
Phosphatidylglycerol Supplementation Alters Mitochondrial Morphology and Cardiolipin Composition


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Materials
2.2. Megabase-Scale CRISPR/Cas9 Knockout of TAZ Gene
2.3. Phosphatidylglycerol (18:1)2 Supplementation
2.4. TEM Observation of Mitochondria in HAP1 and ΔTAZ
2.5. Real-Time Quantitative PCR
2.6. Lipid Extraction
2.7. Mass Spectrometry Analysis
2.8. Statistical Analysis
3. Results
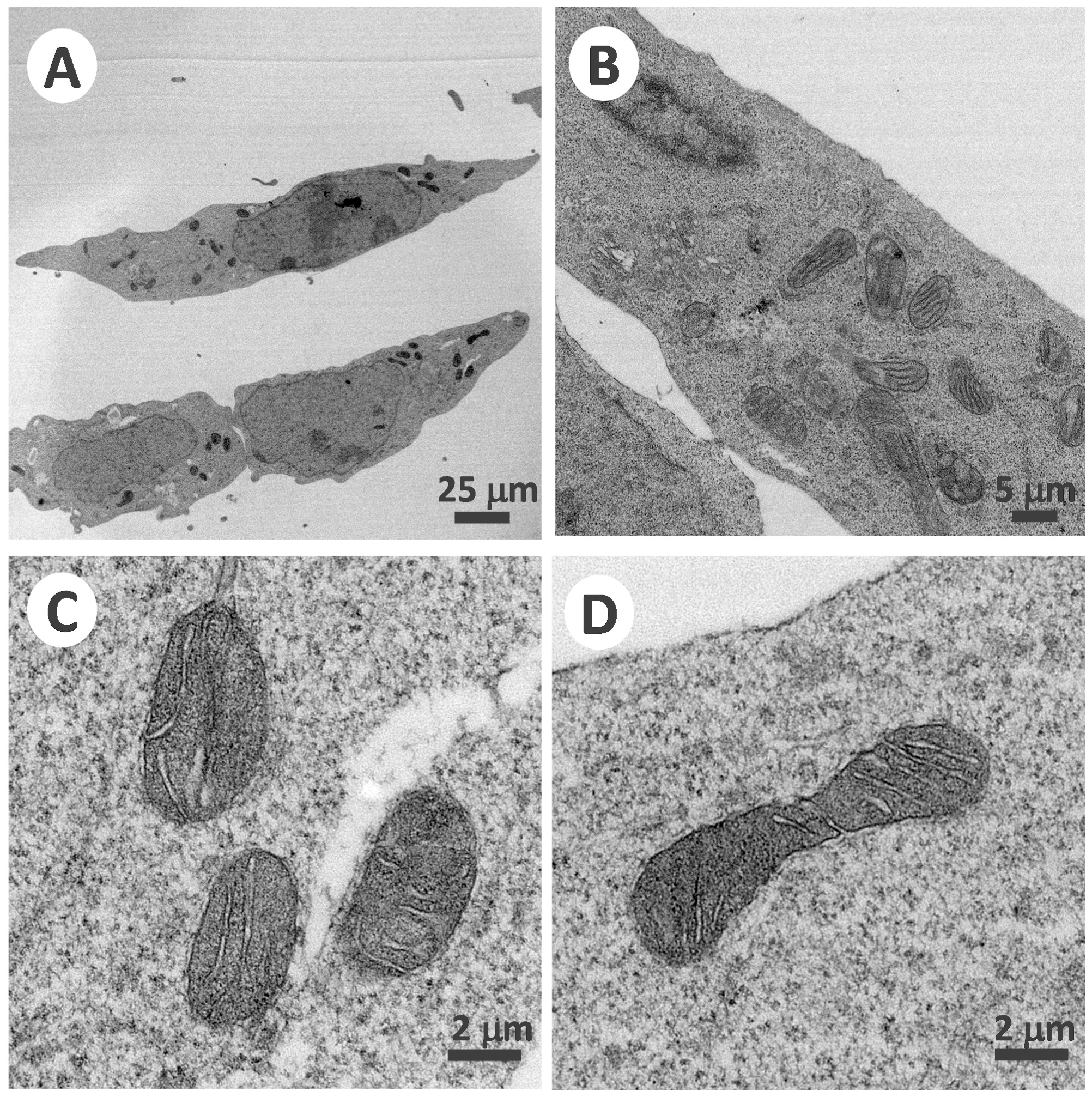
3.1. Genetic TAZ Knockout Effects in the Barth Syndrome Cellular Model
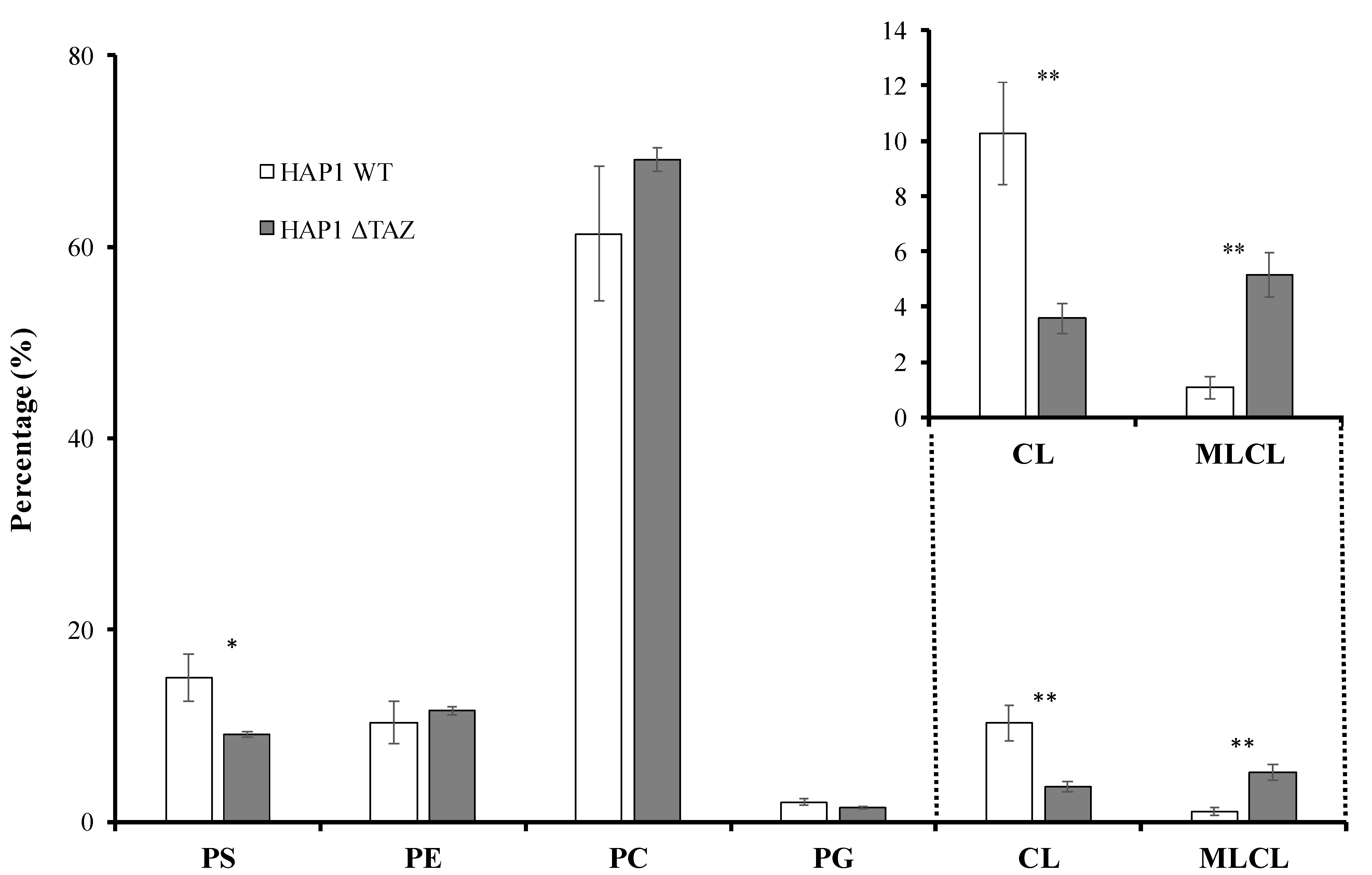
3.2. TAZ Knockout Caused Phospholipid Changes
3.3. CL Species Changes upon TAZ Knockout
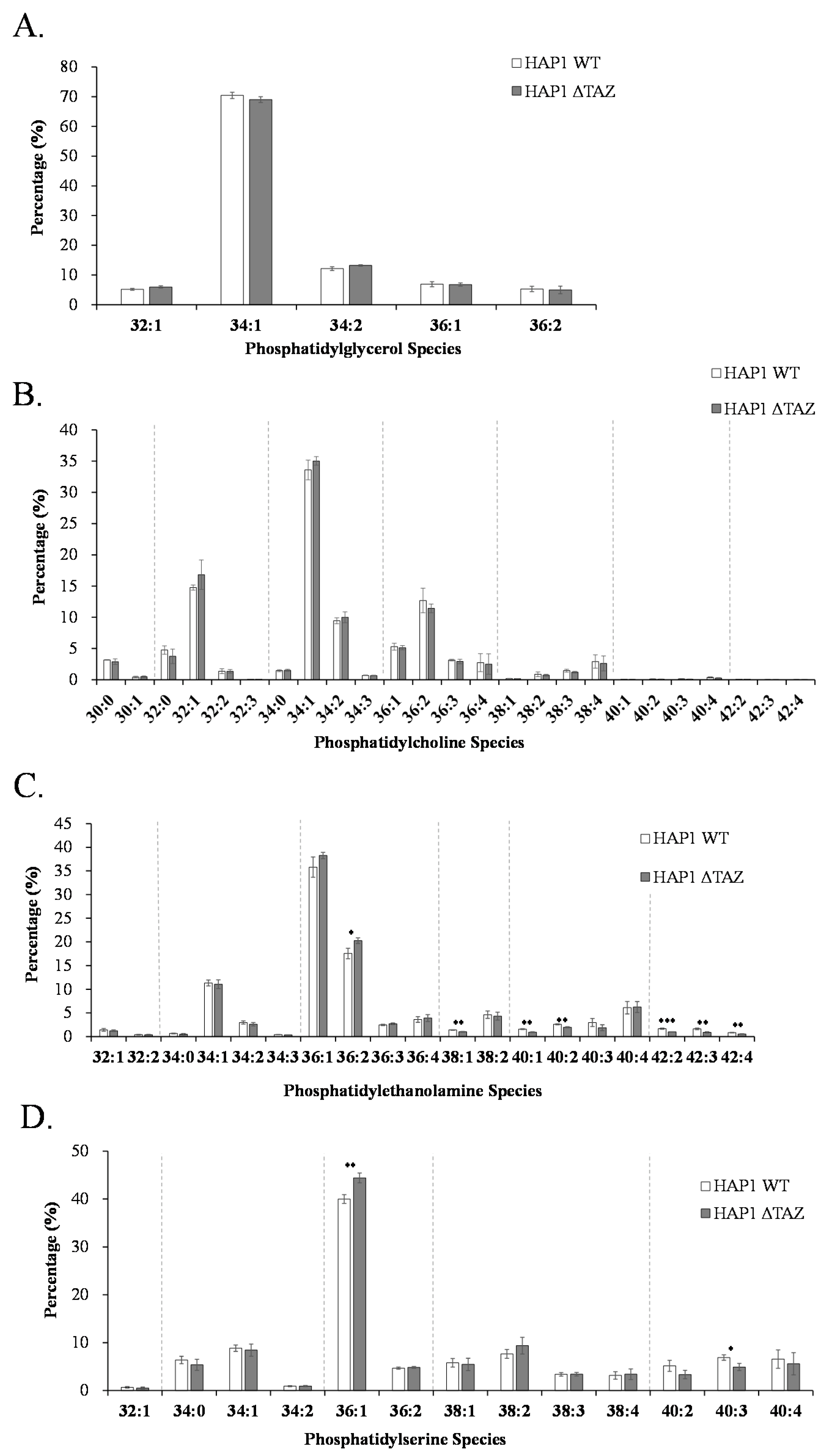
3.4. PG/PC/PE/PS Species Changes after TAZ Knockout
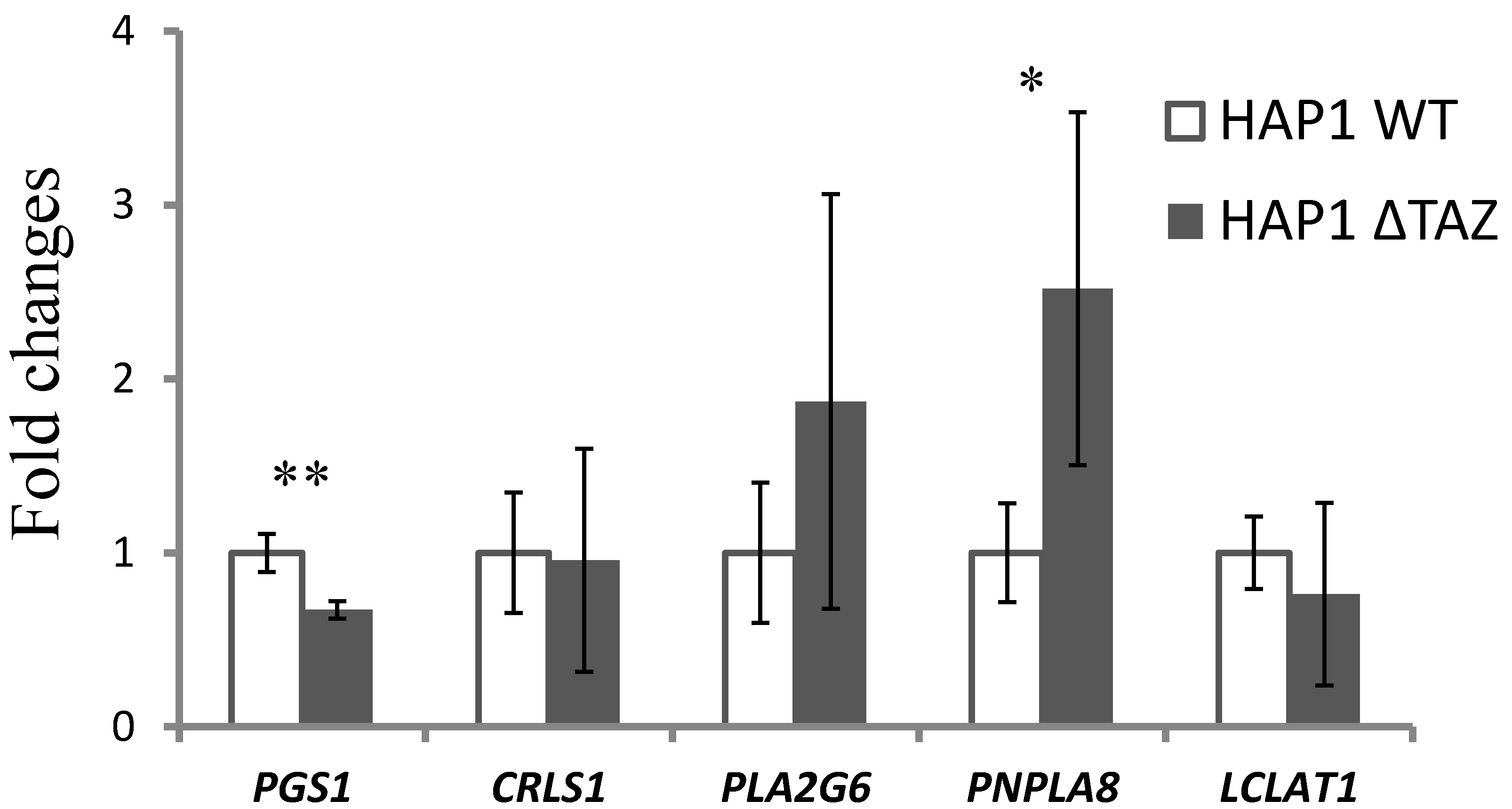
3.5. Gene Expression of CL Synthesis and Remodeling-Related Genes
3.6. PG(18:1)2 Supplementation Effect
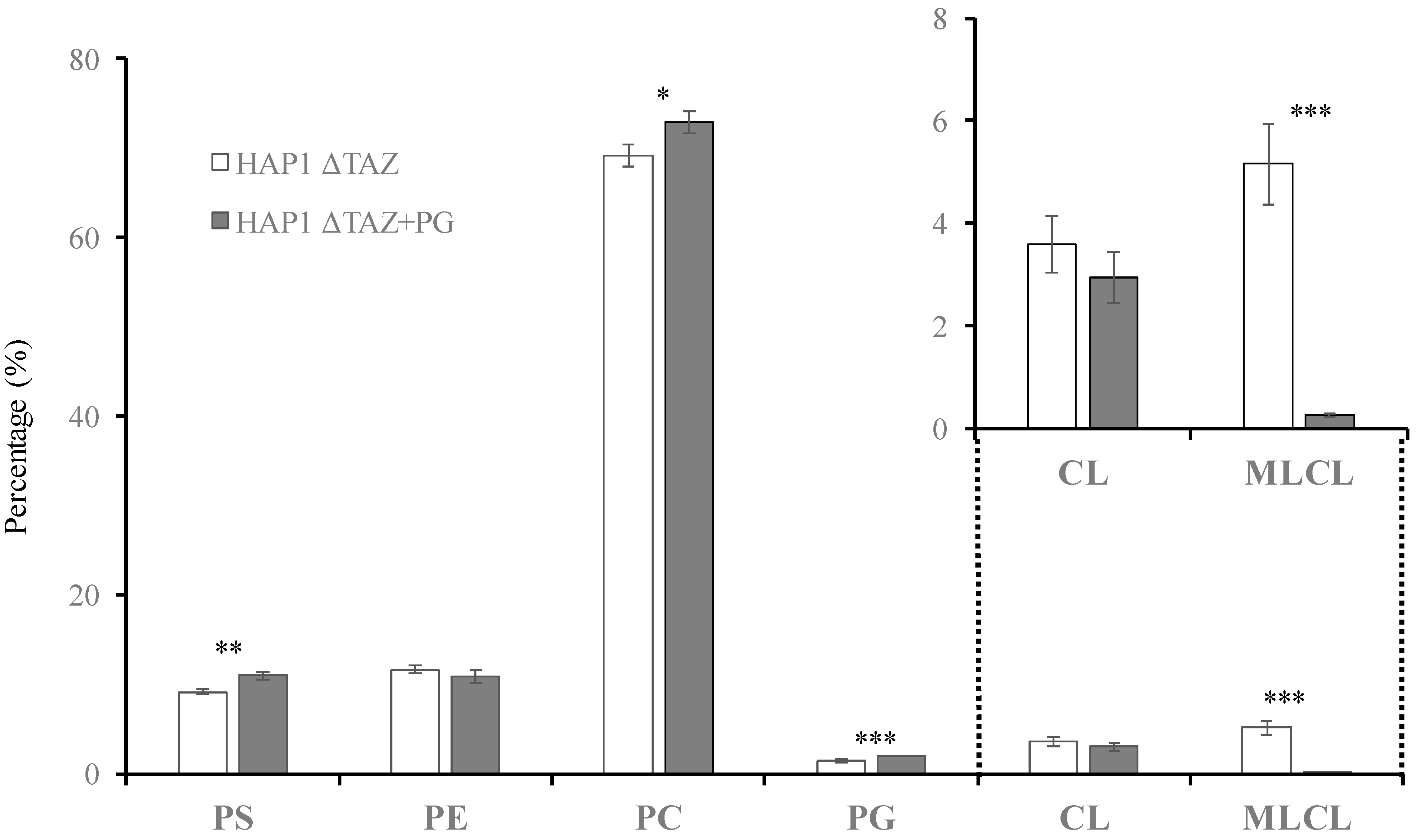
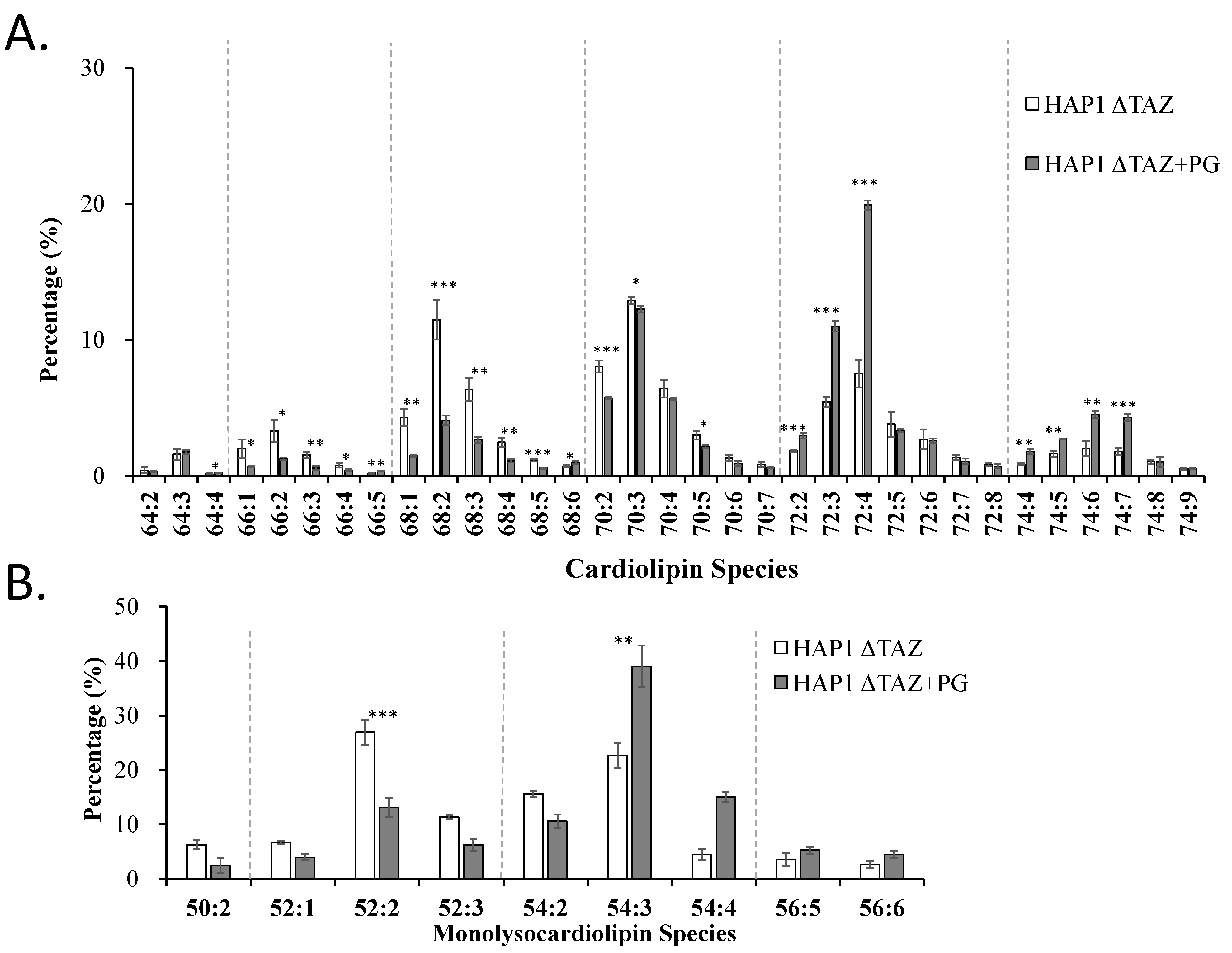
3.7. PG(18:1)2 Supplementation Effects on CL and MLCL
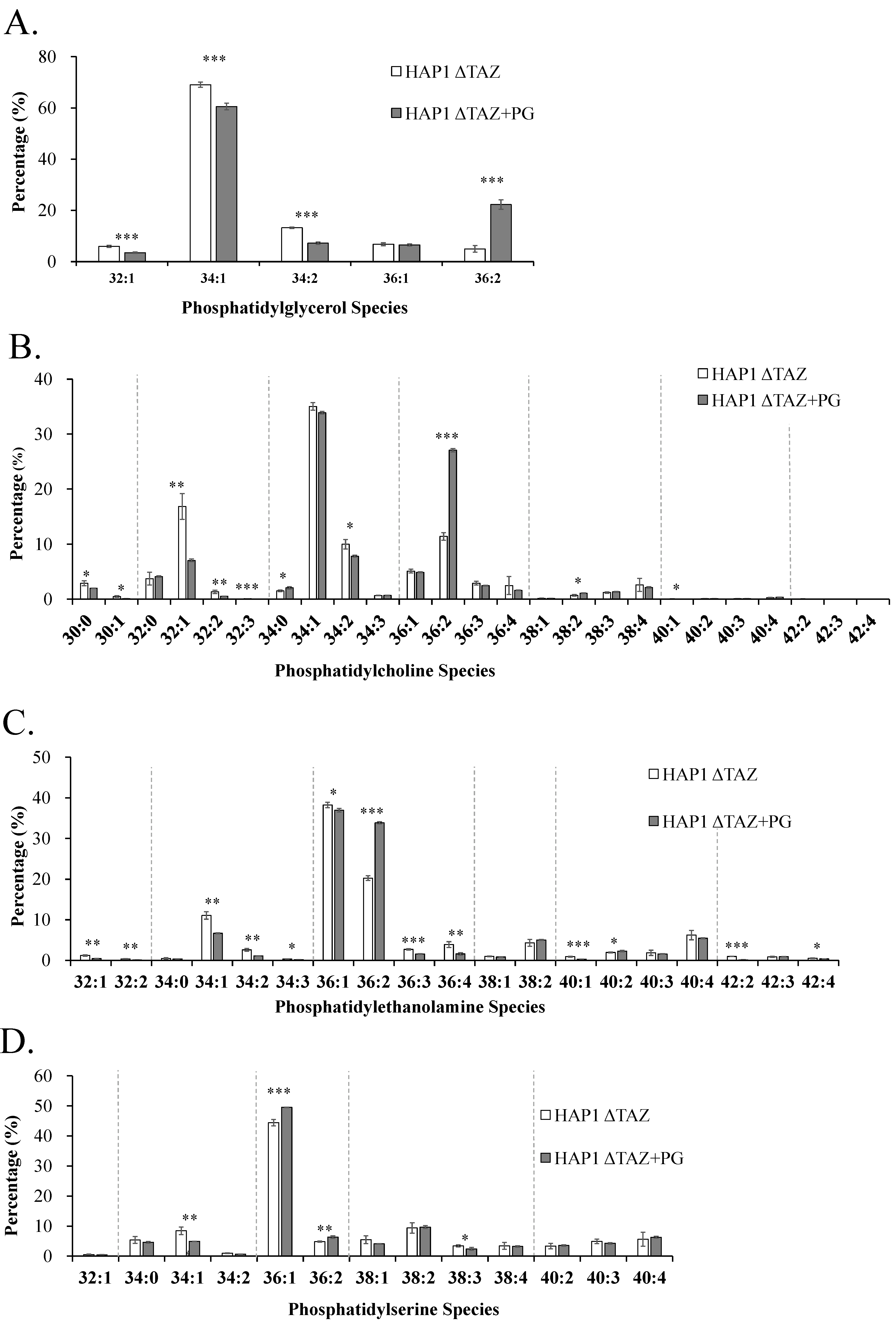
3.8. PG(18:1)2 Supplementation Effects on PG, PC, PE, and PS
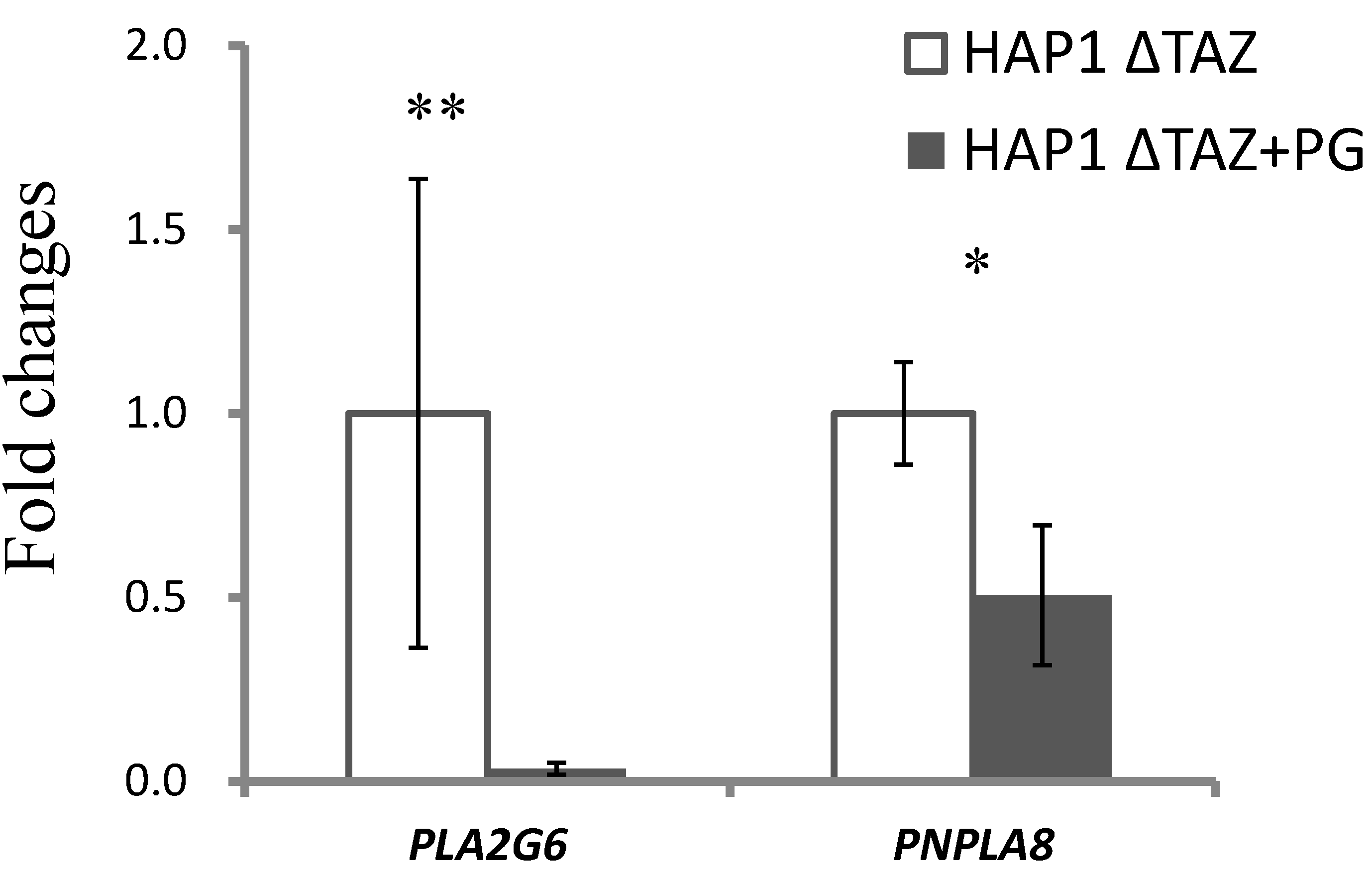
3.9. Gene Expression of Mitochondrial Phospholipases
4. Discussion
4.1. Effects of the Genetic Knock out of TAZ on Cardiolipin
4.2. Effects of the Genetic Knock out of TAZ on Phospholipids
4.3. PG Supplementation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adès, L.C.; Gedeon, A.K.; Wilson, M.J.; Latham, M.; Partington, M.W.; Mulley, J.C.; Nelson, J.; Lui, K.; Sillence, P.D.O. Barth syndrome: Clinical features and confirmation of gene localisation to distal Xq28. Am. J. Med. Genet. 1993, 45, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Mosser, J.; Sarde, C.-O.; Vicaire, S.; Yates, J.R.; Mandel, J.-L. A New Human Gene (DXS1357E) with Ubiquitous Expression, Located in Xq28 Adjacent to the Adrenoleukodystrophy Gene. Genomics 1994, 22, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Neuwald, A.F. Barth syndrome may be due to an acyltransferase deficiency. Curr. Biol. 1997, 7, R462–R466. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Malhotra, A.; Ren, M.; Schlame, M. The Enzymatic Function of Tafazzin. J. Biol. Chem. 2006, 281, 39217–39224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapała, B.; Płatek, T.; Wybrańska, I. A Novel TAZ Gene Mutation and Mosaicism in a Polish Family with Barth Syndrome. Ann. Hum. Genet. 2015, 79, 218–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vreken, P.; Valianpour, F.; Nijtmans, L.G.; Grivell, L.A.; Plecko, B.; Wanders, R.J.; Barth, P.G. Defective Remodeling of Cardiolipin and Phosphatidylglycerol in Barth Syndrome. Biochem. Biophys. Res. Commun. 2000, 279, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.; Rodenburg, R.; Thiels, C.; van Lenthe, H.; Stet, F.; Poll-The, B.T.; Stone, J.E.; Steward, C.; Wanders, R.J.; Smeitink, J.; et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography–mass spectrometry as a diagnostic test for Barth syndrome. Anal. Biochem. 2009, 387, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Kelley, R.I.; Feigenbaum, A.; Towbin, J.A.; Heerdt, P.M.; Schieble, T.; Wanders, R.J.; DiMauro, S.; Blanck, T. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003, 42, 1994–1999. [Google Scholar] [CrossRef] [Green Version]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, R.; Lobasso, S.; Gorgoglione, R.; Bowron, A.; Steward, C.G.; Corcelli, A. Cardiolipin fingerprinting of leukocytes by MALDI-TOF/MS as a screening tool for Barth syndrome. J. Lipid Res. 2015, 56, 1787–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz, F.M.; van Lenthe, H.; Vervaart, M.A.T.; Stet, F.S.; Klinkspoor, J.H.; Vernon, H.J.; Goorden, S.M.I.; Houtkooper, R.H.; Kulik, W.; Wanders, R.J.A. An improved functional assay in blood spot to diagnose Barth syndrome using the monolysocardiolipin/cardiolipin ratio. J. Inherit. Metab. Dis. 2021, 45, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Domingues, R.M.; Tyurin, V.A.; Maciel, E.; Domingues, P.; Amoscato, A.A.; Bayir, H.; Kagan, V.E. Characterization of cardiolipins and their oxidation products by LC–MS analysis. Chem. Phys. Lipids 2014, 179, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, H.-C.; Chen, L.-T.; Chen, J.-Y.; Huang, Y.-L.; Xin, R.-C.; Chan, J.-F.; Hsu, Y.-H.H. Double bonds of unsaturated fatty acids differentially regulate mitochondrial cardiolipin remodeling. Lipids Health Dis. 2019, 18, 53. [Google Scholar] [CrossRef]
