
Reticulocyte Maturation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Overview
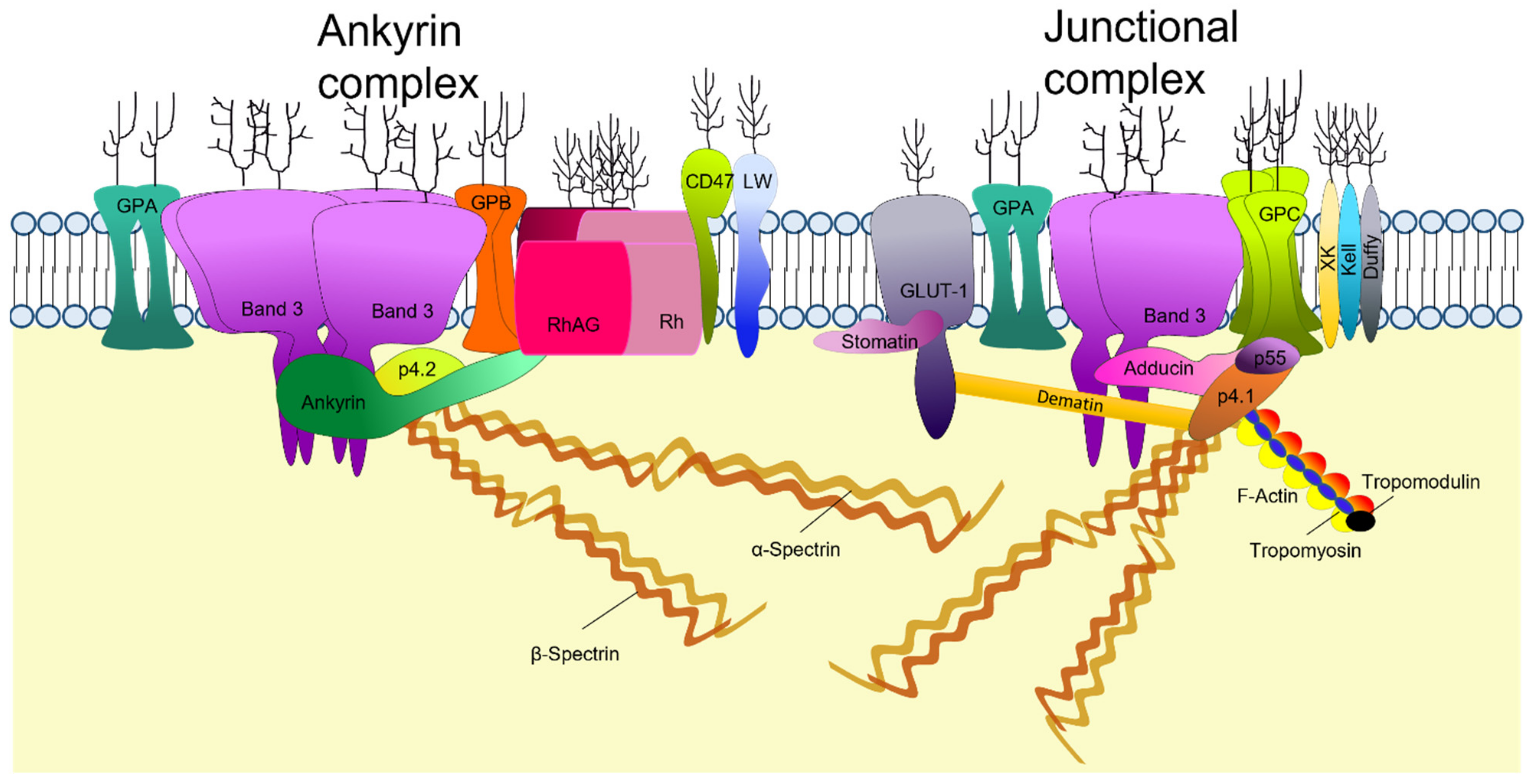
2. Structure of the Mature RBC Membrane
2.1. The Lipid Bilayer
2.2. RBC Membrane Proteins
2.3. Membrane Cytoskeleton
3. Erythropoiesis
3.1. Membrane Protein Changes and Assembly during Terminal Erythroblast Differentiation
3.2. Protein Rearrangement during Erythroblast Enucleation
4. Mechanisms of Reticulocyte Maturation
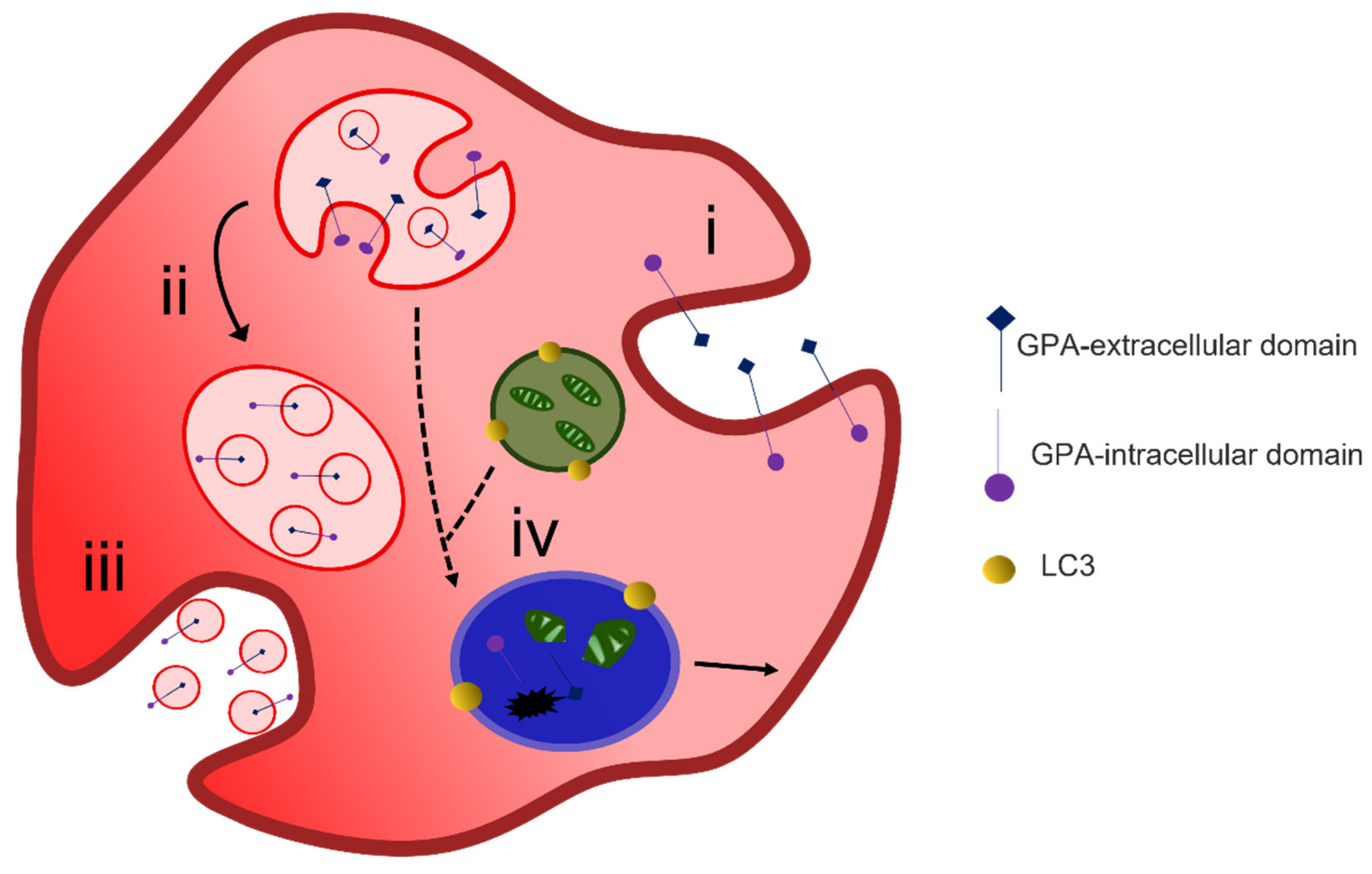
4.1. Protein Removal through Exosome Release
4.2. Alternative Methods of Reticulocyte Maturation
4.3. Organelle Clearance
5. Mature Red Blood Cell versus Immature Red Blood Cell Membrane
6. Red Cell Membrane Variants
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ballas, S.K.; Krasnow, S.H. Structure of erythrocyte membrane and its transport functions. Ann. Clin. Lab. Sci. 1980, 10, 209–219. [Google Scholar] [PubMed]
- Mohandas, N.; Clark, M.R.; Jacobs, M.S.; Shohet, S.B. Analysis of factors regulating erythrocyte deformability. J. Clin. Investig. 1980, 66, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Mel, H.C.; Prenant, M.; Mohandas, N. Reticulocyte motility and form: Studies on maturation and classification. Blood 1977, 49, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Waugh, R.E.; McKenney, J.; Bauserman, R.G.; Brooks, D.M.; Valeri, C.R. Surface Area and Volume Changes During Maturation of Reticulocytes in the Circulation of the Baboon. J. Lab. Clin. Med. 1997, 129, 527–535. [Google Scholar] [CrossRef]
- Wickrema, A.; Dai, C.-H.; Krantz, S.B.; Koury, S.T. Changes in cytoskeletal proteins and their mRNAs during maturation of human erythroid progenitor cells. J. Cell. Physiol. 1994, 160, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Guo, X.; Mohandas, N.; Chasis, J.A.; An, X. Membrane remodeling during reticulocyte maturation. Blood 2010, 115, 2021–2027. [Google Scholar] [CrossRef] [PubMed]
- Diez-Silva, M.; Dao, M.; Han, J.; Lim, C.-T.; Suresh, S. Shape and Biomechanical Characteristics of Human Red Blood Cells in Health and Disease. MRS Bull. 2010, 35, 382–388. [Google Scholar] [CrossRef]
- Virtanen, J.A.; Cheng, K.; Somerharju, P. Phospholipid composition of the mammalian red cell membrane can be rationalized by a superlattice model. Proc. Natl. Acad. Sci. USA 1998, 95, 4964–4969. [Google Scholar] [CrossRef]
- Lorent, J.H.; Levental, K.R.; Ganesan, L.; Rivera-Longsworth, G.; Sezgin, E.; Doktorova, M.D.; Lyman, E.; Levental, I. Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat. Chem. Biol. 2020, 16, 644–652. [Google Scholar] [CrossRef]
- Daleke, D.L. Regulation of transbilayer plasma membrane phospholipid asymmetry. J. Lipid Res. 2003, 44, 233–242. [Google Scholar] [CrossRef]
- Manno, S.; Takakuwa, Y.; Mohandas, N. Identification of a functional role for lipid asymmetry in biological membranes: Phosphatidylserine-skeletal protein interactions modulate membrane stability. Proc. Natl. Acad. Sci. USA 2002, 99, 1943–1948. [Google Scholar] [CrossRef]
- An, X.; Guo, X.; Gratzer, W.; Mohandas, N. Phospholipid binding by proteins of the spectrin family: A comparative study. Biochem. Biophys. Res. Commun. 2005, 327, 794–800. [Google Scholar] [CrossRef]
- Faucherre, A.; Kissa, K.; Nargeot, J.; Mangoni, M.; Jopling, C. Piezo1 plays a role in erythrocyte volume homeostasis. Haematologica 2014, 99, 70–75. [Google Scholar] [CrossRef]
- Wesseling, M.C.; Wagner-Britz, L.; Huppert, H.; Hanf, B.; Hertz, L.; Nguyen, D.B.; Bernhardt, I. Phosphatidylserine Exposure in Human Red Blood Cells Depending on Cell Age. Cell. Physiol. Biochem. 2016, 38, 1376–1390. [Google Scholar] [CrossRef]
- Mankelow, T.J.; Griffiths, R.E.; Trompeter, S.; Flatt, J.F.; Cogan, N.M.; Massey, E.J.; Anstee, D.J. Autophagic vesicles on mature human reticulocytes explain phosphatidylserine-positive red cells in sickle cell disease. Blood 2015, 126, 1831–1834. [Google Scholar] [CrossRef]
