.; Soomets, U.; Langel, Ü. Prediction of Cell-Penetrating Peptides. Int. J. Pept. Res. Ther. 2005, 11, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Deshayes, S.; Konate, K.; Aldrian, G.; Heitz, F.; Divita, G. Interactions of Amphipathic CPPs with Model Membranes. Cell Penetrating Pept. 2010, 683, 41–56. [Google Scholar] [CrossRef]
- Joliot, A.; Prochiantz, A. Transduction peptides: From technology to physiology. Nat. Cell Biol. 2004, 6, 189–196. [Google Scholar] [CrossRef]
- Kardani, K.; Milani, A.; Shabani, S.H.; Bolhassani, A. Cell penetrating peptides: The potent multi-cargo intracellular carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.M.; Horton, K.L.; Kelley, S.O. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008, 6, 2242–2255. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Waizenegger, T.; Köhler, K.; Brock, R. A quantitative validation of fluorophore-labelled cell-permeable peptide conjugates: Fluorophore and cargo dependence of import. Biochim. Biophys. Acta—Biomembr. 2002, 1564, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Säälik, P.; Elmquist, A.; Hansen, M.; Padari, K.; Saar, K.; Viht, K.; Langel, Ü.; Pooga, M. Protein Cargo Delivery Properties of Cell-Penetrating Peptides. A Comparative Study. Bioconjugate Chem. 2004, 15, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.-H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Taylor, R.E.; Zahid, M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics 2020, 12, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, P.; El-Andaloussi, S.; Sutlu, T.; Johansson, H.; Langel, Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of Cell-Penetrating Peptides with Nanoparticles for Therapeutic Application: A Review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecher, J.C.; Nowak, S.; McMurry, J.L. Breaking in and busting out: Cell-penetrating peptides and the endosomal escape problem. Biomol. Concepts 2017, 8, 131–141. [Google Scholar] [CrossRef]
- Oikawa, K.; Tateishi, A.; Odahara, M.; Kodama, Y.; Numata, K. Imaging of the Entry Pathway of a Cell-Penetrating Peptide–DNA Complex From the Extracellular Space to Chloroplast Nucleoids Across Multiple Membranes in Arabidopsis Leaves. Front. Plant Sci. 2021, 12, 759871. [Google Scholar] [CrossRef]
- El Boujnouni, N.; van Asbeck, A.H.; Dieker, J.; Wansink, D.G.; Brock, R. Imaging of CPP Delivery Mechanisms of Oligonucleotides. Cell Penetrating Pept. 2021, 2383, 197–210. [Google Scholar] [CrossRef]
- Richard, J.-P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating Peptides: A REEVALUATION OF THE MECHANISM OF CELLULAR UPTAKE. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef] [Green Version]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef]
- Wimley, W.C.; Hristova, K. The Mechanism of Membrane Permeabilization by Peptides: Still an Enigma. Aust. J. Chem. 2020, 73, 96–103. [Google Scholar] [CrossRef]
- Patel, L.N.; Zaro, J.L.; Shen, W.-C. Cell Penetrating Peptides: Intracellular Pathways and Pharmaceutical Perspectives. Pharm. Res. 2007, 24, 1977–1992. [Google Scholar] [CrossRef]
- Jiao, C.-Y.; Delaroche, D.; Burlina, F.; Alves, I.; Chassaing, G.; Sagan, S. Translocation and Endocytosis for Cell-penetrating Peptide Internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef] [Green Version]
- Melikov, K.; Chernomordik, L.V. Arginine-rich cell penetrating peptides: From endosomal uptake to nuclear delivery. Cell. Mol. Life Sci. 2005, 62, 2739–2749. [Google Scholar] [CrossRef]
- Åmand, H.L.; Fant, K.; Nordén, B.; Esbjörner, E.K. Stimulated endocytosis in penetratin uptake: Effect of arginine and lysine. Biochem. Biophys. Res. Commun. 2008, 371, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Köhler, K.; Fotin-Mleczek, M.; Brock, R. A Stepwise Dissection of the Intracellular Fate of Cationic Cell-penetrating Peptides. J. Biol. Chem. 2004, 279, 12625–12635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundin, P.; Johansson, H.; Guterstam, P.; Holm, T.; Hansen, M.; Langel, Ü.; EL Andaloussi, S. Distinct Uptake Routes of Cell-Penetrating Peptide Conjugates. Bioconjug. Chem. 2008, 19, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Gestin, M.; Dowaidar, M.; Langel, Ü. Uptake Mechanism of Cell-Penetrating Peptides. Adv. Exp. Med. Biol. 2017, 1030, 255–264. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Abadi, A.T.B.; Rizvanov, A.A.; Haertlé, T.; Blatt, N.L. World Health Organization Report: Current Crisis of Antibiotic Resistance. BioNanoScience 2019, 9, 778–788. [Google Scholar] [CrossRef]
- Steiner, H.; Hultmark, D.; Engström, Å.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins. Natural peptide antibiotics of human neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [Green Version]
- Boman, H.G. Peptide Antibiotics and Their Role in Innate Immunity. Annu. Rev. Immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed]
- Wu, M.; Hancock, R. Interaction of the Cyclic Antimicrobial Cationic Peptide Bactenecin with the Outer and Cytoplasmic Membrane. J. Biol. Chem. 1999, 274, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, N.G.; Li, W.; Hossain, M.A.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. (Re)Defining the Proline-Rich Antimicrobial Peptide Family and the Identification of Putative New Members. Front. Chem. 2020, 8, 607769. [Google Scholar] [CrossRef] [PubMed]
- Casteels, P.; Ampe, C.; Jacobs, F.; Vaeck, M.; Tempst, P. Apidaecins: Antibacterial peptides from honeybees. EMBO J. 1989, 8, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, V.; Bukhteeva, I.; Kit, O.Y.; Nesmelova, I.V. Plant Defensins from a Structural Perspective. Int. J. Mol. Sci. 2020, 21, 5307. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.M.; Gonçalves, S.; Santos, N.C. Defensins: Antifungal lessons from eukaryotes. Front. Microbiol. 2014, 5, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2015, 44, D1104–D1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Zasloff, M. Antibiotic peptides as mediators of innate immunity. Curr. Opin. Immunol. 1992, 4, 3–7. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Sugishita, K.; Fujii, N.; Miyajima, K. Molecular Basis for Membrane Selectivity of an Antimicrobial Peptide, Magainin 2. Biochemistry 1995, 34, 3423–3429. [Google Scholar] [CrossRef]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta—Biomembr. 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Maturana, P.; Martinez, M.; Noguera, M.; Santos, N.; Disalvo, E.; Semorile, L.; Maffia, P.; Hollmann, A. Lipid selectivity in novel antimicrobial peptides: Implication on antimicrobial and hemolytic activity. Colloids Surfaces B Biointerfaces 2017, 153, 152–159. [Google Scholar] [CrossRef]
- Chou, S.; Wang, J.; Shang, L.; Akhtar, M.U.; Wang, Z.; Shi, B.; Feng, X.; Shan, A. Short, symmetric-helical peptides have narrow-spectrum activity with low resistance potential and high selectivity. Biomater. Sci. 2019, 7, 2394–2409. [Google Scholar] [CrossRef]
- Nehls, C.; Böhling, A.; Podschun, R.; Schubert, S.; Grötzinger, J.; Schromm, A.; Fedders, H.; Leippe, M.; Harder, J.; Kaconis, Y.; et al. Influence of disulfide bonds in human beta defensin-3 on its strain specific activity against Gram-negative bacteria. Biochim. Biophys. Acta—Biomembr. 2020, 1862, 183273. [Google Scholar] [CrossRef]