- Chao, Y.-J.; Wu, W.-H.; Balazova, M.; Wu, T.-Y.; Lin, J.; Liu, Y.-W.; Hsu, Y.-H.H. Chlorella diet alters mitochondrial cardiolipin contents differentially in organs of Danio rerio analyzed by a lipidomics approach. PLoS ONE 2018, 13, e0193042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.-J.; Chan, J.-F.; Hsu, Y.-H.H. Chemotherapy Drug Induced Discoordination of Mitochondrial Life Cycle Detected by Cardiolipin Fluctuation. PLoS ONE 2016, 11, e0162457. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.-C.; Chao, Y.-J.; Hsu, Y.-H.H. Polyunsaturated fatty acids incorporation into cardiolipin in H9c2 cardiac myoblast. J. Nutr. Biochem. 2015, 26, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Renner, L.D.; Weibel, D.B. Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc. Natl. Acad. Sci. USA 2011, 108, 6264–6269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elías-Wolff, F.; Lindén, M.; Lyubartsev, A.P.; Brandt, E.G. Curvature sensing by cardiolipin in simulated buckled membranes. Soft Matter 2019, 15, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288. [Google Scholar] [CrossRef]
- Schlame, M.; Ren, M.; Xu, Y.; Greenberg, M.L.; Haller, I. Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 2005, 138, 38–49. [Google Scholar] [CrossRef]
- Schlame, M.; Brody, S.; Hostetler, K.Y. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur. J. Biochem. 1993, 212, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Bayır, H.; Kagan, V.E. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: Implications for Parkinson disease. Autophagy 2014, 10, 376–378. [Google Scholar] [CrossRef] [Green Version]
