Abstract
Compartmentalization, together with transbilayer and lateral asymmetries, provide the structural foundation for functional specializations at the cell surface, including the active role of the lipid microenvironment in the modulation of membrane-bound proteins. The chemical synapse, the site where neurotransmitter-coded signals are decoded by neurotransmitter receptors, adds another layer of complexity to the plasma membrane architectural intricacy, mainly due to the need to accommodate a sizeable number of molecules in a minute subcellular compartment with dimensions barely reaching the micrometer. In this review, we discuss how nature has developed suitable adjustments to accommodate different types of membrane-bound receptors and scaffolding proteins via membrane microdomains, and how this “effort-sharing” mechanism has evolved to optimize crosstalk, separation, or coupling, where/when appropriate. We focus on a fast ligand-gated neurotransmitter receptor, the nicotinic acetylcholine receptor, and a second-messenger G-protein coupled receptor, the cannabinoid receptor, as a paradigmatic example.
1. Introduction
Current studies on biological membranes support the notion that they constitute highly specialized structures with compartmentalized functional regions of varying thickness, lipid composition, and functional properties. Signaling pathways require a highly regulated membrane organization since these processes are characteristically dynamic and occur in separated spatio-temporal discrete domains. Of great relevance to cell physiology are the lateral membrane heterogeneities enriched in cholesterol, sphingolipids, and phospholipids with saturated fatty acid acyl chains. Their chemical composition determines their engrossed thickness and their physicochemical properties are akin to those of the liquid-ordered (Lo) domains observed in artificial membranes. These membrane regions, also known as lipid rafts [1,2], play a crucial role in manifold cellular processes by providing the structural substrate for functional compartmentalization. Distinct cellular signaling mechanisms, in particular those operating at the cell-surface membrane, often rely on these discrete lateral domains to perform in an isolated manner in close but separate locations of the membrane. Interactions between domains harboring different signaling proteins can result in the dynamic regulation or modulation of these signaling mechanisms in their synergic potentiation, or their silencing. One example of such an interacting signaling system is discussed in this short review: the endocannabinoid (EC) system and the nicotinic acetylcholine receptors (nAChRs).
The EC system comprises a wide variety of lipid-signaling molecules, enzymes, and receptors. Endocannabinoids (ECs) are important homeostatic modulators of neuronal activity in the central nervous system (CNS). nAChRs are a Cys-loop gene family of neurotransmitter receptors, belonging to the superfamily of pentameric ligand-gated cation channels (pLGIC). In the mammalian brain, there is ample combinatorial diversity of nAChR subtypes that allows a diversity of functional responses to the endogenous neurotransmitter, acetylcholine (ACh), and to a much broader spectrum of chemical compounds that modulate this receptor, such as positive and negative allosteric modulators (PAMs and NAMs, respectively), general anesthetics, fatty acids, and cholesterol, to mention the most important ones.
ECs and EC receptors anatomically and functionally interact with nAChRs in brain areas such as the midbrain, the hippocampus, and the amygdala [3,4,5]. ECs regulate cholinergic afferents on the ventral tegmental area (VTA) dopaminergic neuronal cells, and therefore play an important role in modulating reward stimuli and addiction. Indeed, through multiple interactions nAChR and EC receptors crosstalk in the CNS and induce neuroadaptations in response to diverse stimuli. All these molecular processes including the down or upregulation of selective nAChR subtypes, the modulation of neurotransmitter release, and neuronal scaffolding proteins take place within or in the proximity of the plasma membrane.
In this review, we focus on the heterogeneous composition of the plasma membrane and the significance of compartmentalization for the efficiency of synaptic signaling involving nAChRs and their modulation by the EC system. In addition, we discuss the structural and functional roles played by lipids and proteins in such membrane nanodomains, and their relationship with the EC system and the nAChRs.