- Sherman, I.W.; Prudhomme, J.; Tait, J.F. Altered membrane phospholipid asymmetry in plasmodium falciparum-infected erythrocytes. Parasitol. Today 1997, 13, 242–243. [Google Scholar] [CrossRef]
- Bütikofer, P.; Kuypers, F.A.; Xu, C.M.; Chiu, D.T.; Lubin, B. Enrichment of two glycosyl-phosphatidylinositol-anchored proteins, acetylcholinesterase and decay accelerating factor, in vesicles released from human red blood cells. Blood 1989, 74, 1481–1485. [Google Scholar] [CrossRef]
- Pasini, E.M.; Kirkegaard, M.; Mortensen, P.; Lutz, H.U.; Thomas, A.W.; Mann, M. In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood 2006, 108, 791–801. [Google Scholar] [CrossRef]
- Ravenhill, B.J.; Kanjee, U.; Ahouidi, A.; Nobre, L.; Williamson, J.; Goldberg, J.M.; Antrobus, R.; Dieye, T.; Duraisingh, M.T.; Weekes, M.P. Quantitative comparative analysis of human erythrocyte surface proteins between individuals from two genetically distinct populations. Commun. Biol. 2019, 2, 350. [Google Scholar] [CrossRef]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef]
- Rybicki, A.C.; Schwartz, R.S.; Hustedt, E.J.; Cobb, C.E. Increased rotational mobility and extractability of band 3 from protein 4.2-deficient erythrocyte membranes: Evidence of a role for protein 4.2 in strengthening the band 3-cytoskeleton linkage. Blood 1996, 88, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.L.; Alloisio, N.; Almeida, H.; Gomes, C.; Texier, P.; Lemos, C.; Mimoso, G.; Morlé, L.; Bey-Cabet, F.; Rudigoz, R.C.; et al. Severe hereditary spherocytosis and distal renal tubular acidosis associated with the total absence of band 3. Blood 2000, 96, 1602–1604. [Google Scholar] [PubMed]
- Bruce, L.; Beckmann, R.; Ribeiro, M.L.; Peters, L.; Chasis, J.A.; Delaunay, J.; Mohandas, N.; Anstee, D.J.; Tanner, M.J. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood 2003, 101, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, E.; Satchwell, T.J.; Williamson, R.C.; Toye, A.M. Band 3 multiprotein complexes in the red cell membrane; of mice and men. Blood Cells Mol. Dis. 2010, 45, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Burton, N.M.; Bruce, L.J. Modelling the structure of the red cell membrane. Biochem. Cell Biol. 2011, 89, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Lux, S.E. Anatomy of the red cell membrane skeleton: Unanswered questions. Blood 2016, 127, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Matsuoka, Y.; Bennett, V. Adducin Preferentially Recruits Spectrin to the Fast Growing Ends of Actin Filaments in a Complex Requiring the MARCKS-related Domain and a Newly Defined Oligomerization Domain. J. Biol. Chem. 1998, 273, 19329–19338. [Google Scholar] [CrossRef]
- Liu, S.C.; Derick, L.H.; Palek, J. Visualization of the hexagonal lattice in the erythrocyte membrane skeleton. J. Cell Biol. 1987, 104, 527–536. [Google Scholar] [CrossRef]
- Byers, T.J.; Branton, D. Visualization of the protein associations in the erythrocyte membrane skeleton. Proc. Natl. Acad. Sci. USA 1985, 82, 6153–6157. [Google Scholar] [CrossRef]
- Ciana, A.; Achilli, C.; Minetti, G. Spectrin and Other Membrane-Skeletal Components in Human Red Blood Cells of Different Age. Cell. Physiol. Biochem. 2017, 42, 1139–1152. [Google Scholar] [CrossRef]
- Stabach, P.R.; Simonović, I.; Ranieri, M.A.; Aboodi, M.S.; Steitz, T.A.; Simonović, M.; Morrow, J.S. The structure of the ankyrin-binding site of beta-spectrin reveals how tandem spectrin-repeats generate unique ligand-binding properties. Blood 2009, 113, 5377–5384. [Google Scholar] [CrossRef]
- Gimm, J.A.; An, X.; Nunomura, W.; Mohandas, N. Functional Characterization of Spectrin-Actin-Binding Domains in 4.1 Family of Proteins. Biochemistry 2002, 41, 7275–7282. [Google Scholar] [CrossRef]
- McGough, A.M.; Josephs, R. On the structure of erythrocyte spectrin in partially expanded membrane skeletons. Proc. Natl. Acad. Sci. USA 1990, 87, 5208–5212. [Google Scholar] [CrossRef]
- Alaarg, A.; Schiffelers, R.; van Solinge, W.W.; Van Wijk, R. Red blood cell vesiculation in hereditary hemolytic anemia. Front. Physiol. 2013, 4, 365. [Google Scholar] [CrossRef]
- Pasternack, G.R.; Anderson, R.A.; Leto, T.L.; Marchesi, V.T. Interactions between protein 4.1 and band 3. An alternative binding site for an element of the membrane skeleton. J. Biol. Chem. 1985, 260, 3676–3683. [Google Scholar] [CrossRef]
- An, X.; Lecomte, M.C.; Chasis, J.A.; Mohandas, N.; Gratzer, W.; Kumagai, J.; Hsu, S.Y.; Matsumi, H.; Roh, J.-S.; Fu, P.; et al. Shear-Response of the Spectrin Dimer-Tetramer Equilibrium in the Red Blood Cell Membrane. J. Biol. Chem. 2002, 277, 31796–31800. [Google Scholar] [CrossRef]
- Shen, B.W.; Josephs, R.; Steck, T.L. Ultrastructure of the intact skeleton of the human erythrocyte membrane. J. Cell Biol. 1986, 102, 997–1006. [Google Scholar] [CrossRef]
- Ohanian, V.; Wolfe, L.C.; John, K.M.; Pinder, J.C.; Lux, S.E.; Gratzer, W.B. Analysis of the ternary interaction of the red cell membrane skeletal proteins, spectrin, actin, and 4.1. Biochemistry 1984, 23, 4416–4420. [Google Scholar] [CrossRef]
- Valent, P.; Büsche, G.; Theurl, I.; Uras, I.Z.; Germing, U.; Stauder, R.; Sotlar, K.; Füreder, W.; Bettelheim, P.; Pfeilstöcker, M.; et al. Normal and pathological erythropoiesis in adults: From gene regulation to targeted treatment concepts. Haematologica 2018, 103, 1593–1603. [Google Scholar] [CrossRef]
- Barminko, J.; Reinholt, B.; Baron, M.H. Development and differentiation of the erythroid lineage in mammals. Dev. Comp. Immunol. 2016, 58, 18–29. [Google Scholar] [CrossRef]
- Nandakumar, S.; Ulirsch, J.; Sankaran, V.G. Advances in understanding erythropoiesis: Evolving perspectives. Br. J. Haematol. 2016, 173, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Klei, T.R.L.; Meinderts, S.M.; van den Berg, T.K.; Van Bruggen, R. From the Cradle to the Grave: The Role of Macrophages in Erythropoiesis and Erythrophagocytosis. Front. Immunol. 2017, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Zivot, A.; Lipton, J.M.; Narla, A.; Blanc, L. Erythropoiesis: Insights into pathophysiology and treatments in 2017. Mol. Med. 2018, 24, 11. [Google Scholar] [CrossRef] [PubMed]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From stem cell to red cell: Regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef] [PubMed]
- Dzierzak, E.; Philipsen, S. Erythropoiesis: Development and Differentiation. Cold Spring Harb. Perspect. Med. 2013, 3, a011601. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Huang, X.; Cheng, L. Concise Review: Stem Cell-Based Approaches to Red Blood Cell Production for Transfusion. STEM CELLS Transl. Med. 2014, 3, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Langer, P.J.; Lodish, H.F. Asynchronous synthesis of erythrocyte membrane proteins. Proc. Natl. Acad. Sci. USA 1976, 73, 3206–3210. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.; White, R.A.; Birkenmeier, C.S.; Bloom, M.L.; Lux, S.E.; Barker, J.E. Changing patterns in cytoskeletal mRNA expression and protein synthesis during murine erythropoiesis in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 5749–5753. [Google Scholar] [CrossRef]
- Lodish, H.F. Biosynthesis of Reticulocyte Membrane Proteins by Membrane-Free Polyribosomes. Proc. Natl. Acad. Sci. USA 1973, 70, 1526–1530. [Google Scholar] [CrossRef]
- Koury, M.J.; Sawyer, S.T.; Bondurant, M.C. Splenic erythroblasts in anemia-inducing friend disease: A source of cells for studies of erythropoietin-mediated differentiation. J. Cell. Physiol. 1984, 121, 526–532. [Google Scholar] [CrossRef]
- Hanspal, M.; Palek, J. Synthesis and assembly of membrane skeletal proteins in mammalian red cell precursors. J. Cell Biol. 1987, 105, 1417–1424. [Google Scholar] [CrossRef]
- Southcott, M.J.; Tanner, M.J.; Anstee, D.J. The expression of human blood group antigens during erythropoiesis in a cell culture system. Blood 1999, 93, 4425–4435. [Google Scholar] [CrossRef]
- Bony, V.; Gane, P.; Bailly, P.; Cartron, J.-P. Time-course expression of polypeptides carrying blood group antigens during human erythroid differentiation. Br. J. Haematol. 1999, 107, 263–274. [Google Scholar] [CrossRef]
- Montel-Hagen, A.; Kinet, S.; Manel, N.; Mongellaz, C.; Prohaska, R.; Battini, J.-L.; Delaunay, J.; Sitbon, M.; Taylor, N. Erythrocyte Glut1 Triggers Dehydroascorbic Acid Uptake in Mammals Unable to Synthesize Vitamin C. Cell 2008, 132, 1039–1048. [Google Scholar] [CrossRef]
- Koury, M.J.; Bondurant, M.C.; Atkinson, J.B. Erythropoietin control of terminal erythroid differentiation: Maintenance of cell viability, production of hemoglobin, and development of the erythrocyte membrane. Blood Cells 1987, 13, 217–226. [Google Scholar]
- Satchwell, T.J.; Bell, A.J.; Pellegrin, S.; Kupzig, S.; Ridgwell, K.; Daniels, G.; Anstee, D.J.; Akker, E.V.D.; Toye, A.M. Critical band 3 multiprotein complex interactions establish early during human erythropoiesis. Blood 2011, 118, 182–191. [Google Scholar] [CrossRef][Green Version]
- Ghosh, S.; Cox, K.H.; Cox, J.V. Chicken Erythroid AE1 Anion Exchangers Associate with the Cytoskeleton During Recycling to the Golgi. Mol. Biol. Cell 1999, 10, 455–469. [Google Scholar] [CrossRef]
- Gomez, S.; Morgans, C. Interaction between band 3 and ankyrin begins in early compartments of the secretory pathway and is essential for band 3 processing. J. Biol. Chem. 1993, 268, 19593–19597. [Google Scholar] [CrossRef]
- Thomson-Luque, R.; Wang, C.; Ntumngia, F.B.; Xu, S.; Szekeres, K.; Conway, A.; Adapa, S.R.; Barnes, S.J.; Adams, J.H.; Jiang, R.H. In-depth phenotypic characterization of reticulocyte maturation using mass cytometry. Blood Cells Mol. Dis. 2018, 72, 22–33. [Google Scholar] [CrossRef]
- Hu, J.; Liu, J.; Xue, F.; Halverson, G.; Reid, M.; Guo, A.; Chen, L.; Raza, A.; Galili, N.; Jaffray, J.; et al. Isolation and functional characterization of human erythroblasts at distinct stages: Implications for understanding of normal and disordered erythropoiesis in vivo. Blood 2013, 121, 3246–3253. [Google Scholar] [CrossRef]
- Wong, P.; Hattangadi, S.M.; Cheng, A.W.; Frampton, G.M.; Young, R.A.; Lodish, H.F. Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood 2011, 118, e128–e138. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Murata-Hori, M.; Lodish, H.F. Formation of mammalian erythrocytes: Chromatin condensation and enucleation. Trends Cell Biol. 2011, 21, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Sangiorgi, F.; Woods, C.; Lazarides, E. Vimentin downregulation is an inherent feature of murine erythropoiesis and occurs independently of lineage. Development 1990, 110, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Granger, B.; Repasky, E.A.; Lazarides, E. Synemin and vimentin are components of intermediate filaments in avian erythrocytes. J. Cell Biol. 1982, 92, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Skutelsky, E.; Danon, D. Comparative study of nuclear expulsion from the late erythroblast and cytokinesis. Exp. Cell Res. 1970, 60, 427–436. [Google Scholar] [CrossRef]
- Konstantinidis, D.G.; Pushkaran, S.; Johnson, J.F.; Cancelas, J.A.; Manganaris, S.; Harris, C.E.; Williams, D.A.; Zheng, Y.; Kalfa, T.A. Signaling and cytoskeletal requirements in erythroblast enucleation. Blood 2012, 119, 6118–6127. [Google Scholar] [CrossRef] [PubMed]
- Koury, S.T.; Koury, M.J.; Bondurant, M.C. Cytoskeletal distribution and function during the maturation and enucleation of mammalian erythroblasts. J. Cell Biol. 1989, 109, 3005–3013. [Google Scholar] [CrossRef]