- Nizet, V. Antimicrobial Peptide Resistance Mechanisms of Human Bacterial Pathogens. Curr. Issues Mol. Biol. 2006, 8, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, G.; Rodriguez, A.; Juarez, D.; Corzo, G.; Villegas, E. Improved Protease Stability of the Antimicrobial Peptide Pin2 Substituted with d-Amino Acids. J. Protein Chem. 2013, 32, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Bessalle, R.; Kapitkovsky, A.; Gorea, A.; Shalit, I.; Fridkin, M. All-D-magainin: Chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 1990, 274, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najjar, K.; Erazo-Oliveras, A.; Brock, D.; Wang, T.-Y.; Pellois, J.-P. An l- to d-Amino Acid Conversion in an Endosomolytic Analog of the Cell-penetrating Peptide TAT Influences Proteolytic Stability, Endocytic Uptake, and Endosomal Escape. J. Biol. Chem. 2017, 292, 847–861. [Google Scholar] [CrossRef] [Green Version]
- Eckert, R. Road to clinical efficacy: Challenges and novel strategies for antimicrobial peptide development. Futur. Microbiol. 2011, 6, 635–651. [Google Scholar] [CrossRef]
- Gutsmann, T.; Razquin-Olazarán, I.; Kowalski, I.; Kaconis, Y.; Howe, J.; Bartels, R.; Hornef, M.; Schuerholz, T.; Rössle, M.; Sanchez-Gómez, S.; et al. New Antiseptic Peptides to Protect against Endotoxin-Mediated Shock. Antimicrob. Agents Chemother. 2010, 54, 3817–3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, R.K.; Diep, D.B.; Tønnesen, H.H. Topical antimicrobial peptide formulations for wound healing: Current developments and future prospects. Acta Biomater. 2019, 103, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.C.; Jean, D.; McDermott, A.M. Effect of Preservative-Free Artificial Tears on the Antimicrobial Activity of Human ??-Defensin-2 and Cathelicidin LL-37 In Vitro. Eye Contact Lens Sci. Clin. Pr. 2005, 31, 34–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Serrano, Á.; Gómez, R.; Ortega, P.; De La Mata, F.J. Nanosystems as Vehicles for the Delivery of Antimicrobial Peptides (AMPs). Pharmaceutics 2019, 11, 448. [Google Scholar] [CrossRef] [Green Version]
- Lange, C.F.; Hancock, R.; Samuel, J.; Finlay, W.H. In Vitro Aerosol Delivery and Regional Airway Surface Liquid Concentration of a Liposomal Cationic Peptide. J. Pharm. Sci. 2001, 90, 1647–1657. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef]
- Wildman, K.A.H.; Lee, D.-K.; Ramamoorthy, A. Mechanism of Lipid Bilayer Disruption by the Human Antimicrobial Peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef]
- Ningsih, Z.; Hossain, M.A.; Wade, J.; Clayton, A.H.A.; Gee, M.L. Slow Insertion Kinetics during Interaction of a Model Antimicrobial Peptide with Unilamellar Phospholipid Vesicles. Langmuir 2011, 28, 2217–2224. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [Green Version]
- Baumann, G.; Mueller, P. A molecular model of membrane excitability. J. Supramol. Struct. 1974, 2, 538–557. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-Stave Model or Toroidal Model? A Case Study on Melittin Pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An Antimicrobial Peptide, Magainin 2, Induced Rapid Flip-Flop of Phospholipids Coupled with Pore Formation and Peptide Translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta—Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Pokorny, A.; Birkbeck, T.H.; Almeida, P.F.F. Mechanism and Kinetics of δ-Lysin Interaction with Phospholipid Vesicles. Biochemistry 2002, 41, 11044–11056. [Google Scholar] [CrossRef]
- Rausch, J.M.; Marks, J.R.; Rathinakumar, R.; Wimley, W.C. β-Sheet Pore-Forming Peptides Selected from a Rational Combinatorial Library: Mechanism of Pore Formation in Lipid Vesicles and Activity in Biological Membranes. Biochemistry 2007, 46, 12124–12139. [Google Scholar] [CrossRef] [Green Version]
- Hadley, E.B.; Hancock, R.E. Strategies for the discovery and advancement of novel cationic antimicrobial peptides. Curr. Top. Med. Chem. 2010, 10, 1872–1881. [Google Scholar] [CrossRef]
- Polyansky, A.; Chugunov, A.; Vassilevski, A.; Grishin, E.; Efremov, R. Recent Advances in Computational Modeling of α.-Helical Membrane- Active Peptides. Curr. Protein Pept. Sci. 2012, 13, 644–657. [Google Scholar] [CrossRef]
- Tran, D.P.; Tada, S.; Yumoto, A.; Kitao, A.; Ito, Y.; Uzawa, T.; Tsuda, K. Using molecular dynamics simulations to prioritize and understand AI-generated cell penetrating peptides. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Cardoso, M.H.; Orozco, R.M.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.D.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Fulan, B.M.; Wong, G.C.L.; Ferguson, A.L. Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proc. Natl. Acad. Sci. USA 2016, 113, 13588–13593. [Google Scholar] [CrossRef] [Green Version]
- Giguere, S.; LaViolette, F.; Marchand, M.; Tremblay, D.; Moineau, S.; Liang, X.; Biron, É.; Corbeil, J. Machine Learning Assisted Design of Highly Active Peptides for Drug Discovery. PLoS Comput. Biol. 2015, 11, e1004074. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Wang, X.S.; Zhu, H.; Tropsha, A. Predictive QSAR Modeling: Methods and Applications in Drug Discovery and Chemical Risk Assessment. In Handbook of Computational Chemistry; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1309–1342. [Google Scholar] [CrossRef]
- Waghu, F.; Idicula-Thomas, S. Collection of antimicrobial peptides database and its derivatives: Applications and beyond. Protein Sci. 2019, 29, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. Ab Initio Design of Potent Anti-MRSA Peptides Based on Database Filtering Technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McErlean, E.M.; McCrudden, C.M.; McBride, J.W.; Cole, G.; Kett, V.L.; Robson, T.; Dunne, N.J.; McCarthy, H.O. Rational design and characterisation of an amphipathic cell penetrating peptide for non-viral gene delivery. Int. J. Pharm. 2021, 596, 120223. [Google Scholar] [CrossRef] [PubMed]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef]
- Porto, W.F.; Fensterseifer, I.C.; Ribeiro, S.; Franco, O.L. Joker: An algorithm to insert patterns into sequences for designing antimicrobial peptides. Biochim. Biophys. Acta—Gen. Subj. 2018, 1862, 2043–2052. [Google Scholar] [CrossRef]
- Pillong, M.; Hiss, J.A.; Schneider, P.; Lin, Y.-C.; Posselt, G.; Pfeiffer, B.; Blatter, M.; Müller, A.; Bachler, S.; Neuhaus, C.; et al. Rational Design of Membrane-Pore-Forming Peptides. Small 2017, 13, 1701316. [Google Scholar] [CrossRef]
- Rončević, T.; Vukičević, D.; Krce, L.; Benincasa, M.; Aviani, I.; Maravić, A.; Tossi, A. Selection and redesign for high selectivity of membrane-active antimicrobial peptides from a dedicated sequence/function database. Biochim. Biophys. Acta—Biomembr. 2019, 1861, 827–834. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Zaalishvili, G.; Karapetian, M.; Nasrashvili, T.; Kuljanishvili, N.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Grigolava, M.; et al. De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals 2019, 12, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, E.C.L.; Santana, K.; Josino, L.; Lima e Lima, A.H.L.; Júnior, C.D.S.D.S. Predicting cell-penetrating peptides using machine learning algorithms and navigating in their chemical space. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Gabernet, G.; Gautschi, D.; Müller, A.T.; Neuhaus, C.S.; Armbrecht, L.; Dittrich, P.S.; Hiss, J.A.; Schneider, G. In silico design and optimization of selective membranolytic anticancer peptides. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Porto, W.F.; Irazazabal, L.; Alves, E.S.F.; Ribeiro, S.M.; Matos, C.O.; Pires, Á.S.; Fensterseifer, I.C.M.; Miranda, V.J.; Haney, E.F.; Humblot, V.; et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, A.M.; Wold, S. New Chemical Descriptors Relevant for the Design of Biologically Active Peptides. A Multivariate Characterization of 87 Amino Acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef]
- Beltran, J.A.; Aguilera-Mendoza, L.; Brizuela, C.A. Optimal selection of molecular descriptors for antimicrobial peptides classification: An evolutionary feature weighting approach. BMC Genom. 2018, 19, 672. [Google Scholar] [CrossRef]
- Hedegaard, S.F.; Derbas, M.S.; Lind, T.K.; Kasimova, M.R.; Christensen, M.V.; Michaelsen, M.H.; Campbell, R.; Jorgensen, L.; Franzyk, H.; Cárdenas, M.; et al. Fluorophore labeling of a cell-penetrating peptide significantly alters the mode and degree of biomembrane interaction. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Röckendorf, N.; Borschbach, M.; Frey, A. Molecular Evolution of Peptide Ligands with Custom-Tailored Characteristics for Targeting of Glycostructures. PLoS Comput. Biol. 2012, 8, e1002800. [Google Scholar] [CrossRef]
- Maccari, G.; Di Luca, M.; Nifosì, R.; Cardarelli, F.; Signore, G.; Boccardi, C.; Bifone, A. Antimicrobial Peptides Design by Evolutionary Multiobjective Optimization. PLoS Comput. Biol. 2013, 9, e1003212. [Google Scholar] [CrossRef] [Green Version]
- Krause, T.; Röckendorf, N.; El-Sourani, N.; Ramaker, K.; Henkel, M.; Hauke, S.; Borschbach, M.; Frey, A. Breeding Cell Penetrating Peptides: Optimization of Cellular Uptake by a Function-Driven Evolutionary Process. Bioconjugate Chem. 2018, 29, 4020–4029. [Google Scholar] [CrossRef] [PubMed]
- Ramaker, K.; Henkel, M.; Krause, T.; Röckendorf, N.; Frey, A. Cell penetrating peptides: A comparative transport analysis for 474 sequence motifs. Drug Deliv. 2018, 25, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Kardani, K.; Bolhassani, A. Cppsite 2.0: An Available Database of Experimentally Validated Cell-Penetrating Peptides Predicting their Secondary and Tertiary Structures. J. Mol. Biol. 2020, 433, 166703. [Google Scholar] [CrossRef]
- Röckendorf, N.; Ramaker, K.; Frey, A. Artificial Evolutionary Optimization Process to Improve the Functionality of Cell Penetrating Peptides. Cell Penetrating Pept. 2021, 2383, 45–61. [Google Scholar] [CrossRef]
- Marks, J.R.; Placone, J.; Hristova, K.; Wimley, W.C. Spontaneous Membrane-Translocating Peptides by Orthogonal High-Throughput Screening. J. Am. Chem. Soc. 2011, 133, 8995–9004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauson, A.; He, J.; Wimley, A.W.; Hoffmann, A.R.; Wimley, W.C. Synthetic Molecular Evolution of Pore-Forming Peptides by Iterative Combinatorial Library Screening. ACS Chem. Biol. 2013, 8, 823–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-M.; Ren, J.; Tran, K.M.; Jeon, B.-M.; Park, W.-U.; Kim, H.; Lee, K.E.; Oh, Y.; Choi, M.; Kim, D.-S.; et al. Identification of efficient prokaryotic cell-penetrating peptides with applications in bacterial biotechnology. Commun. Biol. 2021, 4, 1–13. [Google Scholar] [CrossRef]
- Blondelle, S.E. Optimization and High-Throughput Screening of Antimicrobial Peptides. Curr. Pharm. Des. 2010, 16, 3204–3211. [Google Scholar] [CrossRef]
- Rathinakumar, R.; Walkenhorst, W.F.; Wimley, W.C. Broad-Spectrum Antimicrobial Peptides by Rational Combinatorial Design and High-Throughput Screening: The Importance of Interfacial Activity. J. Am. Chem. Soc. 2009, 131, 7609–7617. [Google Scholar] [CrossRef] [Green Version]
- Ashby, M.; Petkova, A.; Gani, J.; Mikut, R.; Hilpert, K. Use of Peptide Libraries for Identification and Optimization of Novel Antimicrobial Peptides. Curr. Top. Med. Chem. 2016, 17, 537–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, S.; Su, W.-C.; Budamagunta, M.; Xiao, W.; Ajena, Y.; Liu, R.; Voss, J.C.; Carney, R.P.; Parikh, A.N.; Lam, K.S. Discovery and mechanistic characterization of a structurally-unique membrane active peptide. Biochim. Biophys. Acta—Biomembr. 2020, 1862, 183394. [Google Scholar] [CrossRef] [PubMed]
- Carney, R.P.; Thillier, Y.; Kiss, Z.; Sahabi, A.; Campos, J.C.H.; Knudson, A.; Liu, R.; Olivos, D.; Saunders, M.; Tian, L.; et al. Combinatorial Library Screening with Liposomes for Discovery of Membrane Active Peptides. ACS Comb. Sci. 2017, 19, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albada, H.B.; Prochnow, P.; Bobersky, S.; Langklotz, S.; Bandow, J.E.; Metzler-Nolte, N. Short Antibacterial Peptides with Significantly Reduced Hemolytic Activity can be Identified by a Systematic l-to-d Exchange Scan of their Amino Acid Residues. ACS Comb. Sci. 2013, 15, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Afshar, S. In Vitro Assays: Friends or Foes of Cell-Penetrating Peptides. Int. J. Mol. Sci. 2020, 21, 4719. [Google Scholar] [CrossRef] [PubMed]
- Oddo, A.; Hansen, P.R. Hemolytic Activity of Antimicrobial Peptides. Antimicrob. Pept. 2016, 1548, 427–435. [Google Scholar] [CrossRef]
- Mercer, D.K.; Torres, M.; Duay, S.S.; Lovie, E.; Simpson, L.; Von Köckritz-Blickwede, M.; De La Fuente-Nunez, C.; O’Neil, D.A.; Angeles-Boza, A.M. Antimicrobial Susceptibility Testing of Antimicrobial Peptides to Better Predict Efficacy. Front. Cell. Infect. Microbiol. 2020, 10, 326. [Google Scholar] [CrossRef] [PubMed]
- López-Pérez, P.M.; Grimsey, E.; Bourne, L.; Mikut, R.; Hilpert, K. Screening and Optimizing Antimicrobial Peptides by Using SPOT-Synthesis. Front. Chem. 2017, 5, 25. [Google Scholar] [CrossRef]
- Anup, N.; Gadeval, A.; Rajpoot, K.; Tekade, R.K. Chapter 24—Software Used in ADME Computation. In Biopharmaceutics and Pharmacokinetics Considerations; Tekade, R.K., Ed.; Advances in Pharmaceutical Product Development and Research; Academic Press: Cambridge, MA, USA, 2021; Volume 1, pp. 699–708. ISBN 978-0-12-814425-1. [Google Scholar]
- Bhhatarai, B.; Walters, W.P.; Hop, C.E.C.A.; Lanza, G.; Ekins, S. Opportunities and challenges using artificial intelligence in ADME/Tox. Nat. Mater. 2019, 18, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Mele, R.; Ingenito, R.; Bianchi, E.; Bonelli, F.; Monteagudo, E.; Orsatti, L. An efficient liquid chromatography-high resolution mass spectrometry approach for the optimization of the metabolic stability of therapeutic peptides. Anal. Bioanal. Chem. 2017, 409, 2685–2696. [Google Scholar] [CrossRef]
- Böttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef] [PubMed]
- Gorris, H.H.; Bade, S.; Röckendorf, N.; Albers, E.; Schmidt, M.A.; Fránek, M.; Frey, A. Rapid Profiling of Peptide Stability in Proteolytic Environments. Anal. Chem. 2009, 81, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Craik, D.J. Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat. Chem. Biol. 2018, 14, 417–427. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Database | Number of Entries | Content | Hyperlink |
|---|---|---|---|
| LAMP2 | 23,253 | AMPs, structure, collection, composition, source, function | http://biotechlab.fudan.edu.cn/database/lamp/index.php |
| DRAMP | 22,259 | AMPs, structure, activity physicochemical-, patent-, clinical data | http://dramp.cpu-bioinfor.org/ |
| DBAASPv3.0 | 17,865 | AMPs, structure, activity | https://dbaasp.org/ |
| CAMPR3 | 8164 | AMPs, Structure, patents, signatures | http://www.camp.bicnirrh.res.in/ |
| Cybase | 4012 | Cyclic proteins, antiviral, insecticidal, antibacterial | http://www.cybase.org.au/ |
| APD3 | 3324 | AMPs, structure, activity | https://aps.unmc.edu/ |
| DAPD | 2571 | Structure, activity, host taxonomy | http://split4.pmfst.hr/dadp/? |
| YADAMP | 2525 | Structure, activity | http://yadamp.unisa.it/default.aspx |
| DAMPD | 1232 | taxonomy, species, AMP family, citations | http://apps.sanbi.ac.za/dampd/ |
| AntiTbPdb | 1010 | Anti-mycobacterial peptides, structure, activity | https://webs.iiitd.edu.in/raghava/antitbpdb/ |
| InverPep | 702 | Invertebrate AMPs, structure, activity, target | https://ciencias.medellin.unal.edu.co/gruposdeinvestigacion/prospeccionydisenobiomoleculas/InverPep/public/home_en |
| ANTISTAPYBASE | 596 | AMPs, structure, activity against MRSA | https://www.antistaphybase.com/ |
| Defensins | 363 | Structure, activity | http://defensins.bii.a-star.edu.sg/ |
| Peptaibols | 317 | Fungal AMPs, non-standard amino acids | http://peptaibol.cryst.bbk.ac.uk/home.shtml |
| PhytAMP | 273 | Plant AMPs | http://phytamp.hammamilab.org/main.php |
| BACTIBASE | 230 | Bacteriocins, structure, function | http://bactibase.hammamilab.org/about.php |
| BaAMPs | 221 | Biofilm active AMPs | http://www.baamps.it/ |
| THIOBASE | 39 | Thiopeptides, structure, activity | https://bioinfo-mml.sjtu.edu.cn/THIOBASE/index.php |
| EnzyBase | N/A | Encybiotics, lysins, lysocymes, bacteriocins | http://biotechlab.fudan.edu.cn/database/EnzyBase/home.php |
| MBPDB | N/A | Milk bioactive peptides, function, species | http://mbpdb.nws.oregonstate.edu/ |
| Name 1 | Conditions | Description |
|---|---|---|
| TAPS-18 | Periodontitis | Cathelicidin based synthetic peptide |
| LEAP2 | Type 2 Diabetes | 39-mer synthetic liver expressed amp |
| LEAP2 | Obesity | 39-mer synthetic liver expressed AMP |
| MSI78 | Diabetic foot infection | broad-spectrum synthetic analogue of magainin |
| LTX109 | Skin Infection, MRSA Inf. | Synthetic peptidomimetic |
| LL37 | Melanoma | Cathelicidin based |
| hlf1-11 | Bacterial Infections, Mycoses | Lactoferrin derived |
| PLG0206 | Joint Infection | Engineered AMP |
| Pxl01 | Surgical adhesions | Lactoferrin derived |
| IB367 | Pneumonia, mucositis | synthetic analogue of Protegrin I |
| Pac113 | Oral Candidiasis | Histatin derived |
| MX-594AN | catheter-related acne | Indolicidin based |
| rBPI21 | meningococcaemia | human bactericidal permeability protein derivative |
| ETD151 | fungal infections | 44 mer variant from lepidopteran Heliothis virescens |
| HB-50 | anti-infective | synthetic natural peptide mimetic of cecropin |
| HB-1345 | broad-spectrum antibiotic | Synthetic Lipohexapeptide |
| CZEN-002 | vulvovaginal candidiasis | synthetic 8-mer from α-melanocyte-stimulating hormone |
| PTX005 | antimicrobial | Synthetic 12 mer |
| Glutoxim | Tuberculosis, NSCL cancer | thiopoietin |
| IMX942 | Nosocomial infections | Synthetic cationic host defense peptide |
| NP213 | Fungal infections | cyclic cationic peptide from NovaBiotics arginine peptide platform |
| OP-145 | Chronic bacterial middle ear infection | Synthetic 24-mer peptide derived from LL-37 |
| CD-NP | Organ failure | Synthetic chimeric 37-mer |
| C16G2 | Treatment of dental subjects | synthetic AMP |
| Sifuvirtide | HIV fusion inhibitor; AIDS | designed based on the 3D structure of the HIV-1 gp41 |
| POL7080 | nosocomial pneumonia | synthetic by amino acid substitution of protegrin I |
| Omiganan | atopic dermatitis, rosacea | Synthetic 12-mer cationic peptide derived from indolicidin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Röckendorf, N.; Nehls, C.; Gutsmann, T. Design of Membrane Active Peptides Considering Multi-Objective Optimization for Biomedical Application. Membranes 2022, 12, 180. https://doi.org/10.3390/membranes12020180
Röckendorf N, Nehls C, Gutsmann T. Design of Membrane Active Peptides Considering Multi-Objective Optimization for Biomedical Application. Membranes. 2022; 12(2):180. https://doi.org/10.3390/membranes12020180
Chicago/Turabian StyleRöckendorf, Niels, Christian Nehls, and Thomas Gutsmann. 2022. "Design of Membrane Active Peptides Considering Multi-Objective Optimization for Biomedical Application" Membranes 12, no. 2: 180. https://doi.org/10.3390/membranes12020180