- Eble, K.S.; Coleman, W.B.; Hantgan, R.R.; Cunningham, C.C. Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J. Biol. Chem. 1990, 265, 19434–19440. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, K.Z.Q.; Chu, C.T. After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Dai, Q.; Chen, J.; Durrant, D.; Freeman, A.; Liu, T.; Grossman, D.; Lee, R.M. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 2003, 1, 892–902. [Google Scholar] [PubMed]
- Schlame, M.; Zhao, M.; Rua, D.; Haldar, D.; Greenberg, M.L. Kinetic analysis of cardiolipin synthase: A membrane enzyme with two glycerophospholipid substrates. Lipids 1995, 30, 633–640. [Google Scholar] [CrossRef]
- Schlame, M.; Ren, M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2080–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandner, K.; Mick, D.; Frazier, A.; Taylor, R.D.; Meisinger, C.; Rehling, P. Taz1, an Outer Mitochondrial Membrane Protein, Affects Stability and Assembly of Inner Membrane Protein Complexes: Implications for Barth Syndrome. Mol. Biol. Cell 2005, 16, 5202–5214. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.A.; Hatch, G.M. Identification of the Human Mitochondrial Linoleoyl-coenzyme A Monolysocardiolipin Acyltransferase (MLCL AT-1). J. Biol. Chem. 2009, 284, 30360–30371. [Google Scholar] [CrossRef] [Green Version]
- Mejia, E.M.; Zegallai, H.; Bouchard, E.; Banerji, V.; Ravandi, A.; Hatch, G.M. Expression of human monolysocardiolipin acyltransferase-1 improves mitochondrial function in Barth syndrome lymphoblasts. J. Biol. Chem. 2018, 293, 7564–7577. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Liu, Y.; Lockwood, J.; Burn, P.; Shi, Y. A Novel Cardiolipin-remodeling Pathway Revealed by a Gene Encoding an Endoplasmic Reticulum-associated Acyl-CoA:Lysocardiolipin Acyltransferase (ALCAT1) in Mouse. J. Biol. Chem. 2004, 279, 31727–31734. [Google Scholar] [CrossRef] [Green Version]
- Schlame, M. Cardiolipin remodeling and the function of tafazzin. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2013, 1831, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Xu, Y.; Ren, M. The Basis for Acyl Specificity in the Tafazzin Reaction. J. Biol. Chem. 2017, 292, 5499–5506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, K.; Hayakawa, T.; Murate, M.; Makino, A.; Ito, K.; Fujisawa, T.; Kobayashi, T. Curvature-dependent recognition of ethanolamine phospholipids by duramycin and cinnamycin. Biophys. J. 2007, 93, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine Deficiency in Mammalian Mitochondria Impairs Oxidative Phosphorylation and Alters Mitochondrial Morphology. J. Biol. Chem. 2013, 288, 4158–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttinger, L.; Horvath, S.E.; Kleinschroth, T.; Hunte, C.; Daum, G.; Pfanner, N.; Becker, T. Phosphatidylethanolamine and Cardiolipin Differentially Affect the Stability of Mitochondrial Respiratory Chain Supercomplexes. J. Mol. Biol. 2012, 423, 677–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenbergen, R.; Nanowski, T.S.; Beigneux, A.; Kulinski, A.; Young, S.; Vance, J.E. Disruption of the Phosphatidylserine Decarboxylase Gene in Mice Causes Embryonic Lethality and Mitochondrial Defects. J. Biol. Chem. 2005, 280, 40032–40040. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin Stabilizes Respiratory Chain Supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.S.; Thompson, M.N.; Fei, N.; Hüttemann, M.; Greenberg, M.L. Cardiolipin and Mitochondrial Phosphatidylethanolamine Have Overlapping Functions in Mitochondrial Fusion in Saccharomyces cerevisiae. J. Biol. Chem. 2012, 287, 17589–17597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, C.; Voelker, D.R.; Langer, T. Making heads or tails of phospholipids in mitochondria. J. Cell Biol. 2011, 192, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Gohil, V.; Ma, L.; Greenberg, M.L. Absence of Cardiolipin Results in Temperature Sensitivity, Respiratory Defects, and Mitochondrial DNA Instability Independent of pet56. J. Biol. Chem. 2004, 279, 32294–32300. [Google Scholar] [CrossRef] [Green Version]
- Tamura, Y.; Endo, T.; Iijima, M.; Sesaki, H. Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J. Cell Biol. 2009, 185, 1029–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, C.; Haag, M.; Potting, C.; Rodenfels, J.; Dip, P.V.; Wieland, F.T.; Brügger, B.; Westermann, B.; Langer, T. The genetic interactome of prohibitins: Coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell Biol. 2009, 184, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, T.; Tani, M.; Moriguchi, A.; Tokunaga, S.; Higuchi, T.; Kitada, S.; Kuge, O. FMP30 is required for the maintenance of a normal cardiolipin level and mitochondrial morphology in the absence of mitochondrial phosphatidylethanolamine synthesis. Mol. Microbiol. 2011, 80, 248–265. [Google Scholar] [CrossRef]
- Horibata, Y.; Sugimoto, H. StarD7 Mediates the Intracellular Trafficking of Phosphatidylcholine to Mitochondria. J. Biol. Chem. 2010, 285, 7358–7365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saita, S.; Tatsuta, T.; Lampe, P.A.; König, T.; Ohba, Y.; Langer, T. PARL partitions the lipid transfer protein STARD7 between the cytosol and mitochondria. EMBO J. 2018, 37, e97909. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Na, C.-L.; Luo, S.; Wu, D.; Hogan, S.; Huang, T.; Weaver, T.E. The Phosphatidylcholine Transfer Protein Stard7 is Required for Mitochondrial and Epithelial Cell Homeostasis. Sci. Rep. 2017, 7, srep46416. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, Y.; Xu, Y.; Ma, Q.; Lin, Z.; Schlame, M.; Bezzerides, V.J.; Strathdee, D.; Pu, W.T. AAV Gene Therapy Prevents and Reverses Heart Failure in a Murine Knockout Model of Barth Syndrome. Circ. Res. 2020, 126, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Gomez, B.; Robinson, N.C. Phospholipase Digestion of Bound Cardiolipin Reversibly Inactivates Bovine Cytochrome bc1. Biochemistry 1999, 38, 9031–9038. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Ryan, M.T.; Schlame, M.; Zhao, M.; Gu, Z.; Klingenberg, M.; Pfanner, N.; Greenberg, M.L. Absence of Cardiolipin in the crd1 Null Mutant Results in Decreased Mitochondrial Membrane Potential and Reduced Mitochondrial Function. J. Biol. Chem. 2000, 275, 22387–22394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.-W.; Chao, Y.-J.; Chang, W.-H.; Chan, J.-F.; Hsu, Y.-H.H. Phosphatidylglycerol Incorporates into Cardiolipin to Improve Mitochondrial Activity and Inhibits Inflammation. Sci. Rep. 2018, 8, 4919. [Google Scholar] [CrossRef] [PubMed]
- Khuchua, Z.; Yue, Z.; Batts, L.; Strauss, A.W. A Zebrafish Model of Human Barth Syndrome Reveals the Essential Role of Tafazzin in Cardiac Development and Function. Circ. Res. 2006, 99, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Valianpour, F.; Chen, S.; Vaz, F.; Hakkaart, G.A.; Wanders, R.J.A.; Greenberg, M.L. Aberrant cardiolipin metabolism in the yeast taz1 mutant: A model for Barth syndrome. Mol. Microbiol. 2003, 51, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Cheng, I.-F.; Balleininger, M.; Vaz, F.M.; Streckfuss-Bömeke, K.; Hübscher, D.; Vukotic, M.; Wanders, R.J.; Rehling, P.; Guan, K. Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 2013, 11, 806–819. [Google Scholar] [CrossRef] [Green Version]
- Acehan, D.; Khuchua, Z.; Houtkooper, R.H.; Malhotra, A.; Kaufman, J.; Vaz, F.M.; Ren, M.; Rockman, H.A.; Stokes, D.L.; Schlame, M. Distinct effects of tafazzin deletion in differentiated and undifferentiated mitochondria. Mitochondrion 2009, 9, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Condell, M.; Plesken, H.; Edelman-Novemsky, I.; Ma, J.; Ren, M.; Schlame, M. A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 11584–11588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acehan, D.; Vaz, F.; Houtkooper, R.; James, J.; Moore, V.; Tokunaga, C.; Kulik, W.; Wansapura, J.; Toth, M.J.; Strauss, A.; et al. Cardiac and Skeletal Muscle Defects in a Mouse Model of Human Barth Syndrome. J. Biol. Chem. 2011, 286, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Chen, Z.; Zhu, M.; Shen, Y.; Leon, L.J.; Chi, L.; Spinozzi, S.; Tan, C.; Gu, Y.; Nguyen, A.; et al. Cardiolipin Remodeling Defects Impair Mitochondrial Architecture and Function in a Murine Model of Barth Syndrome Cardiomyopathy. Circ. Heart Fail. 2021, 14, e008289. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Acehan, D.; Berno, B.; Xu, Y.; Valvo, S.; Ren, M.; Stokes, D.L.; Epand, R.M. The physical state of lipid substrates provides transacylation specificity for tafazzin. Nat. Chem. Biol. 2012, 8, 862–869. [Google Scholar] [CrossRef] [Green Version]
- Stone, S.J.; Cui, Z.; Vance, J.E. Cloning and Expression of Mouse Liver Phosphatidylserine Synthase-1 cDNA: Overexpression in rat hepatoma cells inhibits the CDP-ethanolamine pathway for phosphatidylethanolamine biosynthesis. J. Biol. Chem. 1998, 273, 7293–7302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, S.J.; Vance, J.E. Cloning and expression of murine liver phosphatidylserine synthase (PSS)-2: Differential regulation of phospholipid metabolism by PSS1 and PSS2. Biochem, J. 1999, 342, 57–64. [Google Scholar] [CrossRef]
- Kuge, O.; Saito, K.; Nishijima, M. Control of Phosphatidylserine Synthase II Activity in Chinese Hamster Ovary Cells. J. Biol. Chem. 1999, 274, 23844–23849. [Google Scholar] [CrossRef] [PubMed]
- Kodaki, T.; Nikawa, J.; Hosaka, K.; Yamashita, S. Functional analysis of the regulatory region of the yeast phosphatidylserine synthase gene, PSS. J. Bacteriol. 1991, 173, 7992–7995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, J.E.; Steenbergen, R. Metabolism and functions of phosphatidylserine. Prog. Lipid Res. 2005, 44, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Edelman-Novemsky, I.; Xu, Y.; Plesken, H.; Ma, J.; Schlame, M.; Ren, M. Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc. Natl. Acad. Sci. USA 2009, 106, 2337–2341. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, I.; Chen, Y.-C.; Lai, R.-Y.; Chan, J.-F.; Lee, Y.-H.; Balazova, M.; Hsu, Y.-H.H. Phosphatidylglycerol Supplementation Alters Mitochondrial Morphology and Cardiolipin Composition. Membranes 2022, 12, 383. https://doi.org/10.3390/membranes12040383
Chu I, Chen Y-C, Lai R-Y, Chan J-F, Lee Y-H, Balazova M, Hsu Y-HH. Phosphatidylglycerol Supplementation Alters Mitochondrial Morphology and Cardiolipin Composition. Membranes. 2022; 12(4):383. https://doi.org/10.3390/membranes12040383
Chicago/Turabian StyleChu, I, Ying-Chih Chen, Ruo-Yun Lai, Jui-Fen Chan, Ya-Hui Lee, Maria Balazova, and Yuan-Hao Howard Hsu. 2022. "Phosphatidylglycerol Supplementation Alters Mitochondrial Morphology and Cardiolipin Composition" Membranes 12, no. 4: 383. https://doi.org/10.3390/membranes12040383