- Ji, P.; Jayapal, S.R.; Lodish, H.F. Enucleation of cultured mouse fetal erythroblasts requires Rac GTPases and mDia2. Nat. Cell Biol. 2008, 10, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.-M.; Gimm, J.A.; Lo, A.J.; Koury, M.J.; Krauss, S.W.; Mohandas, N.; Chasis, J.A. Mechanism of protein sorting during erythroblast enucleation: Role of cytoskeletal connectivity. Blood 2004, 103, 1912–1919. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keerthivasan, G.; Small, S.; Liu, H.; Wickrema, A.; Crispino, J.D. Vesicle trafficking plays a novel role in erythroblast enucleation. Blood 2010, 116, 3331–3340. [Google Scholar] [CrossRef]
- Geiduschek, J.B.; Singer, S. Molecular changes in the membranes of mouse erythroid cells accompanying differentiation. Cell 1979, 16, 149–163. [Google Scholar] [CrossRef]
- Chasis, J.A.; Coulombel, L.; Conboy, J.; McGee, S.; Andrews, K.; Kan, Y.W.; Mohandas, N. Differentiation-associated switches in protein 4.1 expression. Synthesis of multiple structural isoforms during normal human erythropoiesis. J. Clin. Investig. 1993, 91, 329–338. [Google Scholar] [CrossRef]
- Bell, A.J.; Satchwell, T.J.; Heesom, K.J.; Hawley, B.R.; Kupzig, S.; Hazell, M.; Mushens, R.; Herman, A.; Toye, A.M. Protein Distribution during Human Erythroblast Enucleation In Vitro. PLoS ONE 2013, 8, e60300. [Google Scholar] [CrossRef]
- Ji, P.; Lodish, H.F. Ankyrin and band 3 differentially affect expression of membrane glycoproteins but are not required for erythroblast enucleation. Biochem. Biophys. Res. Commun. 2012, 417, 1188–1192. [Google Scholar] [CrossRef]
- Keerthivasan, G.; Liu, H.; Gump, J.M.; Dowdy, S.F.; Wickrema, A.; Crispino, J.D. A novel role for survivin in erythroblast enucleation. Haematologica 2012, 97, 1471–1479. [Google Scholar] [CrossRef]
- Gurbuxani, S.; Xu, Y.; Keerthivasan, G.; Wickrema, A.; Crispino, J.D. Differential requirements for survivin in hematopoietic cell development. Proc. Natl. Acad. Sci. USA 2005, 102, 11480–11485. [Google Scholar] [CrossRef]
- Iacopetta, B.J.; Morgan, E.H.; Yeoh, G.C. Receptor-mediated endocytosis of transferrin by developing erythroid cells from the fetal rat liver. J. Histochem. Cytochem. 1983, 31, 336–344. [Google Scholar] [CrossRef]
- Aoto, M.; Iwashita, A.; Mita, K.; Ohkubo, N.; Tsujimoto, Y.; Mitsuda, N. Transferrin receptor 1 is required for enucleation of mouse erythroblasts during terminal differentiation. FEBS Openbio 2019, 9, 291–303. [Google Scholar] [CrossRef]
- Pellegrin, S.; Severn, C.E.; Toye, A.M. Towards manufactured red blood cells for the treatment of inherited anemia. Haematologica 2021, 106, 2304–2311. [Google Scholar] [CrossRef]
- Ney, P.A. Normal and disordered reticulocyte maturation. Curr. Opin. Hematol. 2011, 18, 152–157. [Google Scholar] [CrossRef]
- Vidal, M.; Mangeat, P.; Hoekstra, D. Aggregation reroutes molecules from a recycling to a vesicle-mediated secretion pathway during reticulocyte maturation. J. Cell Sci. 1997, 110, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Koury, M.J.; Koury, S.T.; Kopsombut, P.; Bondurant, M.C. In vitro maturation of nascent reticulocytes to erythrocytes. Blood 2005, 105, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Ovchynnikova, E.; Aglialoro, F.; von Lindern, M.; Van Den Akker, E. The Shape Shifting Story of Reticulocyte Maturation. Front. Physiol. 2018, 9, 829. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, H.S.; Hwang, J.H.; Hoh, J.K.; Cho, Y.-H.; Baek, E.J. The RNA in reticulocytes is not just debris: It is necessary for the final stages of erythrocyte formation. Blood Cells Mol. Dis. 2014, 53, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Bianchini, A.; Teng, K. Reticulocyte maturation and exosome release: Transferrin receptor containing exosomes shows multiple plasma membrane functions. Blood 1989, 74, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.-T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Díaz-Varela, M.; Menezes-Neto, A.; Perez-Zsolt, D.; Gámez-Valero, A.; Barber, J.S.; Izquierdo-Useros, N.; Martinez-Picado, J.; Fernández-Becerra, C.; Del Portillo, H.A. Proteomics study of human cord blood reticulocyte-derived exosomes. Sci. Rep. 2018, 8, 14046. [Google Scholar] [CrossRef] [PubMed]
- Carayon, K.; Chaoui, K.; Ronzier, E.; Lazar, I.; Bertrand-Michel, J.; Roques, V.; Balor, S.; Terce, F.; Lopez, A.; Salomé, L.; et al. Proteolipidic Composition of Exosomes Changes during Reticulocyte Maturation. J. Biol. Chem. 2011, 286, 34426–34439. [Google Scholar] [CrossRef] [PubMed]
- Géminard, C.; de Gassart, A.; Blanc, L.; Vidal, M. Degradation of AP2 During Reticulocyte Maturation Enhances Binding of Hsc70 and Alix to a Common Site on TfR for Sorting into Exosomes. Traffic 2004, 5, 181–193. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Barrès, C.; Blanc, L.; Bette-Bobillo, P.; André, S.; Mamoun, R.; Gabius, H.-J.; Vidal, M. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood 2010, 115, 696–705. [Google Scholar] [CrossRef]
- Griffiths, R.E.; Kupzig, S.; Cogan, N.; Mankelow, T.J.; Betin, V.M.S.; Trakarnsanga, K.; Massey, E.J.; Lane, J.D.; Parsons, S.F.; Anstee, D.J. Maturing reticulocytes internalize plasma membrane in glycophorin A—Containing vesicles that fuse with autophagosomes before exocytosis. Blood 2012, 119, 6296–6306. [Google Scholar] [CrossRef]
- Mankelow, T.; Griffiths, R.E.; Trompeter, S.; Flatt, J.F.; Cogan, N.M.; Massey, E.J.; Anstee, D.J. The ins and outs of reticulocyte maturation revisited: The role of autophagy in sickle cell disease. Autophagy 2016, 12, 590–591. [Google Scholar] [CrossRef][Green Version]
- Griffiths, R.E.; Kupzig, S.; Cogan, N.; Mankelow, T.J.; Betin, V.M.; Trakarnsanga, K.; Massey, E.J.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. The ins and outs of human reticulocyte maturation. Autophagy 2012, 8, 1150–1151. [Google Scholar] [CrossRef]
- Moura, P.; Hawley, B.R.; Mankelow, T.; Griffiths, R.E.; Dobbe, J.; Streekstra, G.J.; Anstee, D.J.; Satchwell, T.J.; Toye, A. Non-muscle myosin II drives vesicle loss during human reticulocyte maturation. Haematologica 2018, 103, 1997–2007. [Google Scholar] [CrossRef]
- Minetti, G.; Bernecker, C.; Dorn, I.; Achilli, C.; Bernuzzi, S.; Perotti, C.; Ciana, A. Membrane Rearrangements in the Maturation of Circulating Human Reticulocytes. Front. Physiol. 2020, 11, 215. [Google Scholar] [CrossRef]
- De Gassart, A.; Géminard, C.; Février, B.; Raposo, G.; Vidal, M. Lipid raft-associated protein sorting in exosomes. Blood 2003, 102, 4336–4344. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, K.; Xiao, X.; Liao, J.; Hu, Q.; Chen, H.; Liu, J.; An, X. Autophagy as a Regulatory Component of Erythropoiesis. Int. J. Mol. Sci. 2015, 16, 4083–4094. [Google Scholar] [CrossRef]
- Honda, S.; Arakawa, S.; Nishida, Y.; Yamaguchi, H.; Ishii, E.; Shimizu, S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat. Commun. 2014, 5, 4004. [Google Scholar] [CrossRef]
- Hammerling, B.C.; Najor, R.H.; Cortez, M.Q.; Shires, S.E.; Leon, L.J.; Gonzalez, E.R.; Boassa, D.; Phan, S.; Thor, A.; Jimenez, R.E.; et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat. Commun. 2017, 8, 14050. [Google Scholar] [CrossRef] [PubMed]
- Grosso, R.; Fader, C.M.; Colombo, M.I. Autophagy: A necessary event during erythropoiesis. Blood Rev. 2017, 31, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.; Colombo, M. Multivesicular Bodies and Autophagy in Erythrocyte Maturation. Autophagy 2006, 2, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Betin, V.M.; Singleton, B.K.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. Autophagy facilitates organelle clearance during differentiation of human erythroblasts: Evidence for a role for ATG4 paralogs during autophagosome maturation. Autophagy 2013, 9, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, V.; Géminard, C.; Reggio, H.; Vidal, M.; Sainte-Marie, J. Fractionation analysis of the endosomal compartment during rat reticulocyte maturation. Cell Biol. Int. 2002, 26, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.-F.; LeDuc, M.; Cochet, S.; Bailly, K.; Lacombe, C.; Mohandas, N.; Guillonneau, F.; El Nemer, W.; Mayeux, P. Absolute proteome quantification of highly purified populations of circulating reticulocytes and mature erythrocytes. Blood Adv. 2018, 2, 2646–2657. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.T.T.; Sinha, A.; Malleret, B.; Suwanarusk, R.; Park, J.E.; Naidu, R.; Das, R.; Dutta, B.; Ong, S.T.; Verma, N.K.; et al. Quantitative mass spectrometry of human reticulocytes reveal proteome-wide modifications during maturation. Br. J. Haematol. 2018, 180, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Trakarnsanga, K.; Heesom, K.J.; Cogan, N.; Green, C.; Toye, A.M.; Parsons, S.F.; Anstee, D.J.; Frayne, J. Comparison of the Proteome of Adult and Cord Erythroid Cells, and Changes in the Proteome Following Reticulocyte Maturation. Mol. Cell Proteom. 2016, 15, 1938–1946. [Google Scholar] [CrossRef]
- Knowles, D.W.; Tilley, L.; Mohandas, N.; Chasis, J.A. Erythrocyte membrane vesiculation: Model for the molecular mechanism of protein sorting. Proc. Natl. Acad. Sci. USA 1997, 94, 12969–12974. [Google Scholar] [CrossRef]
- Ciana, A.; Achilli, C.; Balduini, C.; Minetti, G. On the association of lipid rafts to the spectrin skeleton in human erythrocytes. Biochim. Biophys. Acta (BBA)-Biomembr. 2011, 1808, 183–190. [Google Scholar] [CrossRef][Green Version]
- Chasis, J.A.; Prenant, M.; Leung, A.; Mohandas, N. Membrane assembly and remodeling during reticulocyte maturation. Blood 1989, 74, 1112–1120. [Google Scholar] [CrossRef]
- Malleret, B.; Xu, F.; Mohandas, N.; Suwanarusk, R.; Chu, C.; Leite, J.A.; Low, K.; Turner, C.; Sriprawat, K.; Zhang, R.; et al. Significant Biochemical, Biophysical and Metabolic Diversity in Circulating Human Cord Blood Reticulocytes. PLoS ONE 2013, 8, e76062. [Google Scholar] [CrossRef]
- Li, H.; Lu, L.; Li, X.; Buffet, P.A.; Dao, M.; Karniadakis, G.E.; Suresh, S. Mechanics of diseased red blood cells in human spleen and consequences for hereditary blood disorders. Proc. Natl. Acad. Sci. USA 2018, 115, 9574–9579. [Google Scholar] [CrossRef]
- Fairbanks, G.; Palek, J.; Dino, J.E.; Liu, P.A. Protein kinases and membrane protein phosphorylation in normal and abnormal human erythrocytes: Variation related to mean cell age. Blood 1983, 61, 850–857. [Google Scholar] [CrossRef]
- Moura, P.L.; Iragorri, M.A.L.; Français, O.; Le Pioufle, B.; Dobbe, J.; Streekstra, G.J.; El Nemer, W.; Toye, A.M.; Satchwell, T.J. Reticulocyte and red blood cell deformation triggers specific phosphorylation events. Blood Adv. 2019, 3, 2653–2663. [Google Scholar] [CrossRef]
- Longo, V.; Marrocco, C.; Zolla, L.; Rinalducci, S. Label-free quantitation of phosphopeptide changes in erythrocyte membranes: Towards molecular mechanisms underlying deformability alterations in stored red blood cells. Haematologica 2014, 99, e122–e125. [Google Scholar] [CrossRef][Green Version]
- Park, Y.; Best, C.A.; Badizadegan, K.; Dasari, R.R.; Feld, M.S.; Kuriabova, T.; Henle, M.L.; Levine, A.J.; Popescu, G. Measurement of red blood cell mechanics during morphological changes. Proc. Natl. Acad. Sci. USA 2010, 107, 6731–6736. [Google Scholar] [CrossRef]
- Flatt, J.F.; Bruce, L.J. The Molecular Basis for Altered Cation Permeability in Hereditary Stomatocytic Human Red Blood Cells. Front. Physiol. 2018, 9, 367. [Google Scholar] [CrossRef]
- Flatt, J.F.; Stevens-Hernandez, C.J.; Cogan, N.M.; Eggleston, D.J.; Haines, N.M.; Heesom, K.J.; Picard, V.; Thomas, C.; Bruce, L.J. Expression of South East Asian Ovalocytic Band 3 Disrupts Erythroblast Cytokinesis and Reticulocyte Maturation. Front. Physiol. 2020, 11, 357. [Google Scholar] [CrossRef]
- Stevens-Hernandez, C.J.; Flatt, J.F.; Kupzig, S.; Bruce, L.J. Reticulocyte Maturation and Variant Red Blood Cells. Front. Physiol. 2022, 13. [Google Scholar] [CrossRef]
- Narla, J.; Mohandas, N. Red cell membrane disorders. Int. J. Lab. Hematol. 2017, 39 (Suppl. S1), 47–52. [Google Scholar] [CrossRef] [PubMed]
- Niss, O.; Chonat, S.; Dagaonkar, N.; Almansoori, M.O.; Kerr, K.; Rogers, Z.R.; McGann, P.T.; Quarmyne, M.-O.; Risinger, M.; Zhang, K.; et al. Genotype-phenotype correlations in hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood Cells Mol. Dis. 2016, 61, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, S.; Gallagher, P.G.; Mohandas, N. Hereditary spherocytosis. Lancet 2008, 372, 1411–1426. [Google Scholar] [CrossRef]
- Da Costa, L.; Galimand, J.; Fenneteau, O.; Mohandas, N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013, 27, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Flatt, J.; Bawazir, W.M.; Bruce, L.J. The involvement of cation leaks in the storage lesion of red blood cells. Front. Physiol. 2014, 5, 214. [Google Scholar] [CrossRef] [PubMed]
- Bruce, L.J.; Guizouarn, H.; Burton, N.M.; Gabillat, N.; Poole, J.; Flatt, J.F.; Brady, R.L.; Borgese, F.; Delaunay, J.; Stewart, G.W. The monovalent cation leak in overhydrated stomatocytic red blood cells results from amino acid substitutions in the Rh-associated glycoprotein. Blood 2009, 113, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Fricke, B.; Argent, A.C.; Chetty, M.C.; Pizzey, A.R.; Turner, E.J.; Ho, M.M.; Iolascon, A.; von Düring, M.; Stewart, G.W. The “stomatin” gene and protein in overhydrated hereditary stomatocytosis. Blood 2003, 102, 2268–2277. [Google Scholar] [CrossRef] [PubMed]
- Fricke, B.; Parsons, S.F.; Knöpfle, G.; Düring, M.; Stewart, G.W. Stomatin is mis-trafficked in the erythrocytes of overhydrated hereditary stomatocytosis, and is absent from normal primitive yolk sac-derived erythrocytes. Br. J. Haematol. 2005, 131, 265–277. [Google Scholar] [CrossRef]
- Flatt, J.F.; Guizouarn, H.; Burton, N.M.; Borgese, F.; Tomlinson, R.J.; Forsyth, R.J.; Baldwin, S.A.; Levinson, B.E.; Quittet, P.; Aguilar-Martinez, P.; et al. Stomatin-deficient cryohydrocytosis results from mutations in SLC2A1: A novel form of GLUT1 deficiency syndrome. Blood 2011, 118, 5267–5277. [Google Scholar] [CrossRef] [PubMed]
- Fricke, B.; Jarvis, H.G.; Reid, C.D.L.; Aguilar-Martinez, P.; Robert, A.; Quittet, P.; Chetty, M.; Pizzey, A.; Cynober, T.; Lande, W.F.; et al. Four new cases of stomatin-deficient hereditary stomatocytosis syndrome: Association of the stomatin-deficient cryohydrocytosis variant with neurological dysfunction. Br. J. Haematol. 2004, 125, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Coles, S.E.; Chetty, M.C.; Ho, M.M.; Nicolaou, A.; Kearney, J.W.; Wright, S.D.; Stewart, G.W. Two British families with variants of the ‘cryohydrocytosis’ form of hereditary stomatocytosis. Br. J. Haematol. 1999, 105, 1055–1065. [Google Scholar] [CrossRef]
- Meli, A.; McAndrew, M.; Frary, A.; Rehnstrom, K.; Stevens-Hernandez, C.J.; Flatt, J.F.; Griffiths, A.; Stefanucci, L.; Astle, W.; Anand, R.; et al. Familial pseudohyperkalemia induces significantly higher levels of extracellular potassium in early storage of red cell concentrates without affecting other standard measures of quality: A case control and allele frequency study. Transfusion 2021, 61, 2439–2449. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens-Hernandez, C.J.; Bruce, L.J. Reticulocyte Maturation. Membranes 2022, 12, 311. https://doi.org/10.3390/membranes12030311
Stevens-Hernandez CJ, Bruce LJ. Reticulocyte Maturation. Membranes. 2022; 12(3):311. https://doi.org/10.3390/membranes12030311
Chicago/Turabian StyleStevens-Hernandez, Christian J., and Lesley J. Bruce. 2022. "Reticulocyte Maturation" Membranes 12, no. 3: 311. https://doi.org/10.3390/membranes12030311
APA StyleStevens-Hernandez, C. J., & Bruce, L. J. (2022). Reticulocyte Maturation. Membranes, 12(3), 311. https://doi.org/10.3390/membranes12030311