2. The Nanodomains within the Plasma Membrane
2.1. Cholesterol
The neutral lipid cholesterol plays a key role in membrane compartmentalization [6,7] given that its content and topographical distribution are main determinants of the membrane’s physicochemical properties. Cholesterol increases membrane rigidity and thickness and alters tension within the membrane [8,9,10,11,12]. Thus, cholesterol affects the morphology of the plasma membrane. The cholesterol molecule has a small polar headgroup (-OH) and a hydrophobic region (rigid sterol ring) that changes the physicochemical properties of phospholipid bilayers. Higher lateral pressure (tension) is exerted at the headgroup of the cholesterol molecule, while lower lateral pressure takes place at the hydrophobic regions of the molecule. Cholesterol-containing membranes provide a negative spontaneous curvature which is counterbalanced by the extension of acyl chains of phospholipids that thicken the hydrophobic region of the bilayer, thus compensating, at least in part, for this curvature and reducing lateral pressure (or curvature). Furthermore, the hydrophobic region of the cholesterol molecule restricts the flexible fitting of adjacent protein regions. Cholesterol can induce pressure on transmembrane proteins due to changes in the lateral pressure profile, which varies along the depth of the bilayer [13].
Cholesterol can diffuse in these liquid-ordered (Lo) lipid domains, albeit with slower diffusion rates than in liquid-disordered (Ld), cholesterol-poor regions, where cholesterol can diffuse at the rate of submicroseconds [14]. Cholesterol molecules can also flip-flop between the outer and inner leaflets of the membrane bilayer. Cholesterol distribution within biological membranes is influenced by the affinity of sterol for other lipids and proteins present in the plasmalemma. For instance, cholesterol-phosphatidylcholine and cholesterol-phosphatidylethanolamine associations are common at the inner layer of liquid-ordered domains, whereas the outer layer harbors sphingomyelin-cholesterol associations, thus providing two different liquid phases with different diffusion rates [15,16,17].
Cholesterol interacts with several neurotransmitter receptors. These transmembrane molecules share cholesterol-consensus linear binding sequences, such as the so-called cholesterol recognition/interaction amino acid consensus motifs (CRAC) and its mirror image, CARC [18]. These consensus domains facilitate the incorporation of many membrane proteins into cholesterol-rich domains [19]. Cholesterol was experimentally shown to modulate various pentameric ligand-gated ion channels (pLGICs) [20,21], as well as members of the superfamily of G-protein coupled receptors (GPCRs) [22]. The muscle-type nAChR exhibits a cholesterol-recognition motif (“CRAC”) adjacent to the transmembrane helix M1, and another cholesterol-recognition sequence of opposite orientation (“CARC”) on the M4-facing surface of M1, adjacent to one of the proposed cholesterol-binding cavities [18]. Similarly, the transmembrane helix 7 of human cannabinoid receptor 1 (CB1R) displays a CRAC sequence [23].
Cholesterol interacts with the actin subcortical cytoskeletal meshwork contributing to receptor clustering and compartmentalization [6,24]. The cognitive decline that has been described upon ageing is associated with irreversible loss of membrane cholesterol [25,26]; replenishment of cholesterol in hippocampal slices from aged mice was found to improve learning and memory in those affected [26].
Together with cholesterol, sphingomyelin contributes to the membrane-actin cytoskeleton crosstalk by modulating membrane binding and the activity of the Rho GTPases, a family of small signaling G proteins and subfamily of the Ras superfamily. Sphingomyelin accumulation leads to a decrease in metabotropic glutamate receptors and F-actin content in a Niemann–Pick disease type A mouse model, defective in acid sphingomyelinase, precluding the membrane attachment of RhoA and its effectors ROCK and profilin IIa [27].
2.2. The Complex Array of Proteins in the Synaptic Plasma Membrane
Resident proteins and interacting lipids within the plasma membrane dynamically orchestrate many signaling cascades. Alterations of the membrane lipid composition can result in defective functions of raft-associated proteins, and consequently favor abnormal cell signaling. For example, abnormal cholesterol metabolism has been implicated in the development of neurological disorders such as Alzheimer and Parkinson diseases, Huntington disease, Niemann–Pick type C disease, and schizophrenia spectrum disorders [28,29,30,31,32]. Likewise, alteration of membrane-associated proteins can also hamper many important cell functions and even lead to pathological states. In Alzheimer and Parkinson diseases, loss of plasma membrane integrity in neuronal cells is often observed. This condition favors toxic amyloid-β and α-synuclein aggregation [33,34,35] through lipid-induced conformational changes [34,36,37,38] or mass action [39,40] that, when combined, increase the probability of inter-molecular interactions and promote aggregation [39,40]. During aggregation, amyloid-β oligomers extract phospholipids from the plasma membrane and incorporate these lipid molecules into the growing fibrils, thus causing membrane rupture. Disease proteins can increase membrane permeability by different mechanisms [40], such as protein-induced membrane rigidity [41,42], membrane thinning [43,44] and deformation [43,44,45,46], as well as detergent-like effects [38,41,47], and pore formation [36,37,42,48,49,50]. These pores in the bilayer result in Ca2+ influx from the extracellular compartment and efflux of cytosolic content.
Many proteins at the synapse dynamically modulate synaptic activity, thus influencing a great variety of biochemical states and protein–protein interactions that control protein synthesis and contribute to the reorganization of cytoskeletal architecture [51], which is essential for adequate dendritic spine morphology [52]. For instance, actin filaments can associate with actin-binding proteins (ABPs) to provide the necessary structure to modulate membrane morphology [53]. However, the actin cytoskeleton and its regulatory proteins are not merely structural proteins: they also act as scaffolds for stably positioning and anchoring other scaffolding proteins, ion-channels, neurotransmitter receptors, cell-adhesion proteins, and other macromolecules at the cell surface that contribute to the architectural integrity and function of the postsynaptic density (PSD) [54]. They are also necessary for the trafficking of subcellular structures such as endosomes, and for higher brain functions such as memory formation and extinction [55]. At the PSD, the family of PDZ domain-containing scaffold proteins offers a molecular interface between glutamatergic receptors (GluRs) and the cytoskeleton [56,57]. For example, two proteins that contain PDZ domains, PSD95 and SHANK (SH3 and multiple ankyrin repeat domains), can bind through multiple interactions to NMDA receptors and to the metabotropic glutamate receptor (mGluR) [58,59]. The postsynaptic expression of SHANK was shown to enhance presynaptic function [59], suggesting a key role for the SHANK scaffold in synaptic plasticity.
Another group of proteins that are tethered to membranes by lipid groups attached to their C-termini are the Rab proteins. These proteins belong to the Ras superfamily of small GTPases. They are predominantly present in an active state, whereas the dephosphorylation of GTP by hydrolysis to GDP converts Rab proteins to their inactive form. Rab proteins have been implicated in the facilitation of membrane-associated receptor transport. Some key Rab proteins identified as participating in the internalization and recycling of membrane receptors are Rab 5, Rab 4, and Rab 11. The former is located on the cytoplasmic surfaces of the plasma membrane and is specifically associated with the internalization of clathrin-coated pits and with the fusion of early endosomes [60,61]. Rab 4 is generally described to facilitate the rapid recycling of receptors directly from endosomes and targeted back to the plasma membrane [62,63]. Rab 11 is associated with the slow recycling of GPCRs via the perinuclear recycling compartment [64]. Thus, the regulation of the number and the availability of receptors at the plasma membrane strongly depends on Rab proteins.
3. Endocannabinoids
3.1. The Endocannabinoid System
ECs are involved in a plethora of physiological and pathological processes in mammalian cells, having effects on mood, appetite, reproduction, immunity, memory, and pain perception [65]. The EC system comprises cannabinoid receptors (CBRs), endogenous cannabinoid ligands (endocannabinoids, Ecs), and the enzymes involved in their synthesis and degradation. Ecs are lipid molecules, synthesized de novo by phospholipase action after hydrolyzing the lipid precursors from the cellular membrane [66]. The Ecs anandamide (N-arachidonoylethanolamine, (AN)) and 2-arachidonoylglycerol (2-AG) are lipid messenger molecules. AN is an endogenous lipid neurotransmitter derived from the polyunsaturated fatty acid arachidonic acid and a member of the N-acylethanolamines (NAEs) family [67]. It is a partial agonist of cannabinoid receptor 1 (CB1R) and cannabinoid receptor 2 (CB2R), and a full agonist of the vanilloid receptor 1 (VR1), whereas 2-AG acts as a full agonist of CB1R and CB2R. Additionally, ECs can activate other “non-CB” receptors, such as the G-protein-coupled receptor 55 (GPR55) and peroxisome proliferator-activated receptors (PPARs) [68,69,70,71].
CB1Rs are expressed at very high levels in a subset of GABAergic interneurons, such as the cholecystokinin (CCK)-containing basket cells in the forebrain [72], and at lower levels on many glutamatergic terminals throughout the brain [73]. Within neurons, quantitative electron microscopy studies revealed that CB1Rs are found mainly on the pre-terminal axonal segment and less on more proximal axons, dendrites, or the cell soma [74].
3.2. The Endocannabinoid System in Synaptic Transmission
In contrast to classical neurotransmitters, ECs are produced as needed [75]. Upon postsynaptic depolarization, for example, after activation of metabotropic glutamate, muscarinic, or dopamine D2 receptors [76,77,78], the synthesis of ECs takes place through Ca2+ influx and the activation of the Gq-protein, or via phospholipase C and D activation (Figure 1). However, AN and 2-AG have different synthetic and metabolic pathways [79]. While the former is mainly synthesized from N-acyl-phosphatidylethanolamine (NAPE) by NAPE-specific phospholipase D (NAPE-PLD) and metabolized by fatty acid amidohydrolase (FAAH) [79], the latter is predominantly synthesized from 2-arachidonoyl-containing phospholipids by DAG lipase (DAGL) and metabolized by monoacylglycerol lipase (MAGL). Once synthesized, ECs are released into the synaptic cleft and are able to activate CBRs in presynaptic and/or nearby GABAergic, glutamatergic, and cholinergic neurons [77,80,81,82,83].
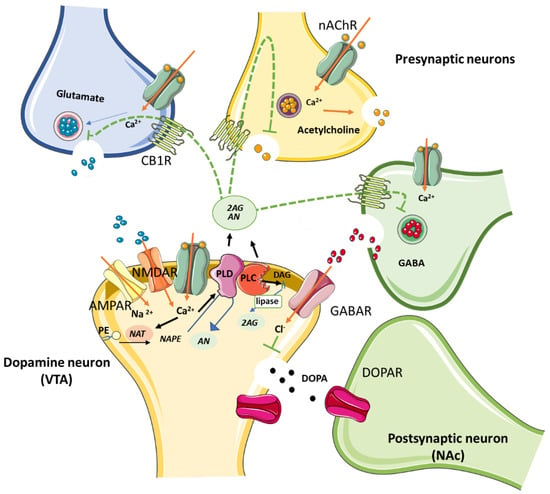
Figure 1.
Schematic illustration of the biosynthetic pathway followed by ECs N-acylethanolamine (AN) and 2-arachidonoylglycerol (2-AG) by NAPE-PLD and DAG lipase, respectively. Both AN and 2-AG bind to presynaptic CB1Rs expressed on GABAergic, glutamatergic and cholinergic terminals, precluding neurotransmitter release. Induction of DOPA secretion by primary inhibition of GABA release is involved in the reward pathway in the brain. Abbreviations: PLD, N-acyl phosphatidylethanolamine phospholipase D; DAG, diacylglycerol; NAT, N-acyltransferase; PE, Phosphatidylethanolamine; nAChRs, nicotinic acetylcholine receptors; CB1R, cannabinoid type 1 receptor; AMPA, amino-3-hydroxy-5-methyl-4-isoazolepropionate receptor; GABA, γ-aminobutyric acid; GABAR, GABA receptor; NMDAR, N-methyl-D-aspartate receptor; DOPA, dopamine; DOPAR, DOPA receptor; VTA, ventral tegmental area; NAc, nucleus accumbens.
The latter retrograde EC modulation of GABA terminals enables the depolarization-induced suppression of inhibition (DSI) through a transient suppression of GABA released onto the postsynaptic neuron, thereby disinhibiting it. EC action on CBRs located on the glutamate terminal, on the other hand, favors a depolarization-induced suppression of excitation (DSE) [84,85] by inhibiting glutamate release, hence activating postsynaptic neurons. However, the role of DSI is reported to predominate over that of DSE due to differences in CB1R sensitivity between inhibitory and excitatory synapses [86]. Importantly, the predominant GABA suppression in the midbrain is suggested to be the mechanism that induces dopamine (DA) release at mesocorticolimbic and nigrostriatal sites [87] associated with reward-based learning and also with addiction [88,89].
The EC system has a central role in the modulation of synaptic transmission throughout the CNS. ECs regulate behavior through reward learning, by modulating mesolimbic reward circuits that include the ventral tegmental area (VTA), the nucleus accumbens (NAc), and the lateral habenula (LHb). It is these CNS functions that have given ECs notoriety in relation to drug abuse and addiction behaviors. Furthermore, EC-like lipid-signaling molecules are increasingly understood as neuromodulators able to regulate dopaminergic transmission, as well as reward and addiction behavior. Thus, upon interacting with different receptors within the plasma membrane, ECs stimulate a great variety of signaling pathways. As we will see, membrane microdomains are key players in modulating signal transduction through the organization of CBRs.
Other nonspecific mechanisms of action, e.g., involving the alteration of the host lipid bilayer, have been suggested for ECs [90]. AN, for instance, was shown to reduce the ionic current of many voltage-gated ion channels in the presence of antagonists of CB1Rs and CB2Rs [91,92,93,94], thus suggesting a receptor-independent mechanism for the modulation of multiple membrane protein functions.
3.3. The Endocannabinoid System Organization in Membrane Microdomains
Once activated by ECs, either CB1R or CB2R trigger a signaling cascade through the inhibitory Gi and Go subtypes of the G proteins (i.e., Gai1, Gai2, Gai3, Gao1, Gao2) [95,96]. Their activation by ECs leads, in turn, to the inhibition of adenylyl cyclase, the activation of mitogen-activated protein kinases, the inhibition of certain voltage-gated calcium channels [97,98], and the activation of G protein-linked inwardly rectifying K+ channels [96,99,100] (Figure 2).
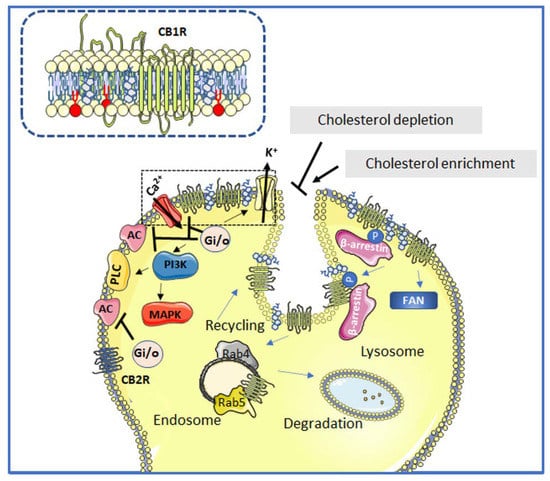
Figure 2.
Schematic depiction of the interactions between cannabinoid receptor signaling and lipid domains. Neuromodulation upon agonist binding to CB1Rs and CB2Rs (unphosphorylated receptors) induces Gi/o-dependent inhibition of adenylyl cyclase (AC) activity and activation of different MAPK cascades. CB1R positively regulates inwardly rectifying K+ channels, whereas it negatively regulates voltage-gated Ca2+ channels. In addition, the CB1R can activate PLC at the plasma membrane and can also signal through non-G proteins such as the adaptor protein FAN. The phosphorylated CB1R is a target of β-arrestin. The attenuation of CB1R signaling at cholesterol-rich lipid domains occurs through a Rab5-dependent endocytic mechanism. Once internalized, the CB1R may either be recycled back to the plasma membrane through a Rab4-dependent mechanism or further targeted to lysosomal degradation. The CB2R does not reside in or interact with Lo lipid platforms. Alteration of ordered lipid domain integrity either by depleting or supplying cholesterol is shown to inhibit or enhance the internalization of CB1R, respectively. The inset depicts a closer view of the cholesterol-rich Lo lipid domain (marked with a dotted line in the main figure). Cholesterol molecule in gray, sphingolipids in red and other phospholipids in yellow.
As shown in Figure 1 and Figure 2, the CB1R can also modulate ion channel proteins [96]. The effects of CBR activation are long lasting because of the several secondary messenger molecules involved in processes that play major roles in neuronal plasticity [101,102]. This makes apparent the diversity of signaling pathways that can be elicited by different CB1R and CB2R agonists. However, the great capacity of CBRs to couple to different subtypes of G proteins does not make them less specific in triggering downstream signaling transduction mechanisms. The plasma membrane plays a major role in confining cannabinoid responses, both spatially and temporally. Cholesterol/sphingomyelin-rich domains provide CBRs with dynamic and organized platforms where assembly of signaling complexes can take place and, in addition, prevent crosstalk between different pathways [103]. Reinforcing the relevance of membrane domains for the EC system signal selectivity, CB1R binding and signaling are demonstrated to be influenced by membrane compartmentalization [103].
Chemicals such as methyl-β-cyclodextrin deplete cholesterol from the plasma membrane (Figure 2) and prevent the onset of cell death by completely blocking the ability of AN to induce superoxide generation, phosphatidylserine exposure, and p38 MAPK activation. However, the use of a CBR antagonist did not prevent cell death in primary hepatic stellate cells [104], thus suggesting a more complex CBR–lipid interface interaction that is disturbed upon cholesterol depletion. In agreement with this observation, the activation of CB1R-dependent adenylate cyclase signaling by AN is reported to be almost doubled by methyl- β-cyclodextrin treatment, pointing to the relevance of these lipid platforms in cell signaling processes [103]. Conversely, upon membrane cholesterol enrichment, CB1R-dependent signaling was shown to be reduced by half in primary cultures and immortalized cell lines [105,106].
CB1Rs are physically associated with ordered lipid domains in a cholesterol-dependent manner (Figure 2). Cholesterol depletion alters AN-induced CB1R endocytosis and its subsequent trafficking to the lysosomal compartment, pointing to the importance of lipid platforms in the intracellular trafficking of the CB1R [107]. No such information is available on CB2Rs. Lipid platforms can physically sequester different signaling components, preventing crosstalk between different pathways [108], or favor internalization of CB1Rs via caveolae-related endocytosis (Figure 2), negatively regulating CB1R function [109]. For example, the desensitization process of CB1R involves phosphorylation of specific serine residues in the intracellular loop III and C terminal regions. Phosphorylation of these serine residues is performed by protein kinase C (PKC) [110] and G-protein-coupled receptor kinase 3 (GRK3) [111], promoting the recruitment of β-arrestin 2 [111]. These three molecules (PKC, GRK3, and β-arrestin 2) are either resident at or dependent on Lo lipid domains [112], which may explain why, upon disruption of these domains, the desensitization process of CB1R fails and signaling through the CB1R is sustained. Once phosphorylated by PKC and GRK3, β-arrestin 2 is recruited and CB1R is targeted for internalization towards either low pH endosomes and subsequent recycling back to the cell surface, or to late endosomes and lysosomes for degradation (Figure 2). The former endocytic and recycling cycle of the CB1R is facilitated by the small GTPases Rab5 and Rab4, respectively (Figure 2) [62].
Although CB1R is present at the plasma membrane, in HEK-293 cells approximately 85% of the heterologously expressed receptors are localized in intracellular vesicles [62], suggesting a predominantly intracellular localization at a steady state. In agreement with these observations, the intracellular localization of CB1R was previously described in AtT20 cells [113] and hippocampal neurons [114]. Intracellularly located CB1Rs cannot interact with ECs or exogenous cannabinoids, thus preventing stimulation. These data support CB1Rs being constitutively endocytosed. Lo lipid domain instability associated with the diminution of cholesterol content at the plasma membrane would lead to a reduction of the endocytic process and an increase in the number of CB1Rs at the plasma membrane available for ligand interaction. This explains why cholesterol depletion from plasma membranes can favor an enhanced cellular response to ECs, by augmenting the availability of such receptors.
CB2R is also regulated by the Rab family. Specifically, Rab 5 has been implicated, as is also the case with CB1R, in the modulation of the endocytic internalization, whereas Rab11 is reported to participate in a slower recycling process via the perinuclear recycling compartment [64].
Agonist-CBR interactions also mediate lipid raft dynamics. If CB1R are activated in a transient manner, sphingomyelin breakdown is initiated, and ceramides accumulate through functional coupling with the adaptor protein FAN [115]. Conversely, if CB1R are continuously stimulated, G protein-dependent de novo ceramide synthesis takes place through activation of serine palmitoyl transferase activity [116].
3.4. Endocannabinoid Interactions with Endocannabinoid Receptors within the Plasma Membrane
The plasma membrane plays a major role in the interaction between AN and CBRs. In order to deliver its biological message, after being synthesized, AN leaves the postsynaptic membrane, crosses the synaptic cleft, and finds its way to CB1Rs at the presynapse. AN is an amphipathic derivative of arachidonic acid with a polar head of an ethanolamine group. It is considered a lipid-derived neurotransmitter molecule with enhanced water solubility. Because of its lipidic nature, however, it is very unlikely that AN interacts with CB1Rs via their extracellular region; the plasma membrane on the other hand provides an adequate microenvironment for AN to interact with the transmembrane helices of the receptor [117,118]. For this to occur, AN has to firstly penetrate the lipid bilayer and, secondly, diffuse in the plasmalemma to find its binding site [117,119]. Cholesterol again is a central player in the process of guiding AN towards its receptor. Cholesterol by itself suffices to act as the AN transporter at the plasma membrane [120]. AN is deemed to exhibit a specificity for cholesterol in biological membranes over other lipids [121]. This interaction is thought to occur through a hydrogen bond between the –OH group of the sterol and the –NH group of AN [121] that, once established, enables cholesterol to trigger the insertion of AN to the plasma membrane through a flip-flop mechanism favoring the development of van der Waals interactions that stabilize the complex [121]. It should be noted that this process is unlikely to occur within Lo lipid domains, where cholesterol is tightly packed with sphingomyelin [122,123] and is hence inaccessible to AN; it is more likely to occur in non-raft, liquid-disordered domains of the plasma membrane with greater accessibility to AN. Since CB1Rs in the brain are mostly found in Lo lipid domains [107,124], to reach its binding site, AN must penetrate these domains either by passive diffusion (owing to its lipid nature) or by a membrane transport system. Again, cholesterol seems to be the molecule that performs these transport and delivery functions.
From an energetic point of view, AN has a much higher affinity for CB1R than for cholesterol (energy of interaction ~ −136 kJ mol−1 vs. −30.3 kJ mol−1) [125]. This thermodynamic characteristic has led to the suggestion that, when the AN-cholesterol complex reaches the CB1R, AN detaches from cholesterol and moves towards the receptor. The AN binding site on the CB1R has been localized between the transmembrane 6 (TMH6) and 7 (TMH7) regions of the GPCR [121]. TMH7 of the CB1R carries a CARC domain [23,121] that is available for cholesterol interaction. It is proposed that cholesterol could attract AN back, contributing to the release of AN from the binding site, and enabling the CB1R to revert to the unbound state after receptor activation. In addition, the cholesterol-AN complex leaves the polar head of the neurotransmitter fully accessible for enzymatic hydrolysis by FAAH [121]. All in all, the plasma membrane provides the EC system with multiple compartments to regulate the synthesis, transport, and even degradation of its neurotransmitters. It harbors metabolic enzymes and further contributes to the enhancement or abolishment of signal transduction.
4. nAChR
nAChRs are excitatory, cationic pLGICs that reside in presynaptic, postsynaptic, and extrasynaptic membranes in the central and peripheral nervous system [126,127,128,129,130]. They are essential players in neurotransmission across different types of synapses and in muscle contraction. nAChRs can be activated by their natural neurotransmitter, ACh), or by a wide variety of ligands [131,132]. nAChRs at the plasma membrane can adopt multiple conformational states. Upon agonist exposure, the closed state of the channel becomes activated and in the “open state”, the influx of small cations can take place. After depolarization of the cell, the channel can adopt a desensitized state that is unable to be activated by ligand binding or return to a closed state.
Seventeen different subunits (α1–α10, β1–β4, γ, ε, and δ) encoded by seventeen genes in vertebrates that combine and form either homo- or hetero-pentameric structures, have been described [133]. The muscle-type nAChR found at the neuromuscular junction is formed from four distinct subunit types organized in an (α)2βγδ pentamer, the fetal γ-subunit being replaced by the ε-subunit in the adult. The subunit composition of the nAChR determines the kinetics of the conformational stages of the channel. For example, while α4β2 nAChRs desensitize rapidly when exposed to the agonist nicotine, α7 nAChRs do not desensitize as fast [134,135,136]. Also, the subunit composition affects the selective cationic permeability of the nAChR and the pharmacological affinities of various agonists [137,138,139,140].
nAChRs influence synaptic plasticity by increasing intracellular Ca2+ release, inducing long-term potentiation (LTP) and favoring a depolarization state. The activation of presynaptically located nAChRs results in the release of many neurotransmitters including dopamine, norepinephrine, GABA, and Glu in a Ca2+-dependent manner. Heteromeric α4β2 and homomeric α7 nAChRs are the most abundant neuronal subtypes [141,142]. The activation of these receptors in hippocampal interneurons indirectly affects neurotransmitters release (Glu or GABA) by activating voltage gated calcium channels [143,144]. Furthermore, presynaptic α4β2 nAChR activation by the agonist nicotine is reported to induce dendritic spine enlargement as a consequence of increased Glu concentration and of glutamatergic neurotransmission [145], thus affecting synaptic plasticity. Activation of other neuronal nAChRs, such as the α3β4 nAChR, at presynaptic sites is shown to stimulate tetrodotoxin-insensitive GABA release via T-type voltage gated calcium channels and Ca2+ from internal stores [146].
At postsynaptic sites, the activation of nAChRs also produces significant inward (depolarizing) currents in neurons in many brain regions and modulates synaptic plasticity. A7 nAChR at postsynaptic sites can regulate glutamate receptors and, hence, glutamatergic signaling [147,148], as well as GABAergic interneuron activity [149,150]. Halff and coworkers proposed a mechanism by which α7 nAChRs would modulate synaptic potentiation independently of fast excitatory transmission [7]. They report that the activation of α7 nAChRs at postsynaptic sites recruits GluA1 receptors from the surface pool of mobile extrasynaptic receptors, this in turn contributing to stabilization of GluA1 receptors in the neuronal spine and producing an increase in the density of this subtype of glutamatergic receptors. However, the participation of functional PSD-95 scaffold protein family members is required to anchor GluA1 macromolecules at the postsynaptic membrane. Both activation and inhibition of α7 nAChRs in the prelimbic cortex result in the induction of LTP [151]. Modulating network excitability by cholinergic signaling must therefore be finely regulated for adequate neural excitability and plasticity to take place.
4.1. nAChR Localization within the Plasma Membrane
The neuronal nAChRