
Polyene Antibiotics Physical Chemistry and Their Effect on Lipid Membranes; Impacting Biological Processes and Medical Applications

Abstract
:
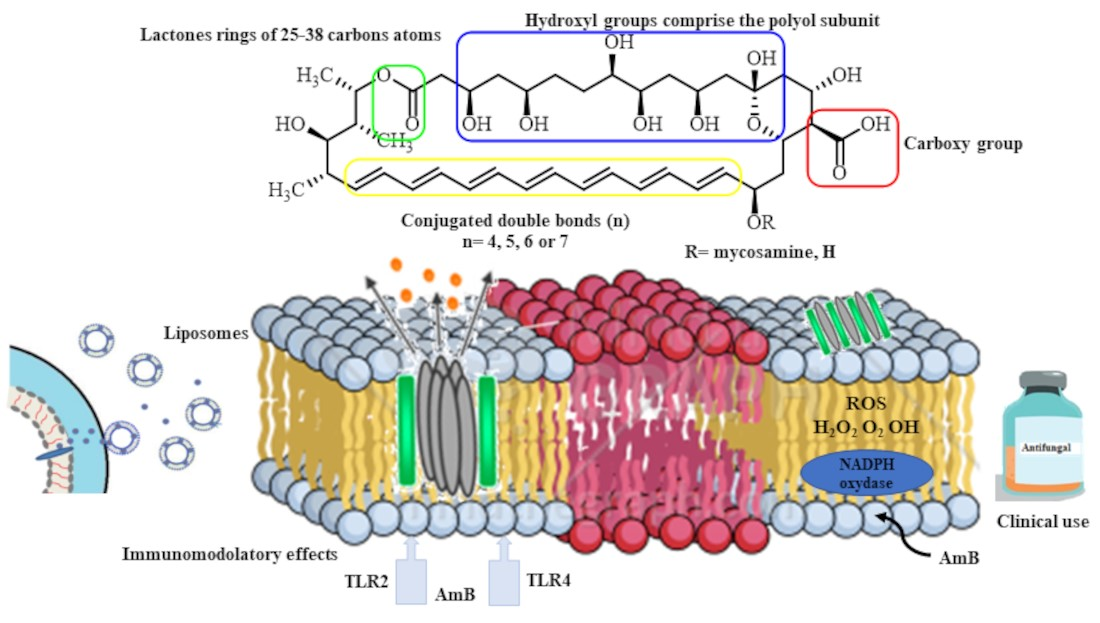{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Polyene Structure and Chemical Behavior
2.1. Chemical Structure of Polyenes
2.2. Oxidation
2.3. Aggregation
3. Mechanisms of Action
4. Alternatives to Reduce Polyene Host-Toxicity
4.1. Polyene Semi-Synthetic Derivatives
4.2. Lipid-Based Formulations
5. Role of Membrane Structure on the Activity of Polyenes
5.1. Binary Lipid Mixtures Containing Sterol
5.2. Sterol-Free Bilayers
5.3. Ternary Lipid Mixtures Containing Sterol
6. Clinical Use of Polyenes
6.1. Recent Advances in Clinical Use
6.2. Pharmacokinetic Changes
6.3. Pharmacodynamic Changes
6.4. Kidney Damage
6.5. Septicemia
6.6. Transplants
6.7. Anti-Parasitic
6.8. Mucormycosis
7. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef]
- Kinsky, S.C. Polyene Antibiotics. In Antibiotics; Gottlieb, D., Shaw, P.D., Eds.; Springer: Berlin/Heidelberg, Germany, 1967; pp. 122–141. [Google Scholar]
- Asher, I.M.; Schwartzman, G. Amphotericin B. Anal. Profiles Drug Subst. 1977, 6, 1–42. [Google Scholar]
- Chong, C.N.; Rickards, R.W. Macrolide Antibiotic Studies. XVI. The Structure of Nystatin. Tetrahedron Lett. 1970, 11, 5145–5148. [Google Scholar] [CrossRef]
- Schaffner, C.P.; Mechlinski, W. Polyene magrolide derivatives. II. J. Antibiot. 1972, 25, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Mazerski, J.; Grzybowska, J.; Borowski, E. Influence of Net Charge on the Aggregation and Solubility Behaviour of Amphotericin B and Its Derivatives in Aqueous Media. Eur. Biophys. J. 1990, 18, 159–164. [Google Scholar] [CrossRef]
- Hamilton-Miller, J.M. Chemistry and Biology of the Polyene Macrolide Antibiotics. Bacteriol. Rev. 1973, 37, 166–196. [Google Scholar] [CrossRef]
- Thomas, A.H. Analysis and Assay of Polyene Antifungal Antibiotics. A Review. Analyst 1976, 101, 321–340. [Google Scholar] [CrossRef]
- Hamilton-Miller, J.M. The Effect of pH and of Temperature on the Stability and Bioactivity of Nystatin and Amphotericin B. J. Pharm. Pharmacol. 1973, 25, 401–407. [Google Scholar] [CrossRef]
- Belkherroubi-Sari, L.; Boucherit, Z.; Chéron, M.; Boucherit, K.; Benyoucef, M.; Belbraouet, S. Modulation of the Polyene Antibiotic Amphotericin B Selective Toxicity by pH Change of the Stock Solutions. Afr. J. Microbiol. Res. 2008, 2, 242–246. [Google Scholar] [CrossRef]
- Cass, A.; Finkelstein, A.; Krespi, V. The Ion Permeability Induced in Thin Lipid Membranes by the Polyene Antibiotics Nystatin and Amphotericin B. J. Gen. Physiol. 1970, 56, 100–124. [Google Scholar] [CrossRef] [Green Version]
- Czernel, G.; Typek, R.; Klimek, K.; Czuryło, A.; Dawidowicz, A.L.; Gagoś, M. Catalytic Effect of Free Iron Ions and Heme-Iron on Chromophore Oxidation of a Polyene Antibiotic Amphotericin B. J. Mol. Struct. 2016, 1111, 69–75. [Google Scholar] [CrossRef]
- Kılıç Yıldırım, G.; Yarar, C.; Şeker Yılmaz, B.; Ceylaner, S. Niemann-Pick Type C Disease with a Novel Intronic Mutation: Three Turkish Cases from the Same Family. J. Pediatr. Endocrinol. Metab. 2021, 35, 535–541. [Google Scholar] [CrossRef]
- Gallelli, J.F. Assay and Stability of Amphotericin B in Aqueous Solutions. Drug Intell. Clin. Pharm. 1967, 1, 102–105. [Google Scholar] [CrossRef]
- Rabek, J.F.; Rånby, B.; Östensson, B.; Flodin, P. Oxidation of Polyene Structures in Poly(vinyl Chloride) by Molecular Oxygen and Singlet Oxygen. J. Appl. Polym. Sci. 1979, 24, 2407–2413. [Google Scholar] [CrossRef]
- Szczeblewski, P.; Laskowski, T.; Bałka, A.; Borowski, E.; Milewski, S. Light-Induced Transformation of the Aromatic Heptaene Antifungal Antibiotic Candicidin D into Its All-Trans Isomer. J. Nat. Prod. 2018, 81, 1540–1545. [Google Scholar] [CrossRef]
- Bolard, J. How Do the Polyene Macrolide Antibiotics Affect the Cellular Membrane Properties? Biochim. Biophys. Acta 1986, 864, 257–304. [Google Scholar] [CrossRef]
- Andreoli, T.E.; Monahan, M. The Interaction of Polyene Antibiotics with Thin Lipid Membranes. J. Gen. Physiol. 1968, 52, 300–325. [Google Scholar] [CrossRef] [Green Version]
- Norman, A.W.; Spielvogel, A.M.; Wong, R.G. Polyene Antibiotic–sterol Interaction1 1Supported in Part by United States Public Health Service Grants AM-09012 and AM-14,750. In Advances in Lipid Research; Elsevier: Amsterdam, The Netherlands, 1976; pp. 127–170. ISBN 9780120249145. [Google Scholar]
- Te Welscher, Y.M.; van Leeuwen, M.R.; de Kruijff, B.; Dijksterhuis, J.; Breukink, E. Polyene Antibiotic That Inhibits Membrane Transport Proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 11156–11159. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Affordability WHO Model List of Essential Medicines—22nd List, 2021. Available online: https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2021.02 (accessed on 1 February 2022).
- Dutcher, J.D. The Discovery and Development of Amphotericin B. Dis. Chest 1968, 54, 296–298. [Google Scholar] [CrossRef]
- Dutcher, J.D.; William, G.; Pagano, J.F.; John, V. Amphotericin B, Its Production, and Its Salts. US Patent US478014A, 13 October 1959. [Google Scholar]
- Cavassin, F.B.; Baú-Carneiro, J.L.; Vilas-Boas, R.R.; Queiroz-Telles, F. Sixty Years of Amphotericin B: An Overview of the Main Antifungal Agent Used to Treat Invasive Fungal Infections. Infect. Dis. Ther. 2021, 10, 115–147. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, B.; Shen, Z.-Y.; Cai, X.; Liu, Z.-Q.; Zheng, Y.-G. Enhanced Amphotericin B Production by Genetically Engineered Streptomyces Nodosus. Microbiol. Res. 2021, 242, 126623. [Google Scholar] [CrossRef]
- Caffrey, P.; Hogan, M.; Song, Y. New Glycosylated Polyene Macrolides: Refining the Ore from Genome Mining. Antibiotics 2022, 11, 334. [Google Scholar] [CrossRef]
- Paila, Y.D.; Saha, B.; Chattopadhyay, A. Amphotericin B Inhibits Entry of Leishmania Donovani into Primary Macrophages. Biochem. Biophys. Res. Commun. 2010, 399, 429–433. [Google Scholar] [CrossRef]
- Ramos, H.; Valdivieso, E.; Gamargo, M.; Dagger, F.; Cohen, B.E. Amphotericin B Kills Unicellular Leishmanias by Forming Aqueous Pores Permeable to Small Cations and Anions. J. Membr. Biol. 1996, 152, 65–75. [Google Scholar] [CrossRef]
- Mangé, A.; Nishida, N.; Milhavet, O.; McMahon, H.E.; Casanova, D.; Lehmann, S. Amphotericin B Inhibits the Generation of the Scrapie Isoform of the Prion Protein in Infected Cultures. J. Virol. 2000, 74, 3135–3140. [Google Scholar] [CrossRef] [Green Version]
- Mangé, A.; Milhavet, O.; McMahon, H.E.M.; Casanova, D.; Lehmann, S. Effect of Amphotericin B on Wild-Type and Mutated Prion Proteins in Cultured Cells. J. Neurochem. 2001, 74, 754–762. [Google Scholar] [CrossRef]
- Pleskoff, O.; Seman, M.; Alizon, M. Amphotericin B Derivative Blocks Human Immunodeficiency Virus Type 1 Entry after CD4 Binding: Effect on Virus-Cell Fusion but Not on Cell-Cell Fusion. J. Virol. 1995, 69, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Konopka, K.; Guo, L.S.; Düzgüneş, N. Anti-HIV Activity of Amphotericin B-Cholesteryl Sulfate Colloidal Dispersion in Vitro. Antivir. Res. 1999, 42, 197–209. [Google Scholar] [CrossRef]
- Kessler, H.A.; Dixon, J.; Howard, C.R.; Tsiquaye, K.; Zuckerman, A.J. Effects of Amphotericin B on Hepatitis B Virus. Antimicrob. Agents Chemother. 1981, 20, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Fanos, V.; Cataldi, L. Renal Transport of Antibiotics and Nephrotoxicity: A Review. J. Chemother. 2001, 13, 461–472. [Google Scholar] [CrossRef]
- Utz, J.P.; Treger, A.; McCULLOUGH, N.B.; Emmons, C.W. Amphotericin B: Intravenous Use in 21 Patients with Systemic Fungal Diseases. Antibiot. Annu. 1958, 6, 628–634. [Google Scholar]
- Hazen, E.L.; Brown, R. Fungicidin, an Antibiotic Produced by a Soil Actinomycete. Proc. Soc. Exp. Biol. Med. 1951, 76, 93–97. [Google Scholar] [CrossRef]
- Beveridge, G.W.; Fairburn, E.; Finn, O.A.; Scott, O.L.; Stewart, T.W.; Summerly, R. A Comparison of Nystatin Cream with Nystatin/triamcinolone Acetonide Combination Cream in the Treatment of Candidal Inflammation of the Flexures. Curr. Med. Res. Opin. 1977, 4, 584–587. [Google Scholar] [CrossRef]
- Keczkes, K.; Leighton, I.; Good, C.S. Topical Treatment of Dermatophytoses and Candidoses. Practitioner 1975, 214, 412–417. [Google Scholar]
- de Wet, P.M.; Rode, H.; van Dyk, A.; Millar, A.J. Perianal Candidosis--a Comparative Study with Mupirocin and Nystatin. Int. J. Dermatol. 1999, 38, 618–622. [Google Scholar] [CrossRef]
- Xiao, Y.; Yuan, P.; Sun, Y.; Xu, Y.; Deng, X.; Wang, X.; Liu, R.; Chen, Q.; Jiang, L. Comparison of Topical Antifungal Agents for Oral Candidiasis Treatment: A Systematic Review and Meta-Analysis. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2021, 133, 282–291. [Google Scholar] [CrossRef]
- Lu, S.-Y. Oral Candidosis: Pathophysiology and Best Practice for Diagnosis, Classification, and Successful Management. J. Fungi 2021, 7, 555. [Google Scholar] [CrossRef]
- Peyron, F.; Favel, A.; Michel-Nguyen, A.; Gilly, M.; Regli, P.; Bolmström, A. Improved Detection of Amphotericin B-Resistant Isolates of Candida Lusitaniae by Etest. J. Clin. Microbiol. 2001, 39, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Desoubeaux, G.; Coste, A.T.; Imbert, C.; Hennequin, C. Overview about Candida Auris: What’s up 12 Years after Its First Description? J. Mycol. Med. 2022, 32, 101248. [Google Scholar] [CrossRef]
- Kamiński, D.M. Recent Progress in the Study of the Interactions of Amphotericin B with Cholesterol and Ergosterol in Lipid Environments. Eur. Biophys. J. 2014, 43, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Hartsel, S.; Bolard, J. Amphotericin B: New Life for an Old Drug. Trends Pharmacol. Sci. 1996, 17, 445–449. [Google Scholar] [CrossRef]
- Mesa-Arango, A.C.; Scorzoni, L.; Zaragoza, O. It Only Takes One to Do Many Jobs: Amphotericin B as Antifungal and Immunomodulatory Drug. Front. Microbiol. 2012, 3, 286. [Google Scholar] [CrossRef] [Green Version]
- Carlson, M.A.; Ferraz, A.A.B.; Condon, R.E. Urinary Adenosine Excretion in Patients Receiving Amphotericin B. Surgery 1997, 121, 190–193. [Google Scholar] [CrossRef]
- Stillman, I.E.; Brezis, M.; Heyman, S.N.; Epstein, F.H.; Spokes, K.; Rosen, S. Effects of Salt Depletion on the Kidney: Changes in Medullary Oxygenation and Thick Ascending Limb Size. J. Am. Soc. Nephrol. 1994, 4, 1538–1545. [Google Scholar] [CrossRef]
- Fanos, V.; Cataldi, L. Amphotericin B-Induced Nephrotoxicity: A Review. J. Chemother. 2000, 12, 463–470. [Google Scholar] [CrossRef]
- Hamill, R.J. Amphotericin B Formulations: A Comparative Review of Efficacy and Toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef]
- Kristanc, L.; Božič, B.; Jokhadar, Š.Z.; Dolenc, M.S.; Gomišček, G. The Pore-Forming Action of Polyenes: From Model Membranes to Living Organisms. Biochim. Biophys. Acta (BBA) Biomembr. 2019, 1861, 418–430. [Google Scholar] [CrossRef]
- Carolus, H.; Pierson, S.; Lagrou, K.; Van Dijck, P. Amphotericin B and Other Polyenes—Discovery, Clinical Use, Mode of Action and Drug Resistance. J. Fungi 2020, 6, 321. [Google Scholar] [CrossRef]
- Baginski, M.; Czub, J. Amphotericin B and Its New Derivatives—Mode of Action. Curr. Drug Metab. 2009, 10, 459–469. [Google Scholar] [CrossRef]
- de Kruijff, B.; Demel, R.A. Polyene Antibiotic-Sterol Interactions in Membranes of Acholeplasma Laidlawii Cells and Lecithin Liposomes. 3. Molecular Structure of the Polyene Antibiotic-Cholesterol Complexes. Biochim. Biophys. Acta 1974, 339, 57–70. [Google Scholar] [CrossRef]
- de Kruijff, B.; Gerritsen, W.J.; Oerlemans, A.; Demel, R.A.; van Deenen, L.L. Polyene Antibiotic-Sterol Interactions in Membranes of Acholeplasma Laidlawii Cells and Lecithin Liposomes. I. Specificity of the Membrane Permeability Changes Induced by the Polyene Antibiotics. Biochim. Biophys. Acta 1974, 339, 30–43. [Google Scholar] [CrossRef]
- Finkelstein, A.; Holz, R. Aqueous Pores Created in Thin Lipid Membranes by the Polyene Antibiotics Nystatin and Amphotericin B. Membranes 1973, 2, 377–408. [Google Scholar]
- Dennis, V.W.; Stead, N.W.; Andreoli, T.E. Molecular Aspects of Polyene- and Sterol-Dependent Pore Formation in Thin Lipid Membranes. J. Gen. Physiol. 1970, 55, 375–400. [Google Scholar] [CrossRef] [Green Version]
- González-Damián, J.; Ortega-Blake, I. Effect of Membrane Structure on the Action of Polyenes II: Nystatin Activity along the Phase Diagram of Ergosterol- and Cholesterol-Containing POPC Membranes. J. Membr. Biol. 2010, 237, 41–49. [Google Scholar] [CrossRef]
- Dos Santos, A.G.; Marquês, J.T.; Carreira, A.C.; Castro, I.R.; Viana, A.S.; Mingeot-Leclercq, M.-P.; de Almeida, R.F.M.; Silva, L.C. The Molecular Mechanism of Nystatin Action Is Dependent on the Membrane Biophysical Properties and Lipid Composition. Phys. Chem. Chem. Phys. 2017, 19, 30078–30088. [Google Scholar] [CrossRef]
- Gray, K.C.; Palacios, D.S.; Dailey, I.; Endo, M.M.; Uno, B.E.; Wilcock, B.C.; Burke, M.D. Amphotericin Primarily Kills Yeast by Simply Binding Ergosterol. Proc. Natl. Acad. Sci. USA 2012, 109, 2234–2239. [Google Scholar] [CrossRef] [Green Version]
- Yilma, S.; Cannon-Sykora, J.; Samoylov, A.; Lo, T.; Liu, N.; Jeffrey Brinker, C.; Neely, W.C.; Vodyanoy, V. Large-Conductance Cholesterol–amphotericin B Channels in Reconstituted Lipid Bilayers. Biosens. Bioelectron. 2007, 22, 1359–1367. [Google Scholar] [CrossRef]
- Ganis, P.; Avitabile, G.; Mechlinski, W.; Schaffner, C.P. Polyene Macrolide Antibiotic Amphotericin B. Crystal Structure of the N-Iodoacetyl Derivative. J. Am. Chem. Soc. 1971, 93, 4560–4564. [Google Scholar] [CrossRef]
- Coutinho, A.; Silva, L.; Fedorov, A.; Prieto, M. Cholesterol and Ergosterol Influence Nystatin Surface Aggregation: Relation to Pore Formation. Biophys. J. 2004, 87, 3264–3276. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, A.; Prieto, M. Self-Association of the Polyene Antibiotic Nystatin in Dipalmitoylphosphatidylcholine Vesicles: A Time-Resolved Fluorescence Study. Biophys. J. 1995, 69, 2541–2557. [Google Scholar] [CrossRef] [Green Version]
- Zielinski, J.; Golik, J.; Pawlak, J.; Borowski, E.; Falkowski, L. The Structure of Nystatin A3, a Component of Nystatin Complex. J. Antibiot. 1988, 41, 1289–1291. [Google Scholar] [CrossRef] [Green Version]
- Borowski, E. Novel Approaches in the Rational Design of Antifungal Agents of Low Toxicity. Il Farm. 2000, 55, 206–208. [Google Scholar] [CrossRef]
- Gagoś, M.; Arczewska, M. Influence of K+ and Na+ Ions on the Aggregation Processes of Antibiotic Amphotericin B: Electronic Absorption and FTIR Spectroscopic Studies. J. Phys. Chem. B 2011, 115, 3185–3192. [Google Scholar] [CrossRef]
- Gagoś, M.; Hereć, M.; Arczewska, M.; Czernel, G.; Dalla Serra, M.; Gruszecki, W.I. Anomalously High Aggregation Level of the Polyene Antibiotic Amphotericin B in Acidic Medium: Implications for the Biological Action. Biophys. Chem. 2008, 136, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zameer Shervani, Hideki Etori, Keijiro Taga, Tadayoshi Yoshida, Hirofumi Okabayashi Aggregation of Polyene Antibiotics as Studied by Electronic Absorption and Circular Dichroism Spectroscopies. Colloids Surf. B Biointerfaces 1996, 7, 31–38. [CrossRef]
- Legrand, P.; Romero, E.A.; Cohen, B.E.; Bolard, J. Effects of Aggregation and Solvent on the Toxicity of Amphotericin B to Human Erythrocytes. Antimicrob. Agents Chemother. 1992, 36, 2518–2522. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Zhang, Z.; Han, X.; Tang, J.; Wang, J.; Dong, S.; Wang, E. Ion Channel Behavior of Amphotericin B in Sterol-Free and Cholesterol- or Ergosterol-Containing Supported Phosphatidylcholine Bilayer Model Membranes Investigated by Electrochemistry and Spectroscopy. Biophys. J. 2002, 83, 3245–3255. [Google Scholar] [CrossRef] [Green Version]
- Antillón, A.; de Vries, A.H.; Espinosa-Caballero, M.; Falcón-González, J.M.; Flores Romero, D.; González-Damián, J.; Jiménez-Montejo, F.E.; León-Buitimea, A.; López-Ortiz, M.; Magaña, R.; et al. An Amphotericin B Derivative Equally Potent to Amphotericin B and with Increased Safety. PLoS ONE 2016, 11, e0162171. [Google Scholar] [CrossRef]
- Palacios, D.S.; Anderson, T.M.; Burke, M.D. A Post-PKS Oxidation of the Amphotericin B Skeleton Predicted to Be Critical for Channel Formation Is Not Required for Potent Antifungal Activity. J. Am. Chem. Soc. 2007, 129, 13804–13805. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhang, J.; Li, X.; Xiao, E.; Lange, J.D.; Rienstra, C.M.; Burke, M.D.; Mitchell, D.A. Sterol Sponge Mechanism Is Conserved for Glycosylated Polyene Macrolides. ACS Cent Sci 2021, 7, 781–791. [Google Scholar] [CrossRef]
- Anderson, T.M.; Clay, M.C.; Cioffi, A.G.; Diaz, K.A.; Hisao, G.S.; Tuttle, M.D.; Nieuwkoop, A.J.; Comellas, G.; Maryum, N.; Wang, S.; et al. Amphotericin Forms an Extramembranous and Fungicidal Sterol Sponge. Nat. Chem. Biol. 2014, 10, 400–406. [Google Scholar] [CrossRef]
- Barwicz, J.; Christian, S.; Gruda, I. Effects of the Aggregation State of Amphotericin B on Its Toxicity to Mice. Antimicrob. Agents Chemother. 1992, 36, 2310–2315. [Google Scholar] [CrossRef] [Green Version]
- Bergy, M.E.; Eble, T.E. The Filipin Complex. Biochemistry 1968, 7, 653–659. [Google Scholar] [CrossRef]
- Ceder, O.; Ryhage, R.; Rodmar, S.; Nihlgård, B.; Nilsson, L. The Structure of Filipin. Acta Chem. Scand. 1964, 18, 558–560. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Richardson, T.I. Relative and Absolute Configuration of Filipin III. Angew. Chem. Int. Ed. Engl. 1995, 34, 1227–1230. [Google Scholar] [CrossRef]
- Norman, A.W.; Demel, R.A.; de Kruyff, B.; van Deenen, L.L. Studies on the Biological Properties of Polyene Antibiotics. Evidence for the Direct Interaction of Filipin with Cholesterol. J. Biol. Chem. 1972, 247, 1918–1929. [Google Scholar] [CrossRef]
- Norman, A.W.; Demel, R.A.; De Kruyff, B.; Van Kessel, W.S.M.G.; Van Deenen, L.L.M. Studies on the Biological Properties of Polyene Antibiotics: Comparison of Other Polyenes with Filipin in Their Ability to Interact Specifically with Sterol. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1972, 290, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kotler-Brajtburg, J.; Price, H.D.; Medoff, G.; Schlessinger, D.; Kobayashi, G.S. Molecular Basis for the Selective Toxicity of Amphotericin B for Yeast and Filipin for Animal Cells. Antimicrob. Agents Chemother. 1974, 5, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Kinsky, S.C. Comparative Responses of Mammalian Erythrocytes and Microbial Protoplasts to Polyene Antibiotics and Vitamin A. Arch. Biochem. Biophys. 1963, 102, 180–188. [Google Scholar] [CrossRef]
- Tingstad, J.E.; Garrett, E.R. Studies on the Stability of Filipin I: Thermal Degradation in the Presence of Air. J. Am. Pharm. Assoc. 1960, 49, 352–355. [Google Scholar] [CrossRef]
- Gimpl, G.; Gehrig-Burger, K. Probes for Studying Cholesterol Binding and Cell Biology. Steroids 2011, 76, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G. The Use and Abuse of Filipin to Localize Cholesterol in Membranes. Cell Biol. Int. Rep. 1984, 8, 519–535. [Google Scholar] [CrossRef]
- Benussi, A.; Cotelli, M.S.; Padovani, A.; Borroni, B. Recent Neuroimaging, Neurophysiological, and Neuropathological Advances for the Understanding of NPC. F1000Research 2018, 7, 194. [Google Scholar] [CrossRef] [Green Version]
- Barreales, E.G.; Rumbero, Á.; Payero, T.D.; de Pedro, A.; Jambrina, E.; Aparicio, J.F. Structural and Bioactivity Characterization of Filipin Derivatives from Engineered Strains Reveals Clues for Reduced Haemolytic Action. Antibiotics 2020, 9, 413. [Google Scholar] [CrossRef] [PubMed]
- Delves-Broughton, J. PRESERVATIVES|Permitted Preservatives—Natamycin. Encycl. Food Microbiol. 2014, 87–91. [Google Scholar]
- Brik, H. Natamycin. In Analytical Profiles of Drug Substances; Academic Press: Cambridge, MA, USA, 1981; pp. 513–561. [Google Scholar]
- Te Welscher, Y.M.; te Welscher, Y.M.; Jones, L.; van Leeuwen, M.R.; Dijksterhuis, J.; de Kruijff, B.; Eitzen, G.; Breukink, E. Natamycin Inhibits Vacuole Fusion at the Priming Phase via a Specific Interaction with Ergosterol. Antimicrob. Agents Chemother. 2010, 54, 2618–2625. [Google Scholar] [CrossRef] [Green Version]
- Te Welscher, Y.M.; ten Napel, H.H.; Balagué, M.M.; Souza, C.M.; Riezman, H.; de Kruijff, B.; Breukink, E. Natamycin Blocks Fungal Growth by Binding Specifically to Ergosterol without Permeabilizing the Membrane. J. Biol. Chem. 2008, 283, 6393–6401. [Google Scholar] [CrossRef] [Green Version]
- Russell, N.J.; Gould, G.W. Food Preservatives; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2003; ISBN 9780306477362. [Google Scholar]
- Gagoś, M.; Czernel, G. Oxidized Forms of Polyene Antibiotic Amphotericin B. Chem. Phys. Lett. 2014, 598, 5–9. [Google Scholar] [CrossRef]
- Klimek, K.; Strubińska, J.; Czernel, G.; Ginalska, G.; Gagoś, M. In Vitro Evaluation of Antifungal and Cytotoxic Activities as Also the Therapeutic Safety of the Oxidized Form of Amphotericin B. Chem. Biol. Interact. 2016, 256, 47–54. [Google Scholar] [CrossRef]
- Brajtburg, J.; Gruda, I.; Daigle, I.; Medoff, G. Concentration Dependent Dual Effect of the Monolauryl Ester of Sucrose on the Antifungal Activity and Absorption Spectra of Amphotericin B (Fungizone). Biochim. Biophys. Acta (BBA) Biomembr. 1989, 985, 307–312. [Google Scholar] [CrossRef]
- Ernst, C.; Grange, J.; Rinnert, H.; Dupont, G.; Lematre, J. Structure of Amphotericin B Aggregates as Revealed by UV and CD Spectroscopies. Biopolymers 1981, 20, 1575–1588. [Google Scholar] [CrossRef]
- Kagan, S.; Ickowicz, D.E.; Domb, A.J.; Dagan, A.; Polacheck, I. Unique Aggregation of Conjugated Amphotericin B and Its Interaction with Lipid Membranes. Med. Mycol. 2017, 55, 414–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diezi, T.A.; Kwon, G. Amphotericin B/sterol Co-Loaded PEG-Phospholipid Micelles: Effects of Sterols on Aggregation State and Hemolytic Activity of Amphotericin B. Pharm. Res. 2012, 29, 1737–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajtár, M.; Vikmon, M.; Morlin, E.; Szejtli, J. Aggregation of Amphotericin B in the Presence of Gamma-Cyclodextrin. Biopolymers 1989, 28, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Belhachemi, M.H.; Boucherit-Otmani, Z.; Boucherit, K.; Belmir, S. Influence of Ascorbic Acid and α-Tocopherol on the Autoxidation and in Vitro Antifungal Activity of Amphotericin B. Curr. Med. Mycol. 2021, 7, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Lambing, H.E.; Wolf, B.D.; Hartsel, S.C. Temperature Effects on the Aggregation State and Activity of Amphotericin B. Biochim. Biophys. Acta 1993, 1152, 185–188. [Google Scholar] [CrossRef]
- Mazerski, J.; Borowski, E. Molecular Dynamics of Amphotericin B. II. Dimer in Water. Biophys. Chem. 1996, 57, 205–217. [Google Scholar] [CrossRef]
- Zielińska, J.; Wieczór, M.; Bączek, T.; Gruszecki, M.; Czub, J. Thermodynamics and Kinetics of Amphotericin B Self-Association in Aqueous Solution Characterized in Molecular Detail. Sci. Rep. 2016, 6, 19109. [Google Scholar] [CrossRef] [Green Version]
- Milhaud, J.; Ponsinet, V.; Takashi, M.; Michels, B. Interactions of the Drug Amphotericin B with Phospholipid Membranes Containing or Not Ergosterol: New Insight into the Role of Ergosterol. Biochim. Biophys. Acta 2002, 1558, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Szomek, M.; Reinholdt, P.; Petersen, D.; Caci, A.; Kongsted, J.; Wüstner, D. Direct Observation of Nystatin Binding to the Plasma Membrane of Living Cells. Biochim. Biophys. Acta (BBA) Biomembr. 2021, 1863, 183528. [Google Scholar] [CrossRef]
- Brajtburg, J.; Elberg, S.; Schwartz, D.R.; Vertut-Croquin, A.; Schlessinger, D.; Kobayashi, G.S.; Medoff, G. Involvement of Oxidative Damage in Erythrocyte Lysis Induced by Amphotericin B. Antimicrob. Agents Chemother. 1985, 27, 172–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol-Anderson, M.; Sligh, J.E., Jr.; Elberg, S.; Brajtburg, J.; Kobayashi, G.S.; Medoff, G. Role of Cell Defense against Oxidative Damage in the Resistance of Candida Albicans to the Killing Effect of Amphotericin B. Antimicrob. Agents Chemother. 1988, 32, 702–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol-Anderson, M.L.; Brajtburg, J.; Medoff, G. Amphotericin B-Induced Oxidative Damage and Killing of Candida Albicans. J. Infect. Dis. 1986, 154, 76–83. [Google Scholar] [CrossRef]
- Sangalli-Leite, F.; Scorzoni, L.; Mesa-Arango, A.C.; Casas, C.; Herrero, E.; Gianinni, M.J.S.; Rodríguez-Tudela, J.L.; Cuenca-Estrella, M.; Zaragoza, O. Amphotericin B Mediates Killing in Cryptococcus Neoformans through the Induction of a Strong Oxidative Burst. Microbes Infect. 2011, 13, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Phillips, A.J.; Sudbery, I.; Ramsdale, M. Apoptosis Induced by Environmental Stresses and Amphotericin B in Candida Albicans. Proc. Natl. Acad. Sci. USA 2003, 100, 14327–14332. [Google Scholar] [CrossRef] [Green Version]
- Chapman, H.A., Jr.; Hibbs, J.B., Jr. Modulation of Macrophage Tumoricidal Capability by Polyene Antibiotics: Support for Membrane Lipid as a Regulatory Determinant of Macrophage Function. Proc. Natl. Acad. Sci. USA 1978, 75, 4349–4353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.; Thorson, L.; Speert, D.P. Enhancement of Macrophage Superoxide Anion Production by Amphotericin B. Antimicrob. Agents Chemother. 1991, 35, 796–800. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, S.A.A.; Robson, G.D. Oxidative and Amphotericin B-Mediated Cell Death in the Opportunistic Pathogen Aspergillus Fumigatus Is Associated with an Apoptotic-like Phenotype. Microbiology 2004, 150, 1937–1945. [Google Scholar] [CrossRef] [Green Version]
- Blum, G.; Perkhofer, S.; Haas, H.; Schrettl, M.; Würzner, R.; Dierich, M.P.; Lass-Flörl, C. Potential Basis for Amphotericin B Resistance in Aspergillus Terreus. Antimicrob. Agents Chemother. 2008, 52, 1553–1555. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Manoharlal, R.; Negi, A.S.; Prasad, R. Synergistic Anticandidal Activity of Pure Polyphenol Curcumin I in Combination with Azoles and Polyenes Generates Reactive Oxygen Species Leading to Apoptosis. FEMS Yeast Res. 2010, 10, 570–578. [Google Scholar] [CrossRef]
- Faustino; Faustino; Pinheiro Lipid Systems for the Delivery of Amphotericin B in Antifungal Therapy. Pharmaceutics 2020, 12, 29. [CrossRef] [PubMed] [Green Version]
- Arning, M.; Kliche, K.O.; Heer-Sonderhoff, A.H.; Wehmeier, A. Infusion-Related Toxicity of Three Different Amphotericin B Formulations and Its Relation to Cytokine Plasma Levels. Mycoses 1995, 38, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Simitsopoulou, M.; Roilides, E.; Dotis, J.; Dalakiouridou, M.; Dudkova, F.; Andreadou, E.; Walsh, T.J. Differential Expression of Cytokines and Chemokines in Human Monocytes Induced by Lipid Formulations of Amphotericin B. Antimicrob. Agents Chemother. 2005, 49, 1397–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.M.; Sugar, I.P.; Chong, P.L. Role of the Sterol Superlattice in the Partitioning of the Antifungal Drug Nystatin into Lipid Membranes. Biochemistry 1998, 37, 11797–11805. [Google Scholar] [CrossRef]
- Tang, D.; Chong, P.L. E/M Dips. Evidence for Lipids Regularly Distributed into Hexagonal Super-Lattices in Pyrene-PC/DMPC Binary Mixtures at Specific Concentrations. Biophys. J. 1992, 63, 903–910. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.L. Evidence for Regular Distribution of Sterols in Liquid Crystalline Phosphatidylcholine Bilayers. Proc. Natl. Acad. Sci. USA 1994, 91, 10069–10073. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.L.; Liu, F.; Wang, M.M.; Truong, K.; Sugar, I.P.; Brown, R.E. Fluorescence Evidence for Cholesterol Regular Distribution in Phosphatidylcholine and in Sphingomyelin Lipid Bilayers. J. Fluoresc. 1996, 6, 221–230. [Google Scholar] [CrossRef]
- Chong, P.L.-G. Effects of Sterol Mole Fraction on Membrane Lateral Organization: Linking Fluorescence Signals to Sterol Superlattices. Perspect. Fluoresc. 2016, 17, 179–196. [Google Scholar]
- Santos, N.C.; Ter-Ovanesyan, E.; Zasadzinski, J.A.; Prieto, M.; Castanho, M.A. Filipin-Induced Lesions in Planar Phospholipid Bilayers Imaged by Atomic Force Microscopy. Biophys. J. 1998, 75, 1869–1873. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, J.C.; Saslowsky, D.E.; Edwardson, J.M.; Henderson, R.M. Real-Time Analysis of the Effects of Cholesterol on Lipid Raft Behavior Using Atomic Force Microscopy. Biophys. J. 2003, 84, 1827–1832. [Google Scholar] [CrossRef] [Green Version]
- Castanho, M.A.; Prieto, M.J. Fluorescence Study of the Macrolide Pentaene Antibiotic Filipin in Aqueous Solution and in a Model System of Membranes. Eur. J. Biochem. 1992, 207, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, T.E.; Dennis, V.W.; Weigl, A.M. The Effect of Amphotericin B on the Water and Nonelectrolyte Permeability of Thin Lipid Membranes. J. Gen. Physiol. 1969, 53, 133–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreoli, T.E. The Structure and Function of Amphotericin B-Cholesterol Pores in Lipid Bilayer Membranes. Ann. N. Y. Acad. Sci. 1974, 235, 448–468. [Google Scholar] [CrossRef] [PubMed]
- Holz, R.; Finkelstein, A. The Water and Nonelectrolyte Permeability Induced in Thin Lipid Membranes by the Polyene Antibiotics Nystatin and Amphotericin B. J. Gen. Physiol. 1970, 56, 125–145. [Google Scholar] [CrossRef] [Green Version]
- Marty, A.; Finkelstein, A. Pores Formed in Lipid Bilayer Membranes by Nystatin, Differences in Its One-Sided and Two-Sided Action. J. Gen. Physiol. 1975, 65, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Umegawa, Y.; Tsuchikawa, H.; Hanashima, S.; Matsumori, N.; Funahashi, K.; Seo, S.; Shinoda, W.; Murata, M. The Amphotericin B–Ergosterol Complex Spans a Lipid Bilayer as a Single-Length Assembly. Biochemistry 2019, 58, 5188–5196. [Google Scholar] [CrossRef]
- Cohen, B.E. A Sequential Mechanism for the Formation of Aqueous Channels by Amphotericin B in Liposomes. The Effect of Sterols and Phospholipid Composition. Biochim. Biophys. Acta 1992, 1108, 49–58. [Google Scholar] [CrossRef]
- Coutinho, A.; Prieto, M. Cooperative Partition Model of Nystatin Interaction with Phospholipid Vesicles. Biophys. J. 2003, 84, 3061–3078. [Google Scholar] [CrossRef] [Green Version]
- Cotero, B.V.; Rebolledo-Antúnez, S.; Ortega-Blake, I. On the Role of Sterol in the Formation of the Amphotericin B Channel. Biochim. Biophys. Acta 1998, 1375, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Ermishkin, L.N.; Kasumov, K.M.; Potzeluyev, V.M. Single Ionic Channels Induced in Lipid Bilayers by Polyene Antibiotics Amphotericin B and Nystatine. Nature 1976, 262, 698–699. [Google Scholar] [CrossRef]
- Venegas, B.; González-Damián, J.; Celis, H.; Ortega-Blake, I. Amphotericin B Channels in the Bacterial Membrane: Role of Sterol and Temperature. Biophys. J. 2003, 85, 2323–2332. [Google Scholar] [CrossRef] [Green Version]
- Baginski, M.; Resat, H.; Borowski, E. Comparative Molecular Dynamics Simulations of Amphotericin B-Cholesterol/ergosterol Membrane Channels. Biochim. Biophys. Acta 2002, 1567, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Cohen, B.E. Amphotericin B Toxicity and Lethality: A Tale of Two Channels. Int. J. Pharm. 1998, 162, 95–106. [Google Scholar] [CrossRef]
- Silva, L.; Coutinho, A.; Fedorov, A.; Prieto, M. Competitive Binding of Cholesterol and Ergosterol to the Polyene Antibiotic Nystatin. A Fluorescence Study. Biophys. J. 2006, 90, 3625–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeida, R.F.M.; de Almeida, R.F.M.; Fedorov, A.; Prieto, M. Sphingomyelin/Phosphatidylcholine/Cholesterol Phase Diagram: Boundaries and Composition of Lipid Rafts. Biophys. J. 2003, 85, 2406–2416. [Google Scholar] [CrossRef] [Green Version]
- Kristanc, L.; Božič, B.; Gomišček, G. The Role of Sterols in the Lipid Vesicle Response Induced by the Pore-Forming Agent Nystatin. Biochim. Biophys. Acta 2014, 1838, 2635–2645. [Google Scholar] [CrossRef]
- Dong, P.-T.; Zong, C.; Dagher, Z.; Hui, J.; Li, J.; Zhan, Y.; Zhang, M.; Mansour, M.K.; Cheng, J.-X. Polarization-Sensitive Stimulated Raman Scattering Imaging Resolves Amphotericin B Orientation in Candida Membrane. Sci. Adv. 2021, 7, eabd5230. [Google Scholar] [CrossRef]
- Ostroumova, O.S.; Efimova, S.S.; Schagina, L.V. Probing Amphotericin B Single Channel Activity by Membrane Dipole Modifiers. PLoS ONE 2012, 7, e30261. [Google Scholar] [CrossRef]
- Wolf, B.D.; Hartsel, S.C. Osmotic Stress Sensitizes Sterol-Free Phospholipid Bilayers to the Action of Amphotericin B. Biochim. Biophys. Acta (BBA) Biomembr. 1995, 1238, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.; Sadownik, A.; Regen, S.L. Molecular Harpoons: Membrane-Disrupting Surfactants That Recognize Osmotic Stress. J. Am. Chem. Soc. 1992, 114, 4011–4013. [Google Scholar] [CrossRef]
- Naka, K.; Sadownik, A.; Regen, S.L. Molecular Harpoons. Membrane-Disruptive Surfactants That Can Recognize Osmotic Stress in Phospholipid Bilayers. J. Am. Chem. Soc. 1993, 115, 2278–2286. [Google Scholar] [CrossRef]
- Regen, S.L.; Jayasuriya, N.; Fabianowski, W. Supramolecular Surfactants: Amphiphilic Polymers Designed to Disrupt Lipid Membranes. Biochem. Biophys. Res. Commun. 1989, 159, 566–571. [Google Scholar] [CrossRef]
- Nagawa, Y.; Regen, S.L. Membrane-Disrupting Surfactants That Are Highly Selective toward Lipid Bilayers of Varying Cholesterol Content. J. Am. Chem. Soc. 1991, 113, 7237–7240. [Google Scholar] [CrossRef]
- Baginski, M.; Resat, H.; McCammon, J.A. Molecular Properties of Amphotericin B Membrane Channel: A Molecular Dynamics Simulation. Mol. Pharmacol. 1997, 52, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czub, J.; Borowski, E.; Baginski, M. Interactions of Amphotericin B Derivatives with Lipid membranes—A Molecular Dynamics Study. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 2616–2626. [Google Scholar] [CrossRef] [Green Version]
- Czub, J.; Neumann, A.; Borowski, E.; Baginski, M. Influence of a Lipid Bilayer on the Conformational Behavior of Amphotericin B Derivatives—A Molecular Dynamics Study. Biophys. Chem. 2009, 141, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Baginski, M.; Sternal, K.; Czub, J. Molecular Aspects of the Interaction between Amphotericin B and a Phospholipid Bilayer: Molecular Dynamics Studies. J. Mol. Modeling 2004, 10, 223–232. [Google Scholar] [CrossRef]
- Khutorsky, V.E. Structures of Amphotericin B-Cholesterol Complex. Biochim. Biophys. Acta 1992, 1108, 123–127. [Google Scholar] [CrossRef]
- Wu, H.-C.; Yoshioka, T.; Nakagawa, K.; Shintani, T.; Tsuru, T.; Saeki, D.; Shaikh, A.R.; Matsuyama, H. Preparation of Amphotericin B-Ergosterol Structures and Molecular Simulation of Water Adsorption and Diffusion. J. Membr. Sci. 2018, 545, 229–239. [Google Scholar] [CrossRef]
- Grela, E.; Wieczór, M.; Luchowski, R.; Zielinska, J.; Barzycka, A.; Grudzinski, W.; Nowak, K.; Tarkowski, P.; Czub, J.; Gruszecki, W.I. Mechanism of Binding of Antifungal Antibiotic Amphotericin B to Lipid Membranes: An Insight from Combined Single-Membrane Imaging, Microspectroscopy, and Molecular Dynamics. Mol. Pharm. 2018, 15, 4202–4213. [Google Scholar] [CrossRef]
- Matsumori, N.; Tahara, K.; Yamamoto, H.; Morooka, A.; Doi, M.; Oishi, T.; Murata, M. Direct Interaction between Amphotericin B and Ergosterol in Lipid Bilayers As Revealed by 2H NMR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 11855–11860. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Smith, J.C.; Ullmann, G.M. A Molecular Mechanics Force Field for Biologically Important Sterols. J. Comput. Chem. 2005, 26, 1383–1399. [Google Scholar] [CrossRef]
- Zielińska, J.; Wieczór, M.; Chodnicki, P.; Grela, E.; Luchowski, R.; Nierzwicki, Ł.; Bączek, T.; Gruszecki, W.I.; Czub, J. Self-Assembly, Stability and Conductance of Amphotericin B Channels: Bridging the Gap between Structure and Function. Nanoscale 2021, 13, 3686–3697. [Google Scholar] [CrossRef]
- Palacios, D.S.; Dailey, I.; Siebert, D.M.; Wilcock, B.C.; Burke, M.D. Synthesis-Enabled Functional Group Deletions Reveal Key Underpinnings of Amphotericin B Ion Channel and Antifungal Activities. Proc. Natl. Acad. Sci. USA 2011, 108, 6733–6738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Umegawa, Y.; Yamagami, M.; Suzuki, T.; Tsuchikawa, H.; Hanashima, S.; Matsumori, N.; Murata, M. The Perpendicular Orientation of Amphotericin B Methyl Ester in Hydrated Lipid Bilayers Supports the Barrel-Stave Model. Biochemistry 2019, 58, 2282–2291. [Google Scholar] [CrossRef] [PubMed]
- Schneiter, R.; Brügger, B.; Sandhoff, R.; Zellnig, G.; Leber, A.; Lampl, M.; Athenstaedt, K.; Hrastnik, C.; Eder, S.; Daum, G.; et al. Electrospray Ionization Tandem Mass Spectrometry (Esi-Ms/Ms) Analysis of the Lipid Molecular Species Composition of Yeast Subcellular Membranes Reveals Acyl Chain-Based Sorting/Remodeling of Distinct Molecular Species En Route to the Plasma Membrane. J. Cell Biol. 1999, 146, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Delhom, R.; Nelson, A.; Laux, V.; Haertlein, M.; Knecht, W.; Fragneto, G.; Wacklin-Knecht, H.P. The Antifungal Mechanism of Amphotericin B Elucidated in Ergosterol and Cholesterol-Containing Membranes Using Neutron Reflectometry. Nanomaterials 2020, 10, 2439. [Google Scholar] [CrossRef] [PubMed]
- Laniado-Laborín, R.; Cabrales-Vargas, M.N. Amphotericin B: Side Effects and Toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227. [Google Scholar] [CrossRef]
- Ostrosky-Zeichner, L.; Marr, K.A.; Rex, J.H.; Cohen, S.H. Amphotericin B: Time for a New “Gold Standard”. Clin. Infect. Dis. 2003, 37, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Walmsley, S.X. Synthesis and Isolation of Biosynthetic Derivatives of Amphotericin B. Doctoral Dissertation, University of Leicester, Leicester, UK, 2017. [Google Scholar]
- Endo, M.M. Synthesis-Enabled Understanding of the Mechanism of Action of Amphotericin B and the Development of Increased Therapeutic Derivatives. Master’s Thesis, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 2016. [Google Scholar]
- Baghirova, A.A.; Kasumov, K.M. Antifungal Macrocycle Antibiotic Amphotericin B-Its Present and Future. Multidisciplinary Perspective for the Use in the Medical Practice. Biochem. Mosc. Suppl. B Biomed. Chem. 2022, 16, 1–12. [Google Scholar] [CrossRef]
- Olsufyeva, E.N.; Yankovskaya, V.S. Main Trends in the Design of Semi-Synthetic Antibiotics of a New Generation. Russ. Chem. Rev. 2020, 89, 339–378. [Google Scholar] [CrossRef]
- Volmer, A.A.; Szpilman, A.M.; Carreira, E.M. Synthesis and Biological Evaluation of Amphotericin B Derivatives. Nat. Prod. Rep. 2010, 27, 1329. [Google Scholar] [CrossRef] [PubMed]
- Paquet, V.; Carreira, E.M. Significant Improvement of Antifungal Activity of Polyene Macrolides by Bisalkylation of the Mycosamine. Org. Lett. 2006, 8, 1807–1809. [Google Scholar] [CrossRef] [PubMed]
- Preobrazhenskaya, M.N.; Olsufyeva, E.N.; Solovieva, S.E.; Tevyashova, A.N.; Reznikova, M.I.; Luzikov, Y.N.; Terekhova, L.P.; Trenin, A.S.; Galatenko, O.A.; Treshalin, I.D.; et al. Chemical Modification and Biological Evaluation of New Semisynthetic Derivatives of 28,29-Didehydronystatin A1 (S44HP), a Genetically Engineered Antifungal Polyene Macrolide Antibiotic. J. Med. Chem. 2009, 52, 189–196. [Google Scholar] [CrossRef]
- Davis, S.A.; Vincent, B.M.; Endo, M.M.; Whitesell, L.; Marchillo, K.; Andes, D.R.; Lindquist, S.; Burke, M.D. Nontoxic Antimicrobials That Evade Drug Resistance. Nat. Chem. Biol. 2015, 11, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Tevyashova, A.N.; Bychkova, E.N.; Solovieva, S.E.; Zatonsky, G.V.; Grammatikova, N.E.; Isakova, E.B.; Mirchink, E.P.; Treshchalin, I.D.; Pereverzeva, E.R.; Bykov, E.E.; et al. Discovery of Amphamide, a Drug Candidate for the Second Generation of Polyene Antibiotics. ACS Infect. Dis. 2020, 6, 2029–2044. [Google Scholar] [CrossRef]
- Preobrazhenskaya, M.N.; Olsufyeva, E.N.; Tevyashova, A.N.; Printsevskaya, S.S.; Solovieva, S.E.; Reznikova, M.I.; Trenin, A.S.; Galatenko, O.A.; Treshalin, I.D.; Pereverzeva, E.R.; et al. Synthesis and Study of the Antifungal Activity of New Mono- and Disubstituted Derivatives of a Genetically Engineered Polyene Antibiotic 28,29-Didehydronystatin A1 (S44HP). J. Antibiot. 2010, 63, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Tevyashova, A.N.; Olsufyeva, E.N.; Solovieva, S.E.; Printsevskaya, S.S.; Reznikova, M.I.; Trenin, A.S.; Galatenko, O.A.; Treshalin, I.D.; Pereverzeva, E.R.; Mirchink, E.P.; et al. Structure-Antifungal Activity Relationships of Polyene Antibiotics of the Amphotericin B Group. Antimicrob. Agents Chemother. 2013, 57, 3815–3822. [Google Scholar] [CrossRef] [Green Version]
- Wong-Beringer, A.; Jacobs, R.A.; Guglielmo, B.J. Lipid Formulations of Amphotericin B: Clinical Efficacy and Toxicities. Clin. Infect. Dis. 1998, 27, 603–618. [Google Scholar] [CrossRef]
- Ahmed, M.Z.; Rao, T.; Saeed, A.; Mutahir, Z.; Hameed, S.; Inayat, S.; Shahzad, H.; Ullah, N.; Abaid-Ullah, M.; Ibrahim, M.; et al. Antifungal Drugs: Mechanism of Action and Resistance. In Biochemistry of Drug Resistance; Springer: Cham, Switherland, 2021; pp. 143–165. [Google Scholar]
- Chavanet, P.; Clement, C.; Duong, M.; Buisson, M.; D’Athis, P.; Dumas, M.; Bonnin, A.; Portier, H. Toxicity and Efficacy of Conventional Amphotericin B Deoxycholate versus Escalating Doses of Amphotericin B Deoxycholate—fat Emulsion in HIV-Infected Patients with Oral Candidosis. Clin. Microbiol. Infect. 1997, 3, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Richter, A.R.; Feitosa, J.P.A.; Paula, H.C.B.; Goycoolea, F.M.; de Paula, R.C.M. Pickering Emulsion Stabilized by Cashew Gum- Poly-L-Lactide Copolymer Nanoparticles: Synthesis, Characterization and Amphotericin B Encapsulation. Colloids Surf. B Biointerfaces 2018, 164, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.C.O.; Nascimento, A.L.; de Vasconcelos, N.M.; Jerônimo, M.S.; Siqueira, I.M.; R-Santos, L.; Cintra, D.O.S.; Fuscaldi, L.L.; Pires Júnior, O.R.; Titze-de-Almeida, R.; et al. Activity and in Vivo Tracking of Amphotericin B Loaded PLGA Nanoparticles. Eur. J. Med. Chem. 2015, 95, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Aigner, M.; Lass-Flörl, C. Encochleated Amphotericin B: Is the Oral Availability of Amphotericin B Finally Reached? J. Fungi 2020, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- ABELCET (Amphotericin B Lipid Complex). In Antibiotics Manual; Chapter 1; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 1–2.
- Herbrecht, R. Liposome Company Abelcet: Amphotericin B Lipid Complex: Clinical Significance of the Increasing Challenge of Systemic Fungal Infec-tions; Liposome Company: Princeton, NJ, USA, 1995; ISSN 1358-5495. [Google Scholar]
- Janoff, A.S.; Boni, L.T.; Popescu, M.C.; Minchey, S.R.; Cullis, P.R.; Madden, T.D.; Taraschi, T.; Gruner, S.M.; Shyamsunder, E.; Tate, M.W. Unusual Lipid Structures Selectively Reduce the Toxicity of Amphotericin B. Proc. Natl. Acad. Sci. USA 1988, 85, 6122–6126. [Google Scholar] [CrossRef] [Green Version]
- Paterson, D.L.; David, K.; Mrsic, M.; Cetkovsky, P.; Weng, X.-H.; Sterba, J.; Krivan, G.; Boskovic, D.; Lu, M.; Zhu, L.-P.; et al. Pre-Medication Practices and Incidence of Infusion-Related Reactions in Patients Receiving AMPHOTEC: Data from the Patient Registry of Amphotericin B Cholesteryl Sulfate Complex for Injection Clinical Tolerability (PRoACT) Registry. J. Antimicrob. Chemother. 2008, 62, 1392–1400. [Google Scholar] [CrossRef] [Green Version]
- Clemons, K.V.; Stevens, D.A. Comparison of Fungizone, Amphotec, AmBisome, and Abelcet for Treatment of Systemic Murine Cryptococcosis. Antimicrob. Agents Chemother. 1998, 42, 899–902. [Google Scholar] [CrossRef] [Green Version]
- Dietze, R.; Milan, E.P.; Berman, J.D.; Grogl, M.; Falqueto, A.; Feitosa, T.F.; Luz, K.G.; Suassuna, F.A.B.; Marinho, L.A.C.; Ksionski, G. Treatment of Brazilian Kala-Azar with a Short Course of Amphocil (Amphotericin B Cholesterol Dispersion). Clin. Infect. Dis. 1993, 17, 981–986. [Google Scholar] [CrossRef]
- de Marie R Janknegt I A Bakker-Woudenberg, S. Clinical Use of Liposomal and Lipid-Complexed Amphotericin B. J. Antimicrob. Chemother. 1994, 33, 907–916. [Google Scholar]
- Leenders, A.C.; de Marie, S. The Use of Lipid Formulations of Amphotericin B for Systemic Fungal Infections. Leukemia 1996, 10, 1570–1575. [Google Scholar]
- Sperry; SPERRY; CUA; WETZEL; ADLER-MOORE Antimicrobial Activity of AmBisome and Non-Liposomal Amphotericin B Following Uptake of Candida Glabrata by Murine Epidermal Langerhans Cells. Med. Mycol. 2008, 36, 135–141. [CrossRef]
- Schlossberg, D.L.; Rafik Samuel, R. AMBISOME (Liposomal Amphotericin B). In Antibiotics Manual: A Guide to Commonly Used Antimicrobials; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 7–8. [Google Scholar]
- Adler-Moore, J.; Proffitt, R.T. AmBisome: Liposomal Formulation, Structure, Mechanism of Action and Pre-Clinical Experience. J. Antimicrob. Chemother. 2002, 49 (Suppl. S1), 21–30. [Google Scholar] [CrossRef] [PubMed]
- Den Boer, M.; den Boer, M.; Das, A.K.; Akhter, F.; Burza, S.; Ramesh, V.; Ahmed, B.-N.; Zijlstra, E.E.; Ritmeijer, K. Safety and Effectiveness of Short-Course AmBisome in the Treatment of Post–Kala-Azar Dermal Leishmaniasis: A Prospective Cohort Study in Bangladesh. Clin. Infect. Dis. 2018, 67, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wijnant, G.-J.; Van Bocxlaer, K.; Yardley, V.; Harris, A.; Alavijeh, M.; Silva-Pedrosa, R.; Antunes, S.; Mauricio, I.; Murdan, S.; Croft, S.L. Comparative Efficacy, Toxicity and Biodistribution of the Liposomal Amphotericin B Formulations Fungisome and AmBisome in Murine Cutaneous Leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 223–228. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Ali, N. Treatment of Visceral Leishmaniasis: Anomalous Pricing and Distribution of AmBisome and Emergence of an Indigenous Liposomal Amphotericin B, FUNGISOME. J. Parasit. Dis. 2016, 40, 1094–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanath, S.S.; Gogtay, N.J.; Kshirsagar, N.A. Post-Marketing Study to Assess the Safety, Tolerability and Effectiveness of Fungisome: An Indian Liposomal Amphotericin B Preparation. J. Postgrad. Med. 2005, 51 (Suppl. S1), S58–S63. [Google Scholar]
- Kshirsagar, N.; Chadha, A.; Kharkar, V.; Khopkar, U.; Darkase, B.; Patel, S. Treatment of Post Kala-Azar Dermal Leishmaniasis with Fungisome—A Novel Indian Liposomal Amphotericin B. Indian J. Drugs Dermatol. 2020, 6, 28. [Google Scholar] [CrossRef]
- Das, S.; Devarajan, P.V. Enhancing Safety and Efficacy by Altering the Toxic Aggregated State of Amphotericin B in Lipidic Nanoformulations. Mol. Pharm. 2020, 17, 2186–2195. [Google Scholar] [CrossRef]
- Kshirsagar, N.A.; Pandya, S.K.; Kirodian, G.B.; Sanath, S. Liposomal Drug Delivery System from Laboratory to Clinic. J. Postgrad. Med. 2005, 51 (Suppl. S1), S5–S15. [Google Scholar]
- Pupe, C.G.; Villardi, M.; Rodrigues, C.R.; Rocha HV, A.; Maia, L.C.; de Sousa, V.P.; Cabral, L.M. Preparation and Evaluation of Antimicrobial Activity of Nanosystems for the Control of Oral Pathogens Streptococcus Mutans and Candida Albicans. Int. J. Nanomed. 2011, 6, 2581. [Google Scholar]
- Campos, F.F.; Calpena Campmany, A.C.; Delgado, G.R.; Serrano, O.L.; Naveros, B.C. Development and Characterization of a Novel Nystatin-Loaded Nanoemulsion for the Buccal Treatment of Candidosis: Ultrastructural Effects and Release Studies. J. Pharm. Sci. 2012, 101, 3739–3752. [Google Scholar] [CrossRef]
- Khan, M.A.; Aljarbou, A.; Khan, A.; Owais, M. Immune Stimulating and Therapeutic Potential of Tuftsin-Incorporated Nystatin Liposomes against Cryptococcus Neoformans in Leukopenic BALB/C Mice. FEMS Immunol. Med. Microbiol. 2012, 66, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín-Quintero, D.; Fernández-Campos, F.; Calpena-Campmany, A.C.; Montes-López, M.J.; Clares-Naveros, B.; Del Pozo-Carrascosa, A. Formulation Design and Optimization for the Improvement of Nystatin-Loaded Lipid Intravenous Emulsion. J. Pharm. Sci. 2013, 102, 4015–4023. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Campos, F.; Clares Naveros, B.; López Serrano, O.; Alonso Merino, C.; Calpena Campmany, A.C. Evaluation of Novel Nystatin Nanoemulsion for Skin Candidosis Infections. Mycoses 2013, 56, 70–81. [Google Scholar] [CrossRef]
- Adler-Moore, J.; Lewis, R.E.; Brüggemann, R.J.M.; Rijnders, B.J.A.; Groll, A.H.; Walsh, T.J. Preclinical Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Antifungal Activity of Liposomal Amphotericin B. Clin. Infect. Dis. 2019, 68, S244–S259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adedoyin, A.; Bernardo, J.F.; Swenson, C.E.; Bolsack, L.E.; Horwith, G.; DeWit, S.; Kelly, E.; Klasterksy, J.; Sculier, J.P.; DeValeriola, D.; et al. Pharmacokinetic Profile of ABELCET (amphotericin B Lipid Complex Injection): Combined Experience from Phase I and Phase II Studies. Antimicrob. Agents Chemother. 1997, 41, 2201–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adedoyin, A.; Swenson, C.E.; Bolcsak, L.E.; Hellmann, A.; Radowska, D.; Horwith, G.; Janoff, A.S.; Branch, R.A. A Pharmacokinetic Study of Amphotericin B Lipid Complex Injection (Abelcet) in Patients with Definite or Probable Systemic Fungal Infections. Antimicrob. Agents Chemother. 2000, 44, 2900–2902. [Google Scholar] [CrossRef] [Green Version]
- Ayestarán, A.; López, R.M.; Montoro, J.B.; Estíbalez, A.; Pou, L.; Julià, A.; López, A.; Pascual, B. Pharmacokinetics of Conventional Formulation versus Fat Emulsion Formulation of Amphotericin B in a Group of Patients with Neutropenia. Antimicrob. Agents Chemother. 1996, 40, 609–612. [Google Scholar] [CrossRef] [Green Version]
- Weiler, S.; Überlacher, E.; Schöfmann, J.; Stienecke, E.; Dunzendorfer, S.; Joannidis, M.; Bellmann, R. Pharmacokinetics of Amphotericin B Colloidal Dispersion in Critically Ill Patients with Cholestatic Liver Disease. Antimicrob. Agents Chemother. 2012, 56, 5414–5418. [Google Scholar] [CrossRef] [Green Version]
- Amantea, M.A.; Bowden, R.A.; Forrest, A.; Working, P.K.; Newman, M.S.; Mamelok, R.D. Population Pharmacokinetics and Renal Function-Sparing Effects of Amphotericin B Colloidal Dispersion in Patients Receiving Bone Marrow Transplants. Antimicrob. Agents Chemother. 1995, 39, 2042–2047. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, V.; Bosse, D.; Jehn, U.; Kähny, B.; Wachholz, K.; Debus, A.; Scholz, P.; Kolb, H.J.; Wilmanns, W. Pharmacokinetics of Liposomal Amphotericin B (Ambisome) in Critically Ill Patients. Antimicrob. Agents Chemother. 1997, 41, 1275–1280. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.J.; Yeldandi, V.; McEvoy, M.; Gonzalez, C.; Chanock, S.; Freifeld, A.; Seibel, N.I.; Whitcomb, P.O.; Jarosinski, P.; Boswell, G.; et al. Safety, Tolerance, and Pharmacokinetics of a Small Unilamellar Liposomal Formulation of Amphotericin B (AmBisome) in Neutropenic Patients. Antimicrob. Agents Chemother. 1998, 42, 2391–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, T.J.; Goodman, J.L.; Pappas, P.; Bekersky, I.; Buell, D.N.; Roden, M.; Barrett, J.; Anaissie, E.J. Safety, Tolerance, and Pharmacokinetics of High-Dose Liposomal Amphotericin B (AmBisome) in Patients Infected with Aspergillus Species and Other Filamentous Fungi: Maximum Tolerated Dose Study. Antimicrob. Agents Chemother. 2001, 45, 3487–3496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, L.; Sood, P.; Lenardon, M.D.; Milne, G.; Olson, J.; Jensen, G.; Wolf, J.; Casadevall, A.; Adler-Moore, J.; Gow, N.A.R. The Viscoelastic Properties of the Fungal Cell Wall Allow Traffic of AmBisome as Intact Liposome Vesicles. mBio 2018, 9, e02383-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, K.; Takemoto, K. Efficacy of Liposomal Amphotericin B against Four Species of Candida Biofilms in an Experimental Mouse Model of Intravascular Catheter Infection. J. Infect. Chemother. 2018, 24, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Yamagishi, Y.; Mikamo, H. In Vitro Efficacy of Liposomal Amphotericin B, Micafungin and Fluconazole against Non-Albicans Candida Species Biofilms. J. Infect. Chemother. 2015, 21, 647–653. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Long, L.; Kim, H.G.; Ghannoum, M.A. Amphotericin B Lipid Complex Is Efficacious in the Treatment of Candida Albicans Biofilms Using a Model of Catheter-Associated Candida Biofilms. Int. J. Antimicrob. Agents 2009, 33, 149–153. [Google Scholar] [CrossRef]
- Schinabeck, M.K.; Long, L.A.; Hossain, M.A.; Chandra, J.; Mukherjee, P.K.; Mohamed, S.; Ghannoum, M.A. Rabbit Model of Candida Albicans Biofilm Infection: Liposomal Amphotericin B Antifungal Lock Therapy. Antimicrob. Agents Chemother. 2004, 48, 1727–1732. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Teng, F.; Zhou, F.; Zhu, L.; Wen, Y.; Feng, R.; Song, Z. Linolenic Acid-Modified MPEG-PEI Micelles for Encapsulation of Amphotericin B. Future Med. Chem. 2019, 11, 2647–2662. [Google Scholar] [CrossRef]
- Bolard, J.; Vertut-Croquin, A.; Cybulska, B.E.; Gary-Bobo, C.M. Transfer of the Polyene Antibiotic Amphotericin B between Single-Walled Vesicles of Dipalmitoylphosphatidylcholine and Egg-Yolk Phosphatidylcholine. Biochim. Biophys. Acta (BBA) Biomembr. 1981, 647, 241–248. [Google Scholar] [CrossRef]
- Zumbuehl, A.; Stano, P.; Heer, D.; Walde, P.; Carreira, E.M. Amphotericin B as a Potential Probe of the Physical State of Vesicle Membranes. Org. Lett. 2004, 6, 3683–3686. [Google Scholar] [CrossRef]
- Serrano, D.; Ballesteros, M.; Schätzlein, A.; Torrado, J.; Uchegbu, I. Amphotericin B Formulations—The Possibility of Generic Competition. Pharm. Nanotechnol. 2013, 1, 250–258. [Google Scholar] [CrossRef]
- Cullis, P.R.; Chonn, A.; Semple, S.C. Interactions of Liposomes and Lipid-Based Carrier Systems with Blood Proteins: Relation to Clearance Behaviour in Vivo. Adv. Drug Deliv. Rev. 1998, 32, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.R.; Bondurant, B.; McLean, S.D.; McGovern, K.A.; O’Brien, D.F. Liposome−Cell Interactions in Vitro: Effect of Liposome Surface Charge on the Binding and Endocytosis of Conventional and Sterically Stabilized Liposomes. Biochemistry 1998, 37, 12875–12883. [Google Scholar] [CrossRef]
- Azanza, J.R.; Sádada, B.; Reis, J. Liposomal Formulations of Amphotericin B: Differences according to the Scientific Evidence. Rev. Esp. Quimioter. 2015, 28, 275–281. [Google Scholar] [PubMed]
- Campbell, R.B.; Fukumura, D.; Brown, E.B.; Mazzola, L.M.; Izumi, Y.; Jain, R.K.; Torchilin, V.P.; Munn, L.L. Cationic Charge Determines the Distribution of Liposomes between the Vascular and Extravascular Compartments of Tumors. Cancer Res. 2002, 62, 6831–6836. [Google Scholar] [PubMed]
- Rivnay, B.; Wakim, J.; Avery, K.; Petrochenko, P.; Myung, J.H.; Kozak, D.; Yoon, S.; Landrau, N.; Nivorozhkin, A. Critical Process Parameters in Manufacturing of Liposomal Formulations of Amphotericin B. Int. J. Pharm. 2019, 565, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Patere, S.N.; Pathak, P.O.; Kumar Shukla, A.; Singh, R.K.; Kumar Dubey, V.; Mehta, M.J.; Patil, A.G.; Gota, V.; Nagarsenker, M.S. Surface-Modified Liposomal Formulation of Amphotericin B: In Vitro Evaluation of Potential Against Visceral Leishmaniasis. AAPS PharmSciTech 2017, 18, 710–720. [Google Scholar] [CrossRef]
- Zhao, M.; Hu, J.; Zhang, L.; Zhang, L.; Sun, Y.; Ma, N.; Chen, X.; Gao, Z. Study of Amphotericin B Magnetic Liposomes for Brain Targeting. Int. J. Pharm. 2014, 475, 9–16. [Google Scholar] [CrossRef]
- Vanegas, J.M.; Faller, R.; Longo, M.L. Influence of Ethanol on Lipid/Sterol Membranes: Phase Diagram Construction from AFM Imaging. Langmuir 2010, 26, 10415–10418. [Google Scholar] [CrossRef]
- Yuan, C.; Johnston, L.J. Phase Evolution in cholesterol/DPPC Monolayers: Atomic Force Microscopy and near Field Scanning Optical Microscopy Studies. J. Microsc. 2002, 205, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ma, Y.; Hou, S.; Miao, Z.; Ma, Q. Interaction of Amphotericin B and Saturated or Unsaturated Phospholipid Monolayers Containing Cholesterol or Ergosterol at the Air-Water Interface. Biophys. Chem. 2020, 258, 106317. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.-W.; Gilbert, K.; Trandum, C.; Zuckermann, M.; Thewalt, J. The Effect of Ergosterol on Dipalmitoylphosphatidylcholine Bilayers: A Deuterium NMR and Calorimetric Study. Biophys. J. 2005, 88, 1799–1808. [Google Scholar] [CrossRef] [Green Version]
- Shaghaghi, M.; Chen, M.-T.; Hsueh, Y.-W.; Zuckermann, M.J.; Thewalt, J.L. Effect of Sterol Structure on the Physical Properties of 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine Membranes Determined Using 2H Nuclear Magnetic Resonance. Langmuir 2016, 32, 7654–7663. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.-C.; Lee, M.-T.; Chung, H.; Sun, Y.-T.; Chen, H.; Charron, N.E.; Huang, H.W. Comparative Study of the Condensing Effects of Ergosterol and Cholesterol. Biophys. J. 2016, 110, 2026–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pencer, J.; Nieh, M.-P.; Harroun, T.A.; Krueger, S.; Adams, C.; Katsaras, J. Bilayer Thickness and Thermal Response of Dimyristoylphosphatidylcholine Unilamellar Vesicles Containing Cholesterol, Ergosterol and Lanosterol: A Small-Angle Neutron Scattering Study. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2005, 1720, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Récamier, K.S.; Hernández-Gómez, A.; González-Damián, J.; Ortega-Blake, I. Effect of Membrane Structure on the Action of Polyenes: I. Nystatin Action in Cholesterol- and Ergosterol-Containing Membranes. J. Membr. Biol. 2010, 237, 31–40. [Google Scholar] [CrossRef]
- HsuChen, C.C.; Feingold, D.S. Polyene Antibiotic Action on Lecithin Liposomes: Effect of Cholesterol and Fatty Acyl Chains. Biochem. Biophys. Res. Commun. 1973, 51, 972–978. [Google Scholar] [CrossRef]
- Ruckwardt, T.; Scott, A.; Scott, J.; Mikulecky, P.; Hartsel, S.C. Lipid and Stress Dependence of Amphotericin B Ion Selective Channels in Sterol-Free Membranes. Biochim. Biophys. Acta 1998, 1372, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Miñones, J.; Miñones, J.; Rodríguez-Patino, J.M.; Conde, O.; Iribarnegaray, E. Miscibility of Amphotericin B−Dipalmitoyl Phosphatidyl Serine Mixed Monolayers Spread on the Air/Water Interface. J. Phys. Chem. B 2003, 107, 4189–4195. [Google Scholar] [CrossRef]
- Miñones, J.; Miñones, J.; Conde, O.; Rodriguez Patino, J.M.; Dynarowicz-Latka, P. Mixed Monolayers of Amphotericin B−Dipalmitoyl Phosphatidyl Choline: Study of Complex Formation. Langmuir 2002, 18, 2817–2827. [Google Scholar] [CrossRef]
- Arczewska, M.; Gagoś, M. Molecular Organization of Antibiotic Amphotericin B in Dipalmitoylphosphatidylcholine Monolayers Induced by K and Na Ions: The Langmuir Technique Study. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2011, 1808, 2706–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, I.; Barwicz, J.; Tancrède, P. The Structuring Effects of Amphotericin B on Pure and Ergosterol- or Cholesterol-Containing Dipalmitoylphosphatidylcholine Bilayers: A Differential Scanning Calorimetry Study. Biochim. Biophys. Acta 1998, 1373, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Paquet, M.-J.; Fournier, I.; Barwicz, J.; Tancrède, P.; Auger, M. The Effects of Amphotericin B on Pure and Ergosterol- or Cholesterol-Containing Dipalmitoylphosphatidylcholine Bilayers as Viewed by 2H NMR. Chem. Phys. Lipids 2002, 119, 1–11. [Google Scholar] [CrossRef]
- Castanho, M.A.; Prieto, M.; Jameson, D.M. The Pentaene Macrolide Antibiotic Filipin Prefers More Rigid DPPC Bilayers: A Fluorescence Pressure Dependence Study. Biochim. Biophys. Acta 1999, 1419, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Konyakhina, T.M.; Feigenson, G.W. Phase Diagram of a Polyunsaturated Lipid Mixture: Brain sphingomyelin/1-Stearoyl-2-Docosahexaenoyl-Sn-Glycero-3-Phosphocholine/cholesterol. Biochim. Biophys. Acta 2016, 1858, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Veatch, S.L.; Keller, S.L. Miscibility Phase Diagrams of Giant Vesicles Containing Sphingomyelin. Phys. Rev. Lett. 2005, 94, 148101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokorny, A.; Yandek, L.E.; Elegbede, A.I.; Hinderliter, A.; Almeida, P.F.F. Temperature and Composition Dependence of the Interaction of Delta-Lysin with Ternary Mixtures of sphingomyelin/cholesterol/POPC. Biophys. J. 2006, 91, 2184–2197. [Google Scholar] [CrossRef] [Green Version]
- Veatch, S.L.; Soubias, O.; Keller, S.L.; Gawrisch, K. Critical Fluctuations in Domain-Forming Lipid Mixtures. Proc. Natl. Acad. Sci. USA 2007, 104, 17650–17655. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.H.; Clair, J.J.; Juhasz, J. Phase Equilibria in DOPC/DPPC-d62/cholesterol Mixtures. Biophys. J. 2009, 96, 521–539. [Google Scholar] [CrossRef] [Green Version]
- Veatch, S.L.; Gawrisch, K.; Keller, S.L. Closed-Loop Miscibility Gap and Quantitative Tie-Lines in Ternary Membranes Containing Diphytanoyl PC. Biophys. J. 2006, 90, 4428–4436. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wu, J.; Heberle, F.A.; Mills, T.T.; Klawitter, P.; Huang, G.; Costanza, G.; Feigenson, G.W. Phase Studies of Model Biomembranes: Complex Behavior of DSPC/DOPC/cholesterol. Biochim. Biophys. Acta 2007, 1768, 2764–2776. [Google Scholar] [CrossRef] [Green Version]
- Silvius, J.R.; del Giudice, D.; Lafleur, M. Cholesterol at Different Bilayer Concentrations Can Promote or Antagonize Lateral Segregation of Phospholipids of Differing Acyl Chain Length. Biochemistry 1996, 35, 15198–15208. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.-W.; Zhao, J.; Wu, J.; Shimoyama, Y.; Freed, J.H.; Feigenson, G.W. New Method for Determining Tie-Lines in Coexisting Membrane Phases Using Spin-Label ESR. Biochim. Biophys. Acta 2005, 1668, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Galván-Hernández, A.; Kobayashi, N.; Hernández-Cobos, J.; Antillón, A.; Nakabayashi, S.; Ortega-Blake, I. Morphology and Dynamics of Domains in Ergosterol or Cholesterol Containing Membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2020, 1862, 183101. [Google Scholar] [CrossRef] [PubMed]
- Chulkov, E.G.; Efimova, S.S.; Schagina, L.V.; Ostroumova, O.S. Direct Visualization of Solid Ordered Domains Induced by Polyene Antibiotics in Giant Unilamellar Vesicles. Chem. Phys. Lipids 2014, 183, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional Rafts in Cell Membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Aresta-Branco, F.; Cordeiro, A.M.; Marinho, H.S.; Cyrne, L.; Antunes, F.; de Almeida, R.F.M. Gel Domains in the Plasma Membrane of Saccharomyces Cerevisiae: Highly Ordered, Ergosterol-Free, and Sphingolipid-Enriched Lipid Rafts. J. Biol. Chem. 2011, 286, 5043–5054. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, P. Management of Invasive Fungal Infections: A Role for Polyenes. J. Antimicrob. Chemother. 2011, 66, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Campoy, S.; Adrio, J.L. Antifungals. Biochem. Pharmacol. 2017, 133, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.R.H.; Bicanic, T.; Salim, R.; Hope, W. Liposomal Amphotericin B (AmBisome(®)): A Review of the Pharmacokinetics, Pharmacodynamics, Clinical Experience and Future Directions. Drugs 2016, 76, 485–500. [Google Scholar] [CrossRef] [Green Version]
- Galgiani, J.N.; Ampel, N.M.; Blair, J.E.; Catanzaro, A.; Johnson, R.H.; Stevens, D.A.; Williams, P.L. Coccidioidomycosis. Clin. Infect. Dis. 2005, 41, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A. Fungizone Intravenous. In Hawley’s Condensed Chemical Dictionary; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Chapman, S.W.; Dismukes, W.E.; Proia, L.A.; Bradsher, R.W.; Pappas, P.G.; Threlkeld, M.G.; Kauffman, C.A.; Infectious Diseases Society of America. Clinical Practice Guidelines for the Management of Blastomycosis: 2008 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 46, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.F.; Woeltje, K.; Espinel-Ingroff, A.; Stanfield, J.; DiPiro, J.T. Efficacy of a Single Intravenous Dose of Amphotericin B for Candida Urinary Tract Infections: Further Favorable Experience. Clin. Microbiol. Infect. 2003, 9, 1024–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesbit, S.A.; Katz, L.E.; McClain, B.W.; Murphy, D.P. Comparison of Two Concentrations of Amphotericin B Bladder Irrigation in the Treatment of Funguria in Patients with Indwelling Urinary Catheters. Am. J. Health. Syst. Pharm. 1999, 56, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Sorkine, P.; Nagar, H.; Weinbroum, A.; Setton, A.; Israitel, E.; Scarlatt, A.; Silbiger, A.; Rudick, V.; Kluger, Y.; Halpern, P. Administration of Amphotericin B in Lipid Emulsion Decreases Nephrotoxicity: Results of a Prospective, Randomized, Controlled Study in Critically Ill Patients. Crit. Care Med. 1996, 24, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.; Pottage, J.C., Jr.; Weaver, D.C. Candida Krusei Fungemia. Report of 4 Cases and Review of the Literature. Medicine 1993, 72, 143–150. [Google Scholar] [CrossRef]
- Donowitz, L.G.; Hendley, J.O. Short-Course Amphotericin B Therapy for Candidemia in Pediatric Patients. Pediatrics 1995, 95, 888–891. [Google Scholar] [CrossRef]
- Zenker, P.N.; Rosenberg, E.M.; Van Dyke, R.B.; Rabalais, G.P.; Daum, R.S. Successful Medical Treatment of Presumed Candida Endocarditis in Critically Ill Infants. J. Pediatr. 1991, 119, 472–477. [Google Scholar] [CrossRef]
- Hall, K.A.; Sethi, G.K.; Rosado, L.J.; Martinez, J.D.; Huston, C.L.; Copeland, J.G. Coccidioidomycosis and Heart Transplantation. J. Heart Lung Transplant. 1993, 12, 525–526. [Google Scholar]
- Mofenson, L.M.; Brady, M.T.; Danner, S.P.; Dominguez, K.L.; Hazra, R.; Handelsman, E.; Havens, P.; Nesheim, S.; Read, J.S.; Serchuck, L.; et al. Guidelines for the Prevention and Treatment of Opportunistic Infections among HIV-Exposed and HIV-Infected Children: Recommendations from CDC, the National Institutes of Health, the HIV Medicine Association of the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the American Academy of Pediatrics. MMWR Recomm. Rep. 2009, 58, 1–166. [Google Scholar]
- Kaplan, J.E.; Benson, C.; Holmes, K.K.; Brooks, J.T.; Pau, A.; Masur, H.; Centers for Disease Control and Prevention (CDC); National Institutes of Health; HIV Medicine Association of the Infectious Diseases Society of America. Guidelines for Prevention and Treatment of Opportunistic Infections in HIV-Infected Adults and Adolescents: Recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm. Rep. 2009, 58, 1–207, quiz CE1–CE4. [Google Scholar] [PubMed]
- Pappas, P.G.; Chetchotisakd, P.; Larsen, R.A.; Manosuthi, W.; Morris, M.I.; Anekthananon, T.; Sungkanuparph, S.; Supparatpinyo, K.; Nolen, T.L.; Zimmer, L.O.; et al. A Phase II Randomized Trial of Amphotericin B Alone or Combined with Fluconazole in the Treatment of HIV-Associated Cryptococcal Meningitis. Clin. Infect. Dis. 2009, 48, 1775–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicanic, T.; Wood, R.; Meintjes, G.; Rebe, K.; Brouwer, A.; Loyse, A.; Bekker, L.-G.; Jaffar, S.; Harrison, T. High-Dose Amphotericin B with Flucytosine for the Treatment of Cryptococcal Meningitis in HIV-Infected Patients: A Randomized Trial. Clin. Infect. Dis. 2008, 47, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Horst, C.M.; Saag, M.S.; Cloud, G.A.; Hamill, R.J.; Graybill, J.R.; Sobel, J.D.; Johnson, P.C.; Tuazon, C.U.; Kerkering, T.; Moskovitz, B.L.; et al. Treatment of Cryptococcal Meningitis Associated with the Acquired Immunodeficiency Syndrome. National Institute of Allergy and Infectious Diseases Mycoses Study Group and AIDS Clinical Trials Group. N. Engl. J. Med. 1997, 337, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Perfect, J.R.; Dismukes, W.E.; Dromer, F.; Goldman, D.L.; Graybill, J.R.; Hamill, R.J.; Harrison, T.S.; Larsen, R.A.; Lortholary, O.; Nguyen, M.-H.; et al. Clinical Practice Guidelines for the Management of Cryptococcal Disease: 2010 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2010, 50, 291–322. [Google Scholar] [CrossRef] [Green Version]
- Dismukes, W.E.; Cloud, G.; Gallis, H.A.; Kerkering, T.M.; Medoff, G.; Craven, P.C.; Kaplowitz, L.G.; Fisher, J.F.; Gregg, C.R.; Bowles, C.A.; et al. Treatment of Cryptococcal Meningitis with Combination Amphotericin B and Flucytosine for Four as Compared with Six Weeks. N. Engl. J. Med. 1987, 317, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Dismukes, W.E. Management of Cryptococcosis. Clin. Infect. Dis. 1993, 17 (Suppl. S2), S507–S512. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.E.; Dismukes, W.E.; Duma, R.J.; Medoff, G.; Sande, M.A.; Gallis, H.; Leonard, J.; Fields, B.T.; Bradshaw, M.; Haywood, H.; et al. A Comparison of Amphotericin B Alone and Combined with Flucytosine in the Treatment of Cryptoccal Meningitis. N. Engl. J. Med. 1979, 301, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alazraki, N.P.; Fierer, J.; Halpern, S.E.; Becker, R.W. Use of a Hyperbaric Solution for Administration of Intrathecal Amphotericin B. N. Engl. J. Med. 1974, 290, 641–646. [Google Scholar] [CrossRef]
- Camarata, P.J.; Dunn, D.L.; Farney, A.C.; Parker, R.G.; Seljeskog, E.L. Continual Intracavitary Administration of Amphotericin B as an Adjunct in the Treatment of Aspergillus Brain Abscess: Case Report and Review of the Literature. Neurosurgery 1992, 31, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Adler, S. Candidal Osteomyelitis and Arthritis in a Neonate. Arch. Pediatr. Adolesc. Med. 1972, 123, 595. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.D.; Yamauchi, T.; Horlick, S.P. Neonatal Candidiasis, Meningitis, Andarthritis: Observations and a Review of the Literature. J. Pediatr. 1972, 81, 31–34. [Google Scholar] [CrossRef]
- Graybill, J.R.; Ellenbogen, C. Complications with the Ommaya Reservoir in Patients with Granulomatous Meningitis. J. Neurosurg. 1973, 38, 477–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giron, J.M.; Poey, C.G.; Fajadet, P.P.; Balagner, G.B.; Assoun, J.A.; Richardi, G.R.; Haddad, J.H.; Caceres, J.C.; Senac, J.P.; Railhac, J.J. Inoperable Pulmonary Aspergilloma: Percutaneous CT-Guided Injection with Glycerin and Amphotericin B Paste in 15 Cases. Radiology 1993, 188, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Munk, P.L.; Vellet, A.D.; Rankin, R.N.; Müller, N.L.; Ahmad, D. Intracavitary Aspergilloma: Transthoracic Percutaneous Injection of Amphotericin Gelatin Solution. Radiology 1993, 188, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kim, H.T.; Kim, Y.H.; Choe, K.O. Treatment of Hemoptysis in Patients with Cavitary Aspergilloma of the Lung: Value of Percutaneous Instillation of Amphotericin B. AJR Am. J. Roentgenol. 1993, 161, 727–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochrane, L.J.; James Cochrane, L.; Morano, J.U.; Norman, J.R.; Keith Mansel, J. Use of Intracavitary Amphotericin B in a Patient with Aspergilloma and Recurrent Hemoptysis. Am. J. Med. 1991, 90, 654–656. [Google Scholar] [CrossRef]
- Hargis, J.L.; Bone, R.C.; Stewart, J.; Rector, N.; Charles Hiller, F. Intracavitary Amphotericin B in the Treatment of Symptomatic Pulmonary Aspergillomas. Am. J. Med. 1980, 68, 389–394. [Google Scholar] [CrossRef]
- Schwartz, S.; Behre, G.; Heinemann, V.; Wandt, H.; Schilling, E.; Arning, M.; Trittin, A.; Kern, W.V.; Boenisch, O.; Bosse, D.; et al. Aerosolized Amphotericin B Inhalations as Prophylaxis of Invasive Aspergillus Infections during Prolonged Neutropenia: Results of a Prospective Randomized Multicenter Trial. Blood 1999, 93, 3654–3661. [Google Scholar]
- Wheat, L.J.; Joseph Wheat, L.; Freifeld, A.G.; Kleiman, M.B.; Baddley, J.W.; McKinsey, D.S.; Loyd, J.E.; Kauffman, C.A. Clinical Practice Guidelines for the Management of Patients with Histoplasmosis: 2007 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2007, 45, 807–825. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, D.S.; Gupta, M.R.; Driks, M.R.; Smith, D.L.; O’Connor, M. Histoplasmosis in Patients with AIDS: Efficacy of Maintenance Amphotericin B Therapy. Am. J. Med. 1992, 92, 225–227. [Google Scholar] [CrossRef]
- Wheat, J. Prevention of Relapse of Histoplasmosis with Itraconazole in Patients with the Acquired Immunodeficiency Syndrome. Ann. Intern. Med. 1993, 118, 610. [Google Scholar] [CrossRef]
- Temple, M.E.; Brady, M.T.; Koranyi, K.I.; Nahata, M.C. Periorbital Cellulitis Secondary to Conidiobolus Incongruus. Pharmacotherapy 2001, 21, 351–354. [Google Scholar] [CrossRef]
- Sugar, A.M. Mucormycosis. Clin. Infect. Dis. 1992, 14 (Suppl. S1), S126–S129. [Google Scholar] [CrossRef]
- Nussbaum, E.S.; Hall, W.A. Rhinocerebral Mucormycosis: Changing Patterns of Disease. Surg. Neurol. 1994, 41, 152–156. [Google Scholar] [CrossRef]
- Borg, F.T.; Ter Borg, F.; Kuijper, E.J.; Van Der Lelie, H. Fatal Mucormycosis Presenting as an Appendiceal Mass with Metastatic Spread to the Liver during Chemotherapy-Induced Granulocytopenia. Scand. J. Infect. Dis. 1990, 22, 499–501. [Google Scholar] [CrossRef]
- Ishida, M.; Taya, N.; Noiri, T.; Kamihata, T.; Hatta, C.; Matsumoto, T.; Sugiyama, Y.; Yoshihara, W. Five Cases of Mucormycosis in Paranasal Sinuses. Acta Otolaryngol. Suppl. 1993, 501, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Adler, D.E.; Milhorat, T.H.; Miller, J.I. Treatment of Rhinocerebral Mucormycosis with Intravenous Interstitial, and Cerebrospinal Fluid Administration of Amphotericin B: Case Report. Neurosurgery 1998, 42, 644–648; discussion 648–649. [Google Scholar] [CrossRef]
- Morrison, V.A.; McGlave, P.B. Mucormycosis in the BMT Population. Bone Marrow Transplant. 1993, 11, 383–388. [Google Scholar] [PubMed]
- Kauffman, C.A.; Bustamante, B.; Chapman, S.W.; Pappas, P.G. Clinical Practice Guidelines for the Management of Sporotrichosis: 2007 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2007, 45, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Crout, J.E.; Brewer, N.S.; Tompkins, R.B. Sporotrichosis Arthritis: Clinical Features in Seven Patients. Ann. Intern. Med. 1977, 86, 294–297. [Google Scholar] [CrossRef]
- Wong-Beringer, A. Treatment of Funguria. JAMA J. Am. Med. Assoc. 1992, 267, 2780. [Google Scholar] [CrossRef]
- Wise, G.J.; Wainstein, S.; Goldberg, P.; Kozinn, P.J. Candidal Cystitis. Management by Continuous Bladder Irrigation with Amphotericin B. JAMA 1973, 224, 1636–1637. [Google Scholar] [CrossRef]
- Paladino, J.A.; Crass, R.E. Amphotericin B and Flucytosine in the Treatment of Candidal Cystitis. Clin. Pharm. 1982, 1, 349–352. [Google Scholar]
- Michigan, S. Genitourinary Fungal Infections. J. Urol. 1976, 116, 390–397. [Google Scholar] [CrossRef]
- Kunin, C.M. Detection, Prevention, and Management of Urinary Tract Infections: A Manual for the Physician, Nurse, and Allied Health Worker; Lea & Febiger: Philadelphia, PA, USA, 1974. [Google Scholar]
- Wise, G.J.; Ray, B.; Kozinn, P.J. The Serodiagnosis of Significant Genitourinary Candidiasis. J. Urol. 1972, 107, 1043–1046. [Google Scholar] [CrossRef]
- Kantor, H.I.; Kamholz, J.H.; Boulas, S.H. Treatment Trends in Monilial Vaginitis. South. Med. J. 1966, 59, 535–537. [Google Scholar] [CrossRef]
- Davis, J.E.; Frudenfeld, J.H.; Goddard, J.L. Comparative Evaluation of Monistat and Mycostatin in the Treatment of Vulvovaginal Candidiasis. Obstet. Gynecol. 1974, 44, 403–406. [Google Scholar]
- Pinto Reis, C.; Vasques Roque, L.; Baptista, M.; Rijo, P. Innovative Formulation of Nystatin Particulate Systems in Toothpaste for Candidiasis Treatment. Pharm. Dev. Technol. 2016, 21, 282–287. [Google Scholar] [CrossRef]
- Carter, V.H. Haloprogin and Nystatin Therapy for Cutaneous Candidiasis. Comparison of the Efficacy of Haloprogin and Nystatin Therapy. Arch. Dermatol. 1974, 110, 81–82. [Google Scholar] [CrossRef]
- Halverstam, C.; Cohen, S.R. Cutaneous Candidiasis and Chronic Mucocutaneous Candidiasis. Treat. Ski. Dis. 2018, 171–174. [Google Scholar] [CrossRef]
- Kahl, G. The Dictionary of Genomics, Transcriptomics and Proteomics; John Wiley & Sons: Hoboken, NJ, USA, 2015; p. 1. [Google Scholar]
- Patil, A.; Lakhani, P.; Majumdar, S. Current Perspectives on Natamycin in Ocular Fungal Infections. J. Drug Deliv. Sci. Technol. 2017, 41, 206–212. [Google Scholar] [CrossRef]
- Al-Khikani, F.H.O. Amphotericin B as Antiviral Drug: Possible Efficacy against COVID-19. Ann. Thorac. Med. 2020, 15, 118–124. [Google Scholar] [CrossRef]
- Hartsel, S.C.; Weiland, T.R. Amphotericin B Binds to Amyloid Fibrils and Delays Their Formation: A Therapeutic Mechanism? Biochemistry 2003, 42, 6228–6233. [Google Scholar] [CrossRef]
- Casadevall, A. Fungal Diseases in the 21st Century: The Near and Far Horizons. Pathog. Immun. 2018, 3, 183–196. [Google Scholar] [CrossRef]
- Mendonça, A.; Santos, H.; Franco-Duarte, R.; Sampaio, P. Fungal Infections Diagnosis—Past, Present and Future. Res. Microbiol. 2022, 173, 103915. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Sun, T.; Ding, C. Pathogen-Host Interaction Repertoire at Proteome and Posttranslational Modification Levels During Fungal Infections. Front. Cell. Infect. Microbiol. 2021, 11. [Google Scholar] [CrossRef]
- Belakhov, V.V.; Garabadzhiu, A.V.; Chistyakova, T.B. Polyene Macrolide Antibotic Derivatives: Preparation, Overcoming Drug Resistance, and Prospects for Use in Medical Practice (Review). Pharm. Chem. J. 2019, 52, 890–901. [Google Scholar] [CrossRef]
- Borman, A.M.; Johnson, E.M. Erratum for Borman and Johnson, “Name Changes for Fungi of Medical Importance, 2018 to 2019”. J. Clin. Microbiol. 2021, 59, e00331-21. [Google Scholar] [CrossRef]
- Janbon, G.; Quintin, J.; Lanternier, F.; d’Enfert, C. Studying Fungal Pathogens of Humans and Fungal Infections: Fungal Diversity and Diversity of Approaches. Genes Immun. 2019, 20, 403–414. [Google Scholar] [CrossRef]
- Benedict, K.; Richardson, M.; Vallabhaneni, S.; Jackson, B.R.; Chiller, T. Emerging Issues, Challenges, and Changing Epidemiology of Fungal Disease Outbreaks. Lancet Infect. Dis. 2017, 17, e403–e411. [Google Scholar] [CrossRef]
- Brown, E.M.; McTaggart, L.R.; Dunn, D.; Pszczolko, E.; Tsui, K.G.; Morris, S.K.; Stephens, D.; Kus, J.V.; Richardson, S.E. Epidemiology and Geographic Distribution of Blastomycosis, Histoplasmosis, and Coccidioidomycosis, Ontario, Canada, 1990–2015. Emerg. Infect. Dis. 2018, 24, 1257–1266. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Denning, D.W.; Chakrabarti, A. One Health Aspects & Priority Roadmap for Fungal Diseases: A Mini-Review. Indian J. Med. Res. 2021, 153, 311–319. [Google Scholar] [CrossRef]
- Ashraf, N.; Kubat, R.C.; Poplin, V.; Adenis, A.A.; Denning, D.W.; Wright, L.; McCotter, O.; Schwartz, I.S.; Jackson, B.R.; Chiller, T.; et al. Re-Drawing the Maps for Endemic Mycoses. Mycopathologia 2020, 185, 843–865. [Google Scholar] [CrossRef]
- Firacative, C. Invasive Fungal Disease in Humans: Are We Aware of the Real Impact? Mem. Inst. Oswaldo Cruz 2020, 115, e200430. [Google Scholar] [CrossRef]
- Benedict, K.; Jackson, B.R.; Chiller, T.; Beer, K.D. Estimation of Direct Healthcare Costs of Fungal Diseases in the United States. Clin. Infect. Dis. 2019, 68, 1791–1797. [Google Scholar] [CrossRef] [Green Version]
- Benedict, K.; Whitham, H.K.; Jackson, B.R. Economic Burden of Fungal Diseases in the United States. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2022; Volume 9, p. ofac097. [Google Scholar] [CrossRef]
- Lionakis, M.S.; Hohl, T.M. Call to Action: How to Tackle Emerging Nosocomial Fungal Infections. Cell Host Microbe 2020, 27, 859–862. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Nosanchuk, J.D. Fungal Diseases as Neglected Pathogens: A Wake-up Call to Public Health Officials. PLoS Negl. Trop. Dis. 2020, 14, e0007964. [Google Scholar] [CrossRef] [Green Version]
- Bruno, M.; Matzaraki, V.; van de Veerdonk, F.L.; Kumar, V.; Netea, M.G. Challenges and Opportunities in Understanding Genetics of Fungal Diseases: Towards a Functional Genomics Approach. Infect. Immun. 2021, 89, e0000521. [Google Scholar] [CrossRef]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal Evolution: Cellular, Genomic and Metabolic Complexity. Biol. Rev. Camb. Philos. Soc. 2020, 95, 1198–1232. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.E.; Chen, S.C.-A.; Meyer, W.; Halliday, C.L. A New Age in Molecular Diagnostics for Invasive Fungal Disease: Are We Ready? Front. Microbiol. 2019, 10, 2903. [Google Scholar] [CrossRef] [Green Version]
- Shishodia, S.K.; Tiwari, S.; Shankar, J. Resistance Mechanism and Proteins in Species against Antifungal Agents. Mycology 2019, 10, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, B.M.; Lancaster, A.K.; Scherz-Shouval, R.; Whitesell, L.; Lindquist, S. Fitness Trade-Offs Restrict the Evolution of Resistance to Amphotericin B. PLoS Biol. 2013, 11, e1001692. [Google Scholar] [CrossRef]
- Saag, M.S.; Graybill, R.J.; Larsen, R.A.; Pappas, P.G.; Perfect, J.R.; Powderly, W.G.; Sobel, J.D.; Dismukes, W.E. Mycoses Study Group Cryptococcal Subproject Practice Guidelines for the Management of Cryptococcal Disease. Clin. Infect. Dis. 2000, 30, 710–718. [Google Scholar] [CrossRef]
- Waness, A.; Dawsari, G.A.; Al Jahdali, H. The Rise of an Opportunistic Infection Called “Invasive Zygomycosis”. J. Glob. Infect. Dis. 2009, 1, 131–138. [Google Scholar] [CrossRef]
- Ellis, D. Amphotericin B: Spectrum and Resistance. J. Antimicrob. Chemother. 2002, 49 (Suppl. S1), 7–10. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.A.; Della Ripa, L.A.; Hu, L.; Cioffi, A.G.; Pogorelov, T.V.; Rienstra, C.M.; Burke, M.D. C3-OH of Amphotericin B Plays an Important Role in Ion Conductance. J. Am. Chem. Soc. 2015, 137, 15102–15104. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, S.C.; Dockrell, D.H. Improved Outcomes Associated with Advances in Therapy for Invasive Fungal Infections in Immunocompromised Hosts. J. Infect. 2007, 55, 287–299. [Google Scholar] [CrossRef]
- Yardley, V.; Croft, S.L. In Vitro and in Vivo Activity of Amphotericin B-Lipid Formulations against Experimental Trypanosoma Cruzi Infections. Am. J. Trop. Med. Hyg. 1999, 61, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Reuter, S.; Merkle, M.; Brehm, K.; Kern, P.; Manfras, B. Effect of Amphotericin B on Larval Growth of Echinococcus Multilocularis. Antimicrob. Agents Chemother. 2003, 47, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Moné, Y.; Mitta, G.; Duval, D.; Gourbal, B.E.F. Effect of Amphotericin B on the Infection Success of Schistosoma Mansoni in Biomphalaria Glabrata. Exp. Parasitol. 2010, 125, 70–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgos, S.E.F.; Tsan, P.; Sletta, H.; Ellingsen, T.E.; Lancelin, J.-M.; Zotchev, S.B. Probing the Structure-Function Relationship of Polyene Macrolides: Engineered Biosynthesis of Soluble Nystatin Analogues. J. Med. Chem. 2006, 49, 2431–2439. [Google Scholar] [CrossRef] [PubMed]
- Cavell, G. The Problem with Amphotericin. Clin. Drug Investig. 2020, 40, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, U. Comparison of Effects of Amphotericin B Deoxycholate Infused over 4 or 24 Hours: Randomised Controlled. BMJ 2001, 322, 579. [Google Scholar] [CrossRef] [Green Version]
- Nucci, M.; Loureiro, M.; Silveira, F.; Casali, A.R.; Bouzas, L.F.; Velasco, E.; Spector, N.; Pulcheri, W. Comparison of the Toxicity of Amphotericin B in 5% Dextrose with That of Amphotericin B in Fat Emulsion in a Randomized Trial with Cancer Patients. Antimicrob. Agents Chemother. 1999, 43, 1445–1448. [Google Scholar] [CrossRef] [Green Version]
- Mishra, J.; Dey, A.; Singh, N.; Somvanshi, R.; Singh, S. Evaluation of Toxicity & Therapeutic Efficacy of a New Liposomal Formulation of Amphotericin B in a Mouse Model. Indian J. Med. Res. 2013, 137, 767–776. [Google Scholar]
- Bunim, J.J.; Buchanan, W.W.; Wertlake, P.T.; Sokoloff, L.; Bloch, K.J.; Beck, J.S.; Alepa, F.P. Clinical, pathologic, and serologic studies in sjoegren’s syndrome; combined clinical staff conference at the national institutes of health. Ann. Intern. Med. 1964, 61, 509–530. [Google Scholar] [CrossRef]
- Googe, J.H.; Walterspiel, J.N. Arrhythmia Caused by Amphotericin B in a Neonate. Pediatr. Infect. Dis. J. 1988, 7, 73. [Google Scholar] [CrossRef]
- Groot, O.A.; Trof, R.J.; Girbes, A.R.; Swart, N.L.; Beishuizen, A. Acute Refractory Hyperkalaemia and Fatal Cardiac Arrest Related to Administration of Liposomal Amphotericin B. Neth. J. Med. 2008, 66, 433–437. [Google Scholar]
- Moen, M.D.; Lyseng-Williamson, K.A.; Scott, L.J. Liposomal Amphotericin B: A Review of Its Use as Empirical Therapy in Febrile Neutropenia and in the Treatment of Invasive Fungal Infections. Drugs 2009, 69, 361–392. [Google Scholar] [CrossRef]
- Bicanic, T.; Bottomley, C.; Loyse, A.; Brouwer, A.E.; Muzoora, C.; Taseera, K.; Jackson, A.; Phulusa, J.; Hosseinipour, M.C.; van der Horst, C.; et al. Toxicity of Amphotericin B Deoxycholate-Based Induction Therapy in Patients with HIV-Associated Cryptococcal Meningitis. Antimicrob. Agents Chemother. 2015, 59, 7224–7231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R.F.; Nahata, M.C. A Comparative Review of Conventional and Lipid Formulations of Amphotericin B. J. Clin. Pharm. Ther. 1999, 24, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Tonin, F.S.; Steimbach, L.M.; Borba, H.H.; Sanches, A.C.; Wiens, A.; Pontarolo, R.; Fernandez-Llimos, F. Efficacy and Safety of Amphotericin B Formulations: A Network Meta-Analysis and a Multicriteria Decision Analysis. J. Pharm. Pharmacol. 2017, 69, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Ma, J.; Saliba, F.; Dupont, B.; Wingard, J.R.; Hachem, R.Y.; Mattiuzzi, G.N.; Chandrasekar, P.H.; Kontoyiannis, D.P.; Rolston, K.V.; et al. Drug-Induced Nephrotoxicity Caused by Amphotericin B Lipid Complex and Liposomal Amphotericin B: A Review and Meta-Analysis. Medicine 2010, 89, 236–244. [Google Scholar] [CrossRef]
- Wingard, J.R.; White, M.H.; Anaissie, E.; Raffalli, J.; Goodman, J.; Arrieta, A.; L Amph/ABLC Collaborative Study Group. A Randomized, Double-Blind Comparative Trial Evaluating the Safety of Liposomal Amphotericin B versus Amphotericin B Lipid Complex in the Empirical Treatment of Febrile Neutropenia. Clin. Infect. Dis. 2000, 31, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.P.; Crank, C.W.; Leikin, J.B. An Evaluation of Hepatotoxicity and Nephrotoxicity of Liposomal Amphotericin B (L-AMB). J. Med. Toxicol. 2011, 7, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Craddock, C.; Anson, J.; Chu, P.; Dodgson, A.; Duncan, N.; Gomez, C.; Mehta, J.; Sadullah, S.; Subudhi, C.; Yin, J.-L. Best Practice Guidelines for the Management of Adverse Events Associated with Amphotericin B Lipid Complex. Expert Opin. Drug Saf. 2010, 9, 139–147. [Google Scholar] [CrossRef]
- Wade, R.L.; Chaudhari, P.; Natoli, J.L.; Taylor, R.J.; Nathanson, B.H.; Horn, D.L. Nephrotoxicity and Other Adverse Events among Inpatients Receiving Liposomal Amphotericin B or Amphotericin B Lipid Complex. Diagn. Microbiol. Infect. Dis. 2013, 76, 361–367. [Google Scholar] [CrossRef]
- Heidemann, H.T.; Gerkens, J.F.; Spickard, W.A.; Jackson, E.K.; Branch, R.A. Amphotericin B Nephrotoxicity in Humans Decreased by Salt Repletion. Am. J. Med. 1983, 75, 476–481. [Google Scholar] [CrossRef]
- Downes, K.J.; Hayes, M.; Fitzgerald, J.C.; Pais, G.M.; Liu, J.; Zane, N.R.; Goldstein, S.L.; Scheetz, M.H.; Zuppa, A.F. Mechanisms of Antimicrobial-Induced Nephrotoxicity in Children. J. Antimicrob. Chemother. 2020, 75, 1–13. [Google Scholar] [CrossRef]
- Sawaya, B.P.; Weihprecht, H.; Campbell, W.R.; Lorenz, J.N.; Webb, R.C.; Briggs, J.P.; Schnermann, J. Direct Vasoconstriction as a Possible Cause for Amphotericin B-Induced Nephrotoxicity in Rats. J. Clin. Investig. 1991, 87, 2097–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershkovich, P.; Wasan, E.K.; Lin, M.; Sivak, O.; Leon, C.G.; Clement, J.G.; Wasan, K.M. Pharmacokinetics and Biodistribution of Amphotericin B in Rats Following Oral Administration in a Novel Lipid-Based Formulation. J. Antimicrob. Chemother. 2009, 64, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Shaw, P.J.; Nath, C.E.; Yadav, S.P.; Stephen, K.R.; Earl, J.W.; McLachlan, A.J. Population Pharmacokinetics of Liposomal Amphotericin B in Pediatric Patients with Malignant Diseases. Antimicrob. Agents Chemother. 2006, 50, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekersky, I.; Boswell, G.W.; Hiles, R.; Fielding, R.M.; Buell, D.; Walsh, T.J. Safety, Toxicokinetics and Tissue Distribution of Long-Term Intravenous Liposomal Amphotericin B (AmBisome): A 91-Day Study in Rats. Pharm. Res. 2000, 17, 1494–1502. [Google Scholar] [CrossRef]
- Lestner, J.M.; Howard, S.J.; Goodwin, J.; Gregson, L.; Majithiya, J.; Walsh, T.J.; Jensen, G.M.; Hope, W.W. Pharmacokinetics and Pharmacodynamics of Amphotericin B Deoxycholate, Liposomal Amphotericin B, and Amphotericin B Lipid Complex in an in Vitro Model of Invasive Pulmonary Aspergillosis. Antimicrob. Agents Chemother. 2010, 54, 3432–3441. [Google Scholar] [CrossRef] [Green Version]
- Groll, A.H.; Rijnders, B.J.A.; Walsh, T.J.; Adler-Moore, J.; Lewis, R.E.; Brüggemann, R.J.M. Clinical Pharmacokinetics, Pharmacodynamics, Safety and Efficacy of Liposomal Amphotericin B. Clin. Infect. Dis. 2019, 68, S260–S274. [Google Scholar] [CrossRef]
- Wijnant, G.-J.; Van Bocxlaer, K.; Yardley, V.; Harris, A.; Murdan, S.; Croft, S.L. Relation between Skin Pharmacokinetics and Efficacy in AmBisome Treatment of Murine Cutaneous Leishmaniasis. Antimicrob. Agents Chemother. 2018, 62, e02009-17. [Google Scholar] [CrossRef] [Green Version]
- Groll, A.H.; Giri, N.; Petraitis, V.; Petraitiene, R.; Candelario, M.; Bacher, J.S.; Piscitelli, S.C.; Walsh, T.J. Comparative Efficacy and Distribution of Lipid Formulations of Amphotericin B in Experimental Candida Albicans Infection of the Central Nervous System. J. Infect. Dis. 2000, 182, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Hope, W.W.; Goodwin, J.; Felton, T.W.; Ellis, M.; Stevens, D.A. Population Pharmacokinetics of Conventional and Intermittent Dosing of Liposomal Amphotericin B in Adults: A First Critical Step for Rational Design of Innovative Regimens. Antimicrob. Agents Chemother. 2012, 56, 5303–5308. [Google Scholar] [CrossRef] [Green Version]
- Vogelsinger, H.; Weiler, S.; Djanani, A.; Kountchev, J.; Bellmann-Weiler, R.; Wiedermann, C.J.; Bellmann, R. Amphotericin B Tissue Distribution in Autopsy Material after Treatment with Liposomal Amphotericin B and Amphotericin B Colloidal Dispersion. J. Antimicrob. Chemother. 2006, 57, 1153–1160. [Google Scholar] [CrossRef]
- Mouton, J.W.; Theuretzbacher, U.; Craig, W.A.; Tulkens, P.M.; Derendorf, H.; Cars, O. Tissue Concentrations: Do We Ever Learn? J. Antimicrob. Chemother. 2008, 61, 235–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, J.A.; Adler-Moore, J.P.; Schwartz, J.; Jensen, G.M.; Proffitt, R.T. Comparative Efficacies, Toxicities, and Tissue Concentrations of Amphotericin B Lipid Formulations in a Murine Pulmonary Aspergillosis Model. Antimicrob. Agents Chemother. 2006, 50, 2122–2131. [Google Scholar] [CrossRef] [Green Version]
- Bhamra, R.; Sa’ad, A.; Bolcsak, L.E.; Janoff, A.S.; Swenson, C.E. Behavior of Amphotericin B Lipid Complex in Plasma in Vitro and in the Circulation of Rats. Antimicrob. Agents Chemother. 1997, 41, 886–892. [Google Scholar] [CrossRef] [Green Version]
- Wasan, K.M.; Kennedy, A.L.; Cassidy, S.M.; Ramaswamy, M.; Holtorf, L.; Chou, J.W.; Pritchard, P.H. Pharmacokinetics, Distribution in Serum Lipoproteins and Tissues, and Renal Toxicities of Amphotericin B and Amphotericin B Lipid Complex in a Hypercholesterolemic Rabbit Model: Single-Dose Studies. Antimicrob. Agents Chemother. 1998, 42, 3146–3152. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.Y.; Gao, J.; Shin, D.H.; Alvarez, C.; Zhong, W.; Kwon, G.S. Pharmacokinetics and Renal Toxicity of Monomeric Amphotericin B in Rats after a Multiple Dose Regimen. Pharm. Nanotechnol. 2016, 4, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.; Lü, T.M.; Ma, A.D.; Wang, L.; Li, G.L.; Qiu, A.Z.; Pan, Z.Y.; Wang, Y.Y.; Liu, X.J. Comparative Pharmacokinetics of Continuous and Conventional Intrathecal Amphotericin B in Rabbits. Antimicrob. Agents Chemother. 2012, 56, 5253–5257. [Google Scholar] [CrossRef] [Green Version]
- Adler-Moore, J.P.; Gangneux, J.-P.; Pappas, P.G. Comparison between Liposomal Formulations of Amphotericin B. Med. Mycol. 2016, 54, 223–231. [Google Scholar] [CrossRef]
- Walsh, T.J.; Jackson, A.J.; Lee, J.W.; Amantea, M.; Sein, T.; Bacher, J.; Zech, L. Dose-Dependent Pharmacokinetics of Amphotericin B Lipid Complex in Rabbits. Antimicrob. Agents Chemother. 2000, 44, 2068–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autmizguine, J.; Guptill, J.T.; Cohen-Wolkowiez, M.; Benjamin, D.K., Jr.; Capparelli, E.V. Pharmacokinetics and Pharmacodynamics of Antifungals in Children: Clinical Implications. Drugs 2014, 74, 891–909. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.J.; Olson, J.A.; Constable, D.; Schwartz, J.; Proffitt, R.T.; Adler-Moore, J.P. Effects of Dosing Regimen on Accumulation, Retention and Prophylactic Efficacy of Liposomal Amphotericin B. J. Antimicrob. Chemother. 2007, 59, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Strenger, V.; Meinitzer, A.; Donnerer, J.; Hofer, N.; Dornbusch, H.J.; Wanz, U.; Seidel, M.G.; Sperl, D.; Lackner, H.; Schwinger, W.; et al. Amphotericin B Transfer to CSF Following Intravenous Administration of Liposomal Amphotericin B. J. Antimicrob. Chemother. 2014, 69, 2522–2526. [Google Scholar] [CrossRef] [PubMed]
- Groll, A.H.; Mickiene, D.; Piscitelli, S.C.; Walsh, T.J. Distribution of Lipid Formulations of Amphotericin B into Bone Marrow and Fat Tissue in Rabbits. Antimicrob. Agents Chemother. 2000, 44, 408–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.-Q.; Shao, K.; Wang, X.; Wang, R.-Y.; Cao, Y.-H.; Yu, Y.-Q.; Lou, J.-N.; Chen, Y.-Q.; Zhao, H.-Z.; Zhang, Q.-Q.; et al. In Vitro and in Vivo Evidence for Amphotericin B as a P-Glycoprotein Substrate on the Blood-Brain Barrier. Antimicrob. Agents Chemother. 2014, 58, 4464–4469. [Google Scholar] [CrossRef] [Green Version]
- Papastamelos, A.G.; Tunkel, A.R. Antibacterial agents in infections of the central nervous system and eye. Infect. Dis. Clin. N. Am. 1995, 9, 615–637. [Google Scholar] [CrossRef]
- Takemoto, K.; Yamamoto, Y.; Ueda, Y. Influence of the Progression of Cryptococcal Meningitis on Brain Penetration and Efficacy of AmBisome in a Murine Model. Chemotherapy 2006, 52, 271–278. [Google Scholar] [CrossRef]
- Lopez-Berestein, G.; Rosenblum, M.G.; Mehta, R. Altered Tissue Distribution of Amphotericin B by Liposomal Encapsulation: Comparison of Normal Mice to Mice Infected with Candida Albicans. Cancer Drug Deliv. 1984, 1, 199–205. [Google Scholar] [CrossRef]
- Pippa, L.F.; Marques, M.P.; da Silva, A.C.T.; Vilar, F.C.; de Haes, T.M.; da Fonseca, B.A.L.; Martinez, R.; Coelho, E.B.; Wichert-Ana, L.; Lanchote, V.L. Sensitive LC-MS/MS Methods for Amphotericin B Analysis in Cerebrospinal Fluid, Plasma, Plasma Ultrafiltrate, and Urine: Application to Clinical Pharmacokinetics. Front. Chem. 2021, 9, 782131. [Google Scholar] [CrossRef]
- Bekersky, I.; Fielding, R.M.; Dressler, D.E.; Lee, J.W.; Buell, D.N.; Walsh, T.J. Pharmacokinetics, Excretion, and Mass Balance of Liposomal Amphotericin B (AmBisome) and Amphotericin B Deoxycholate in Humans. Antimicrob. Agents Chemother. 2002, 46, 828–833. [Google Scholar] [CrossRef] [Green Version]
- Kagan, L.; Gershkovich, P.; Wasan, K.M.; Mager, D.E. Dual Physiologically Based Pharmacokinetic Model of Liposomal and Nonliposomal Amphotericin B Disposition. Pharm. Res. 2014, 31, 35–45. [Google Scholar] [CrossRef]
- Van Etten, E.W.; Stearne-Cullen, L.E.; ten Kate, M.; Bakker-Woudenberg, I.A. Efficacy of Liposomal Amphotericin B with Prolonged Circulation in Blood in Treatment of Severe Pulmonary Aspergillosis in Leukopenic Rats. Antimicrob. Agents Chemother. 2000, 44, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, M.; Peteherych, K.D.; Kennedy, A.L.; Wasan, K.M. Amphotericin B Lipid Complex or Amphotericin B Multiple-Dose Administration to Rabbits with Elevated Plasma Cholesterol Levels: Pharmacokinetics in Plasma and Blood, Plasma Lipoprotein Levels, Distribution in Tissues, and Renal Toxicities. Antimicrob. Agents Chemother. 2001, 45, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andes, D.; Safdar, N.; Marchillo, K.; Conklin, R. Pharmacokinetic-Pharmacodynamic Comparison of Amphotericin B (AMB) and Two Lipid-Associated AMB Preparations, Liposomal AMB and AMB Lipid Complex, in Murine Candidiasis Models. Antimicrob. Agents Chemother. 2006, 50, 674–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellepeddi, V.K.; Joseph, A.; Nance, E. Pharmacokinetics of Nanotechnology-Based Formulations in Pediatric Populations. Adv. Drug Deliv. Rev. 2019, 151–152, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Kip, A.E.; Schellens, J.H.M.; Beijnen, J.H.; Dorlo, T.P.C. Clinical Pharmacokinetics of Systemically Administered Antileishmanial Drugs. Clin. Pharmacokinet. 2018, 57, 151–176. [Google Scholar] [CrossRef] [Green Version]
- Johansen, H.K.; Gøtzsche, P.C. Amphotericin B Lipid Soluble Formulations versus Amphotericin B in Cancer Patients with Neutropenia. Cochrane Database Syst. Rev. 2014, CD000969. [Google Scholar] [CrossRef] [Green Version]
- Berman, J. Liposomal Amphotericin B Treatment and the Leishmaniases. Am. J. Trop. Med. Hyg. 2019, 101, 727–728. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Dey, S.; Chakraborty, H.; Mukherjee, S.; Halder, A.; Sarkar, A.; Chakraborty, P.; Ghosh, R.; Sarkar, J. Mucormycosis: A New Threat to Coronavirus Disease 2019 with Special Emphasis on India. Clin. Epidemiol. Glob. Health 2022, 15, 101013. [Google Scholar] [CrossRef]
- Adler-Moore, J.P.; Proffitt, R.T. Amphotericin B Lipid Preparations: What Are the Differences? Clin. Microbiol. Infect. 2008, 14 (Suppl. S4), 25–36. [Google Scholar] [CrossRef] [Green Version]
- Proffitt, R.T.; Satorius, A.; Chiang, S.M.; Sullivan, L.; Adler-Moore, J.P. Pharmacology and Toxicology of a Liposomal Formulation of Amphotericin B (AmBisome) in Rodents. J. Antimicrob. Chemother. 1991, 28 (Suppl. B), 49–61. [Google Scholar] [CrossRef]
- Janoff, A.S.; Perkins, W.R.; Saletan, S.L.; Swenson, C.E. Amphotericin B Lipid Complex (ablcTM): A Molecular Rationale for the Attenuation of Amphotericin B Related Toxicities. J. Liposome Res. 1993, 3, 451–471. [Google Scholar] [CrossRef]
- Bellmann, R.; Smuszkiewicz, P. Pharmacokinetics of Antifungal Drugs: Practical Implications for Optimized Treatment of Patients. Infection 2017, 45, 737–779. [Google Scholar] [CrossRef] [PubMed]
- Schiave, L.A.; Nascimento, E.; Gaspar, G.G.; Vilar, F.C.; Martinez, E.Z.; de Gaitani, C.M.; Martinez, R. Minimum Concentration of Amphotericin B in Serum according to the Formulation, Dose, and Daily or Prolonged Intermittent Therapeutic Regimen. Rev. Soc. Bras. Med. Trop. 2020, 53, e20180463. [Google Scholar] [CrossRef] [PubMed]
- Andrew, E.C.; Curtis, N.; Coghlan, B.; Cranswick, N.; Gwee, A. Adverse Effects of Amphotericin B in Children; a Retrospective Comparison of Conventional and Liposomal Formulations. Br. J. Clin. Pharmacol. 2018, 84, 1006–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, D.R.; Lalatsa, A. Oral Amphotericin B: The Journey from Bench to Market. J. Drug Deliv. Sci. Technol. 2017, 42, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Armstrong-James, D.; Koh, M.; Ostermann, M.; Cockwell, P. Optimal Management of Acute Kidney Injury in Critically Ill Patients with Invasive Fungal Infections Being Treated with Liposomal Amphotericin B. BMJ Case Rep. 2020, 13, e233072. [Google Scholar] [CrossRef]
- Alvarez-Lerma, F.; Soriano, M.C.; Rodríguez, M.; Catalán, M.; Llorente, A.M.; Vidart, N.; Garitacelaya, M.; Maraví, E.; Fernández, E.; Alvarado, F.; et al. Impact of Liposomal Amphotericin B on Renal Function in Critically Ill Patients with Renal Function Impairment. Rev. Esp. Quimioter. 2012, 25, 206–215. [Google Scholar]
- Tashiro, M.; Obata, Y.; Takazono, T.; Ota, Y.; Wakamura, T.; Shiozawa, Y.; Tsuyuki, A.; Miyazaki, T.; Nishino, T.; Izumikawa, K. Association between Fluid Infusions and the Recovery from Acute Kidney Injury in Patients Administered Liposomal Amphotericin B: A Nationwide Observational Study. Ren. Fail. 2022, 44, 282–292. [Google Scholar] [CrossRef]
- Takazono, T.; Tashiro, M.; Ota, Y.; Obata, Y.; Wakamura, T.; Miyazaki, T.; Nishino, T.; Izumikawa, K. Factor Analysis of Acute Kidney Injury in Patients Administered Liposomal Amphotericin B in a Real-World Clinical Setting in Japan. Sci. Rep. 2020, 10, 15033. [Google Scholar] [CrossRef]
- Tashiro, M.; Takazono, T.; Ota, Y.; Wakamura, T.; Takahashi, A.; Sato, K.; Miyazaki, T.; Obata, Y.; Nishino, T.; Izumikawa, K. Efficacy of Early Administration of Liposomal Amphotericin B in Patients with Septic Shock: A Nationwide Observational Study. J. Infect. Chemother. 2021, 27, 1471–1476. [Google Scholar] [CrossRef]
- Odysseos, G.; Mayr, U.; Bozsaki, G.; Seidensticker, C.; Ehmer, U.; Schmid, R.M.; Lahmer, T.; Dill, V. Isavuconazole and Liposomal Amphotericin B as Successful Combination Therapy of Refractory Invasive Candidiasis in a Liver Transplant Recipient: A Case Report and Literature Review. Mycopathologia 2022, 187, 113–120. [Google Scholar] [CrossRef]
- Garcia, A.; Adler-Moore, J.P.; Proffitt, R.T. Single-Dose AmBisome (Liposomal Amphotericin B) as Prophylaxis for Murine Systemic Candidiasis and Histoplasmosis. Antimicrob. Agents Chemother. 2000, 44, 2327–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.E.; Albert, N.D.; Kontoyiannis, D.P. Efficacy of Single-Dose Liposomal Amphotericin B or Micafungin Prophylaxis in a Neutropenic Murine Model of Invasive Pulmonary Aspergillosis. Antimicrob. Agents Chemother. 2008, 52, 4178–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehrnbecher, T.; Bochennek, K.; Klingebiel, T.; Gastine, S.; Hempel, G.; Groll, A.H. Extended Dosing Regimens for Fungal Prophylaxis. Clin. Microbiol. Rev. 2019, 32, e00010-19. [Google Scholar] [CrossRef] [PubMed]
- Lestner, J.M.; Groll, A.H.; Aljayyoussi, G.; Seibel, N.L.; Shad, A.; Gonzalez, C.; Wood, L.V.; Jarosinski, P.F.; Walsh, T.J.; Hope, W.W. Population Pharmacokinetics of Liposomal Amphotericin B in Immunocompromised Children. Antimicrob. Agents Chemother. 2016, 60, 7340–7346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obata, Y.; Takazono, T.; Tashiro, M.; Ota, Y.; Wakamura, T.; Takahashi, A.; Sato, K.; Miyazaki, T.; Nishino, T.; Izumikawa, K. The Clinical Usage of Liposomal Amphotericin B in Patients Receiving Renal Replacement Therapy in Japan: A Nationwide Observational Study. Clin. Exp. Nephrol. 2021, 25, 279–287. [Google Scholar] [CrossRef] [PubMed]
- El-Cheikh, J.; Faucher, C.; Fürst, S.; Duran, S.; Berger, P.; Vey, N.; Stoppa, A.-M.; Bouabdallah, R.; Gastaut, J.-A.; Viens, P.; et al. High-Dose Weekly Liposomal Amphotericin B Antifungal Prophylaxis Following Reduced-Intensity Conditioning Allogeneic Stem Cell Transplantation. Bone Marrow Transplant. 2007, 39, 301–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, A.; Nguyen, V.; Hsu, C.; Hyde, B.; Simms-Waldrip, T. Invasive Fungal Infections While on Voriconazole, Liposomal Amphotericin B, or Micafungin for Antifungal Prophylaxis in Pediatric Stem Cell Transplant Patients. J. Pediatr. Pharmacol. Ther. 2019, 24, 220–226. [Google Scholar] [CrossRef]
- Youngs, J.; Low, J.M.; Whitney, L.; Logan, C.; Chase, J.; Yau, T.; Klammer, M.; Koh, M.; Bicanic, T. Safety and Efficacy of Intermittent High-Dose Liposomal Amphotericin B Antifungal Prophylaxis in Haemato-Oncology: An Eight-Year Single-Centre Experience and Review of the Literature. J. Fungi 2020, 6, 385. [Google Scholar] [CrossRef]
- Minodier, P.; Retornaz, K.; Horelt, A.; Garnier, J.-M. Liposomal Amphotericin B in the Treatment of Visceral Leishmaniasis in Immunocompetent Patients. Fundam. Clin. Pharmacol. 2003, 17, 183–188. [Google Scholar] [CrossRef]
- Uliana, S.R.B.; Trinconi, C.T.; Coelho, A.C. Chemotherapy of Leishmaniasis: Present Challenges. Parasitology 2018, 145, 464–480. [Google Scholar] [CrossRef]
- Hervás, J.A.; Martín-Santiago, A.; Hervás, D.; Rojo, E.; Mena, A.; Rocamora, V.; Dueñas, J. Old World Leishmania Infantum Cutaneous Leishmaniasis Unresponsive to Liposomal Amphotericin B Treated with Topical Imiquimod. Pediatr. Infect. Dis. J. 2012, 31, 97–100. [Google Scholar] [CrossRef] [PubMed]
- González, U.; Pinart, M.; Rengifo-Pardo, M.; Macaya, A.; Alvar, J.; Tweed, J.A. Interventions for American Cutaneous and Mucocutaneous Leishmaniasis. Cochrane Database Syst. Rev. 2009, CD004834. [Google Scholar] [CrossRef] [PubMed]
- Shirzadi, M.R. Lipsosomal Amphotericin B: A Review of Its Properties, Function, and Use for Treatment of Cutaneous Leishmaniasis. Res. Rep. Trop. Med. 2019, 10, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naeem, F.; Nathan, K.; Chivinski, J.; Ekmekjian, T.; Libman, M.; Barkati, S. Intravenous Liposomal Amphotericin B Efficacy and Safety for Cutaneous and Mucosal Leishmaniasis: A Systematic Review and Meta-Analysis Protocol. BMJ Open 2021, 11, e045707. [Google Scholar] [CrossRef]
- Rodríguez Galvis, M.C.; Pérez Franco, J.E.; Casas Vargas, M.Y.; Ordoñez Rubiano, M.F. Effectiveness and Safety of Amphotericin B Deoxycholate, Amphotericin B Colloidal Dispersion, and Liposomal Amphotericin B as Third-Line Treatments for Cutaneous and Mucocutaneous Leishmaniasis: A Retrospective Study. Am. J. Trop. Med. Hyg. 2020, 102, 274–279. [Google Scholar] [CrossRef]
- Espinoza Gutierrez, B.J.; Martínez Martínez, I.; de Rodríguez Fragoso, M.L.; Rodríguez López, Y.A.; Regla Contreras, J.I.; López Ortiz, M.; Fernández Zertuche, M.; Ortega Blake, I.; Galván Hernández, A. Pharmaceutical Composition Containing Benznidazole and N-(L)-Histidinamide of Amphotericin B for the Treatment of Trypanosomiasis. Available online: https://worldwide.espacenet.com/publicationDetails/biblio?FT=D&date=20201224&DB=&locale=&CC=WO&NR=2020256537A1&KC=A1&ND=1# (accessed on 10 May 2022).
- Lanternier, F.; Poiree, S.; Elie, C.; Garcia-Hermoso, D.; Bakouboula, P.; Sitbon, K.; Herbrecht, R.; Wolff, M.; Ribaud, P.; Lortholary, O.; et al. Prospective Pilot Study of High-Dose (10 Mg/kg/day) Liposomal Amphotericin B (L-AMB) for the Initial Treatment of Mucormycosis. J. Antimicrob. Chemother. 2015, 70, 3116–3123. [Google Scholar] [CrossRef] [Green Version]
- Chikley, A.; Ben-Ami, R.; Kontoyiannis, D.P. Mucormycosis of the Central Nervous System. J. Fungi 2019, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Molina, K.C.; Gutman, J.A.; Scherger, S.; Lum, J.M.; Mossad, S.B.; Burgess, M.; Cheng, M.P.; Chuang, S.T.; Jacobs, S.E.; et al. Mucormycosis in Hematopoietic Cell Transplant Recipients and in Patients With Hematological Malignancies in the Era of New Antifungal Agents. In Open Forum Infectious Diseases; Oxford University Press: Oxford, UK, 2021; Volume 8, p. ofaa646. [Google Scholar] [CrossRef]
- Trobisch, A.; Marterer, R.; Gorkiewicz, G.; Flaschberger, S.; Lackner, H.; Seidel, M.; Sperl, D.; Karastaneva, A.; Kohlmaier, B.; Egger, M.; et al. Invasive Mucormycosis during Treatment for Acute Lymphoblastic Leukaemia-Successful Management of Two Life-Threatening Diseases. Support. Care Cancer 2020, 28, 2157–2161. [Google Scholar] [CrossRef] [Green Version]
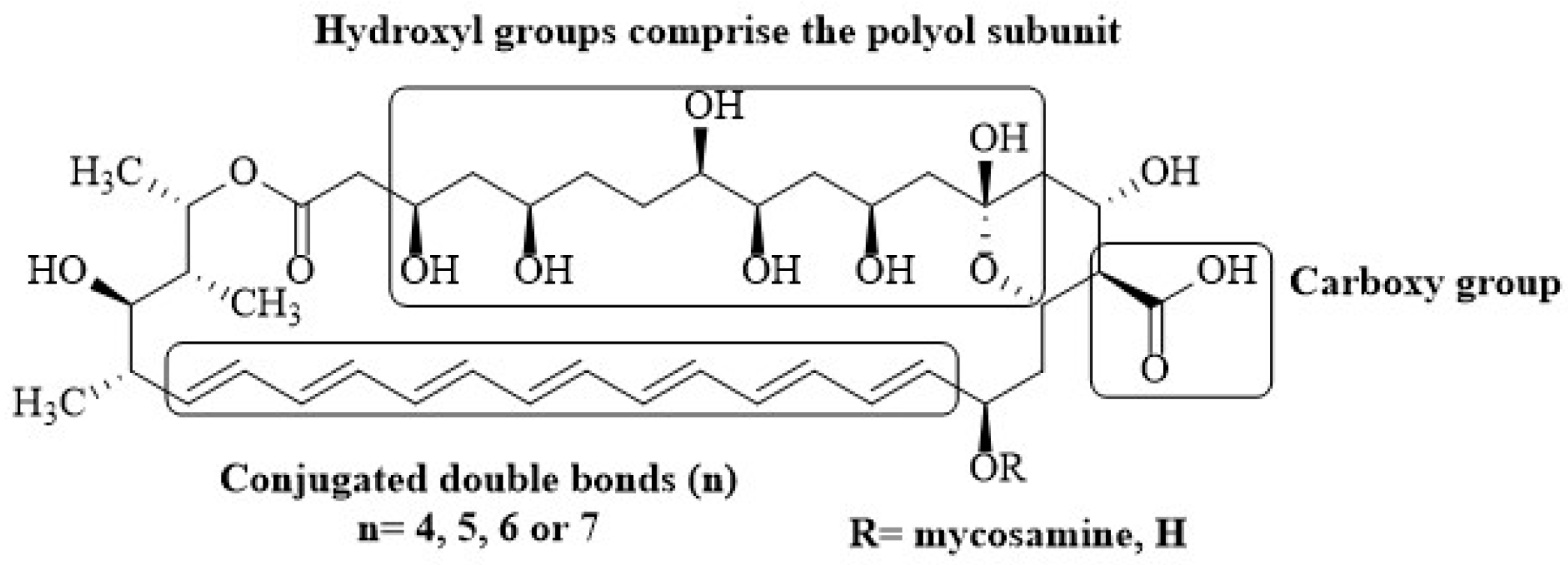
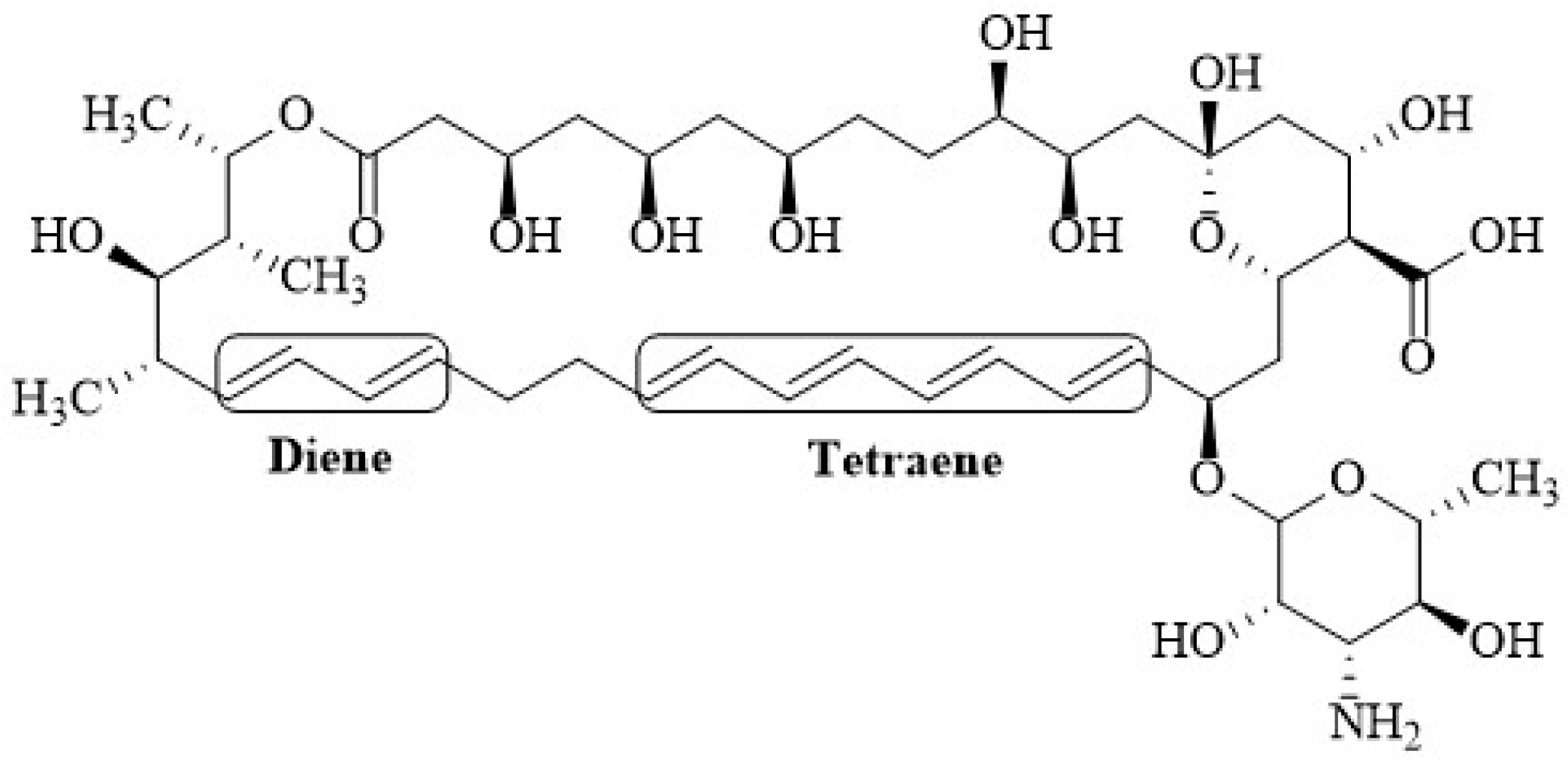
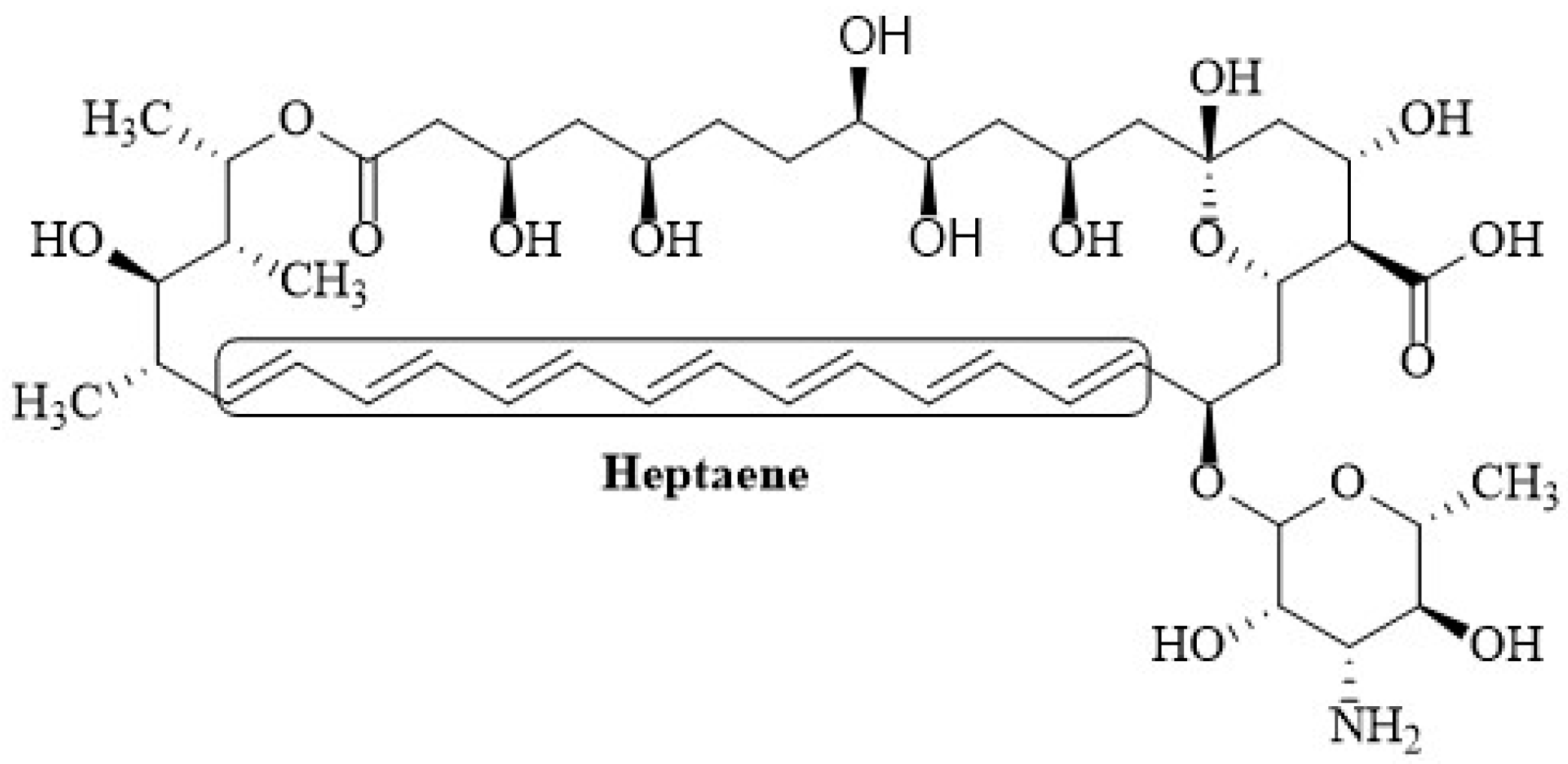
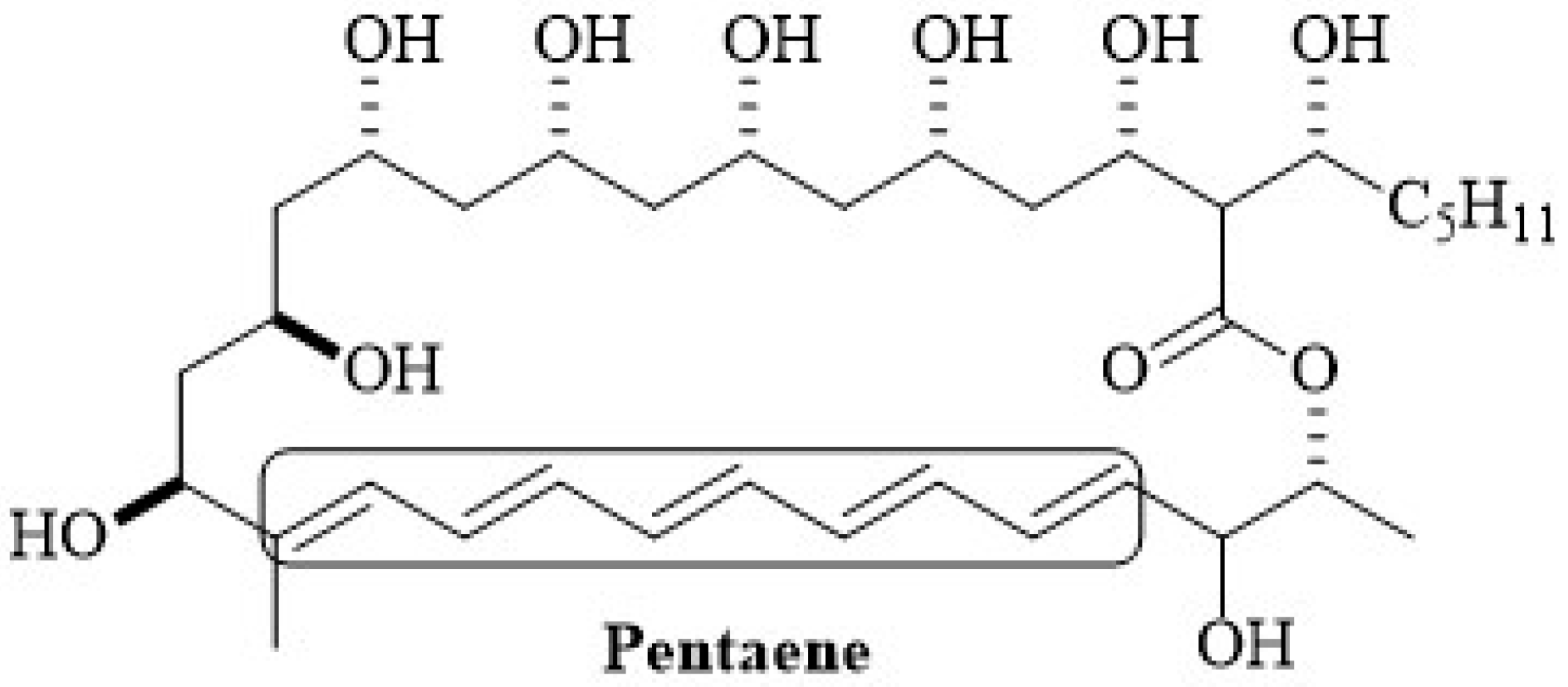
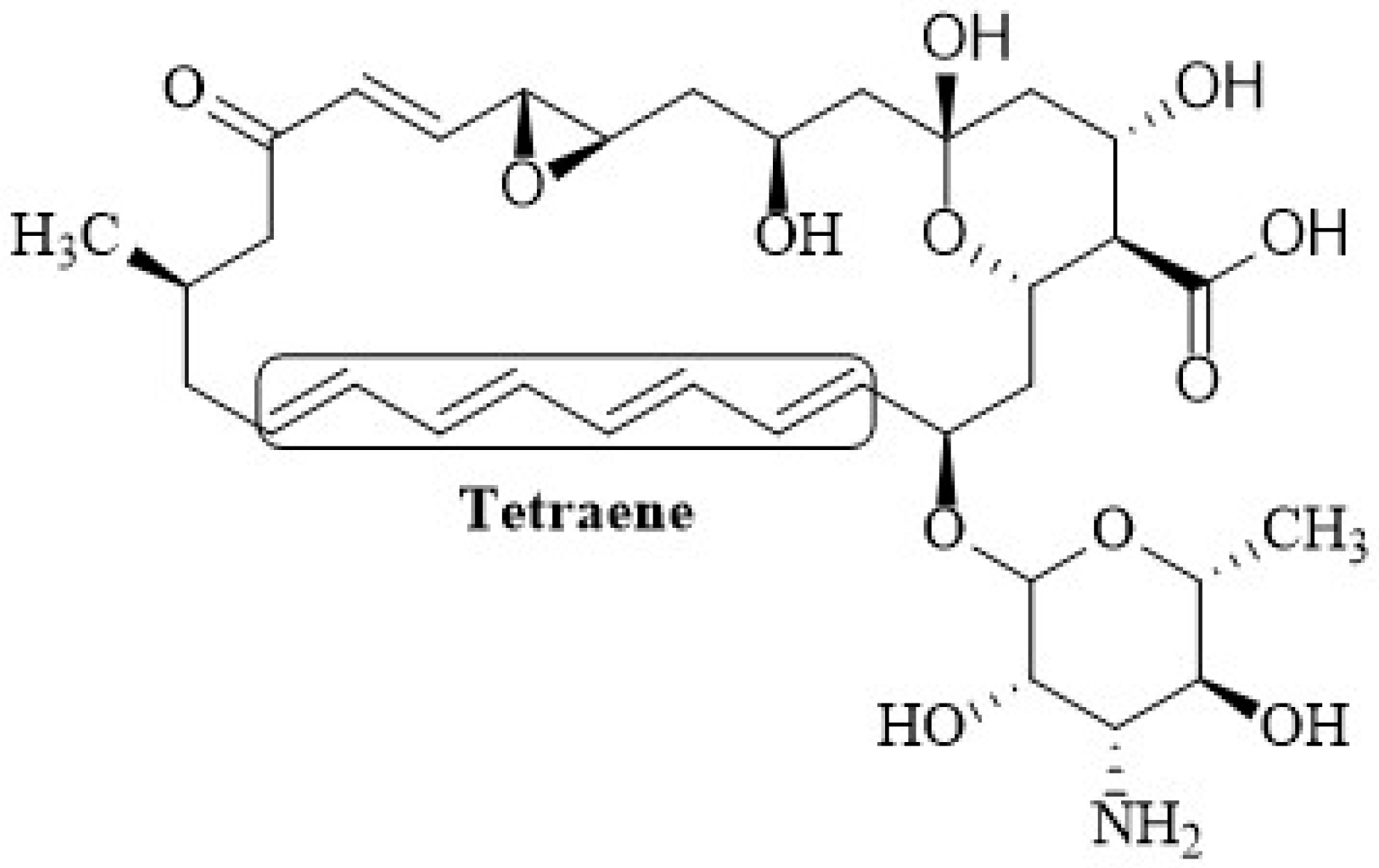
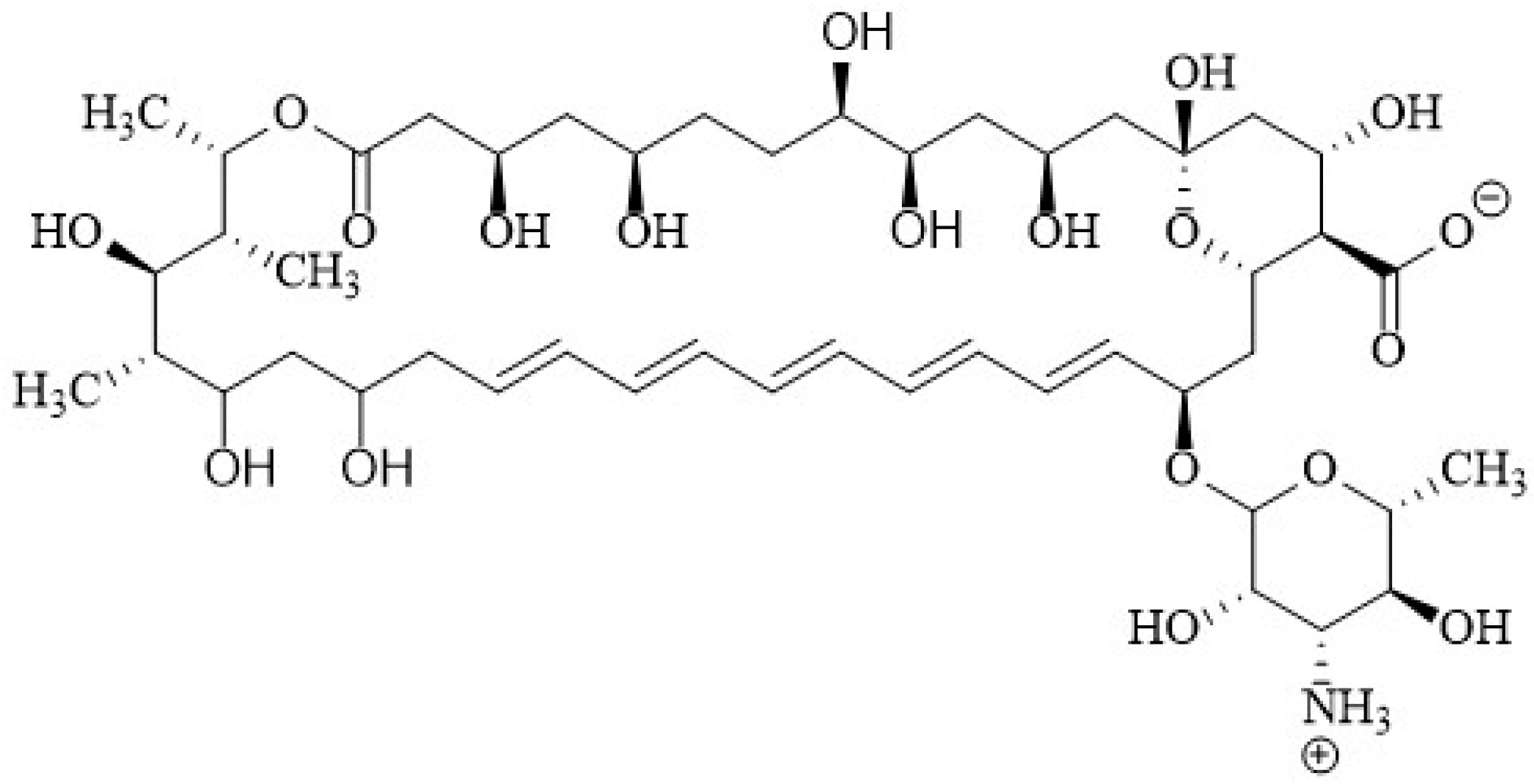
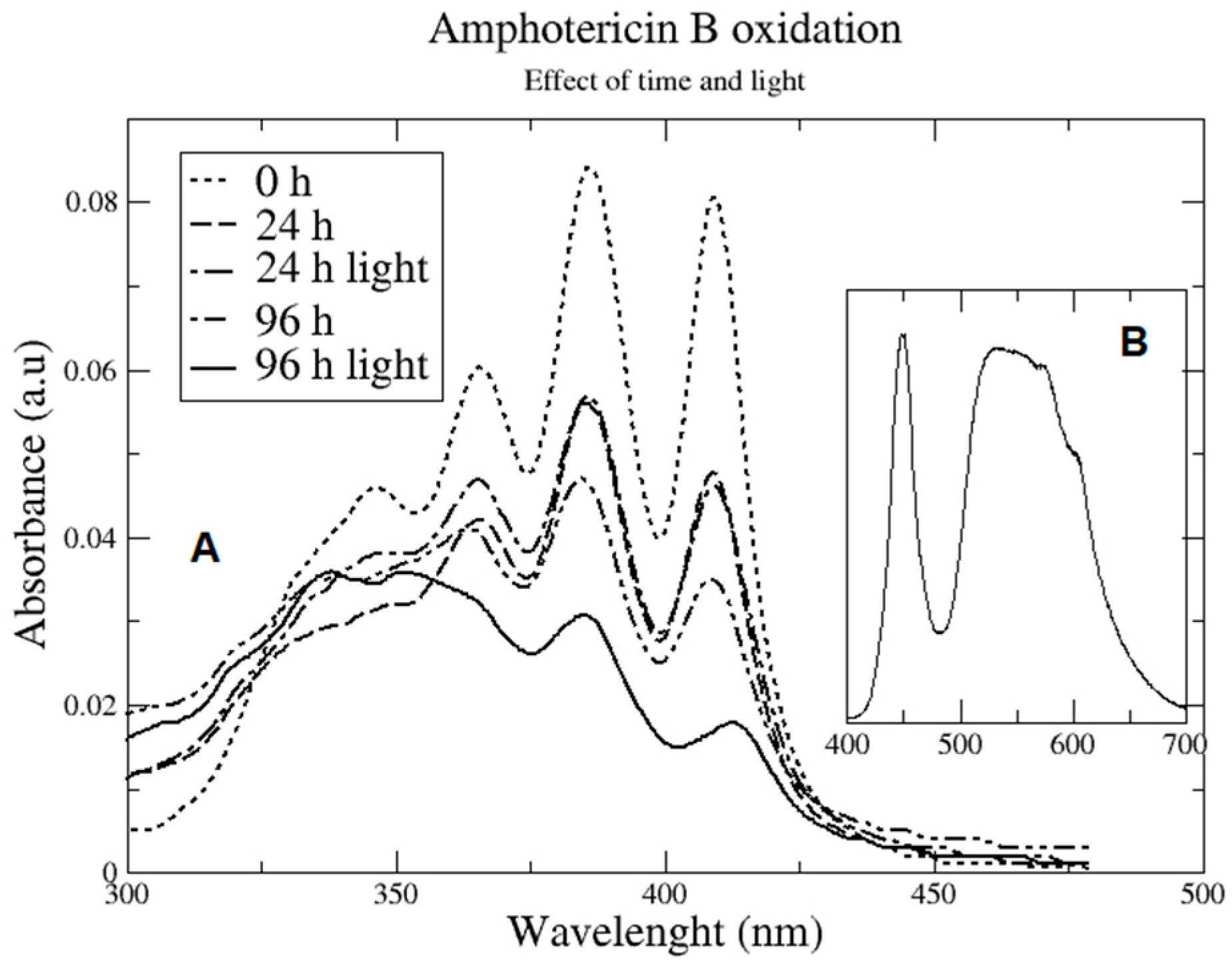
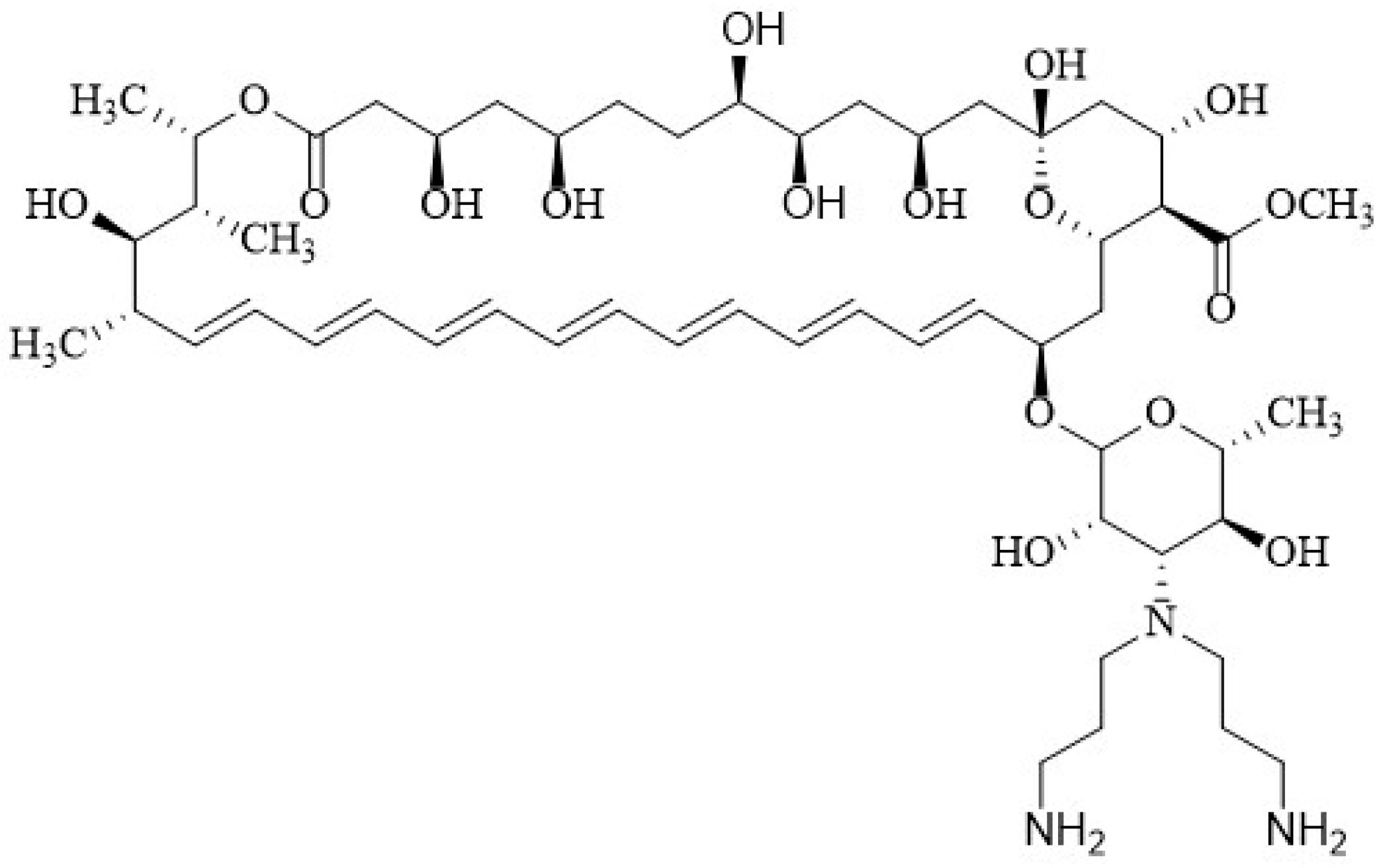




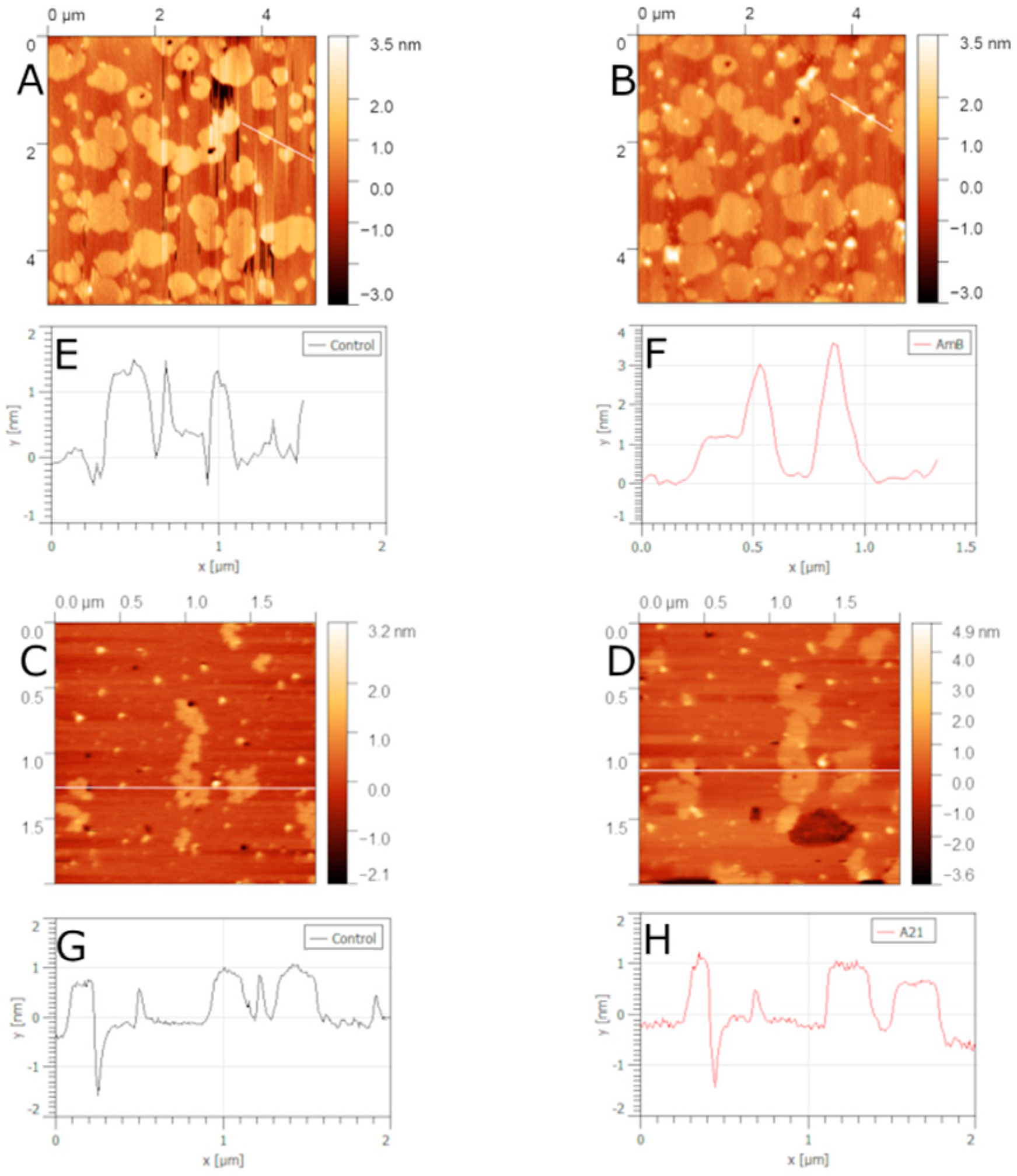

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haro-Reyes, T.; Díaz-Peralta, L.; Galván-Hernández, A.; Rodríguez-López, A.; Rodríguez-Fragoso, L.; Ortega-Blake, I. Polyene Antibiotics Physical Chemistry and Their Effect on Lipid Membranes; Impacting Biological Processes and Medical Applications. Membranes 2022, 12, 681. https://doi.org/10.3390/membranes12070681
Haro-Reyes T, Díaz-Peralta L, Galván-Hernández A, Rodríguez-López A, Rodríguez-Fragoso L, Ortega-Blake I. Polyene Antibiotics Physical Chemistry and Their Effect on Lipid Membranes; Impacting Biological Processes and Medical Applications. Membranes. 2022; 12(7):681. https://doi.org/10.3390/membranes12070681
Chicago/Turabian StyleHaro-Reyes, Tammy, Lucero Díaz-Peralta, Arturo Galván-Hernández, Anahi Rodríguez-López, Lourdes Rodríguez-Fragoso, and Iván Ortega-Blake. 2022. "Polyene Antibiotics Physical Chemistry and Their Effect on Lipid Membranes; Impacting Biological Processes and Medical Applications" Membranes 12, no. 7: 681. https://doi.org/10.3390/membranes12070681
APA StyleHaro-Reyes, T., Díaz-Peralta, L., Galván-Hernández, A., Rodríguez-López, A., Rodríguez-Fragoso, L., & Ortega-Blake, I. (2022). Polyene Antibiotics Physical Chemistry and Their Effect on Lipid Membranes; Impacting Biological Processes and Medical Applications. Membranes, 12(7), 681. https://doi.org/10.3390/membranes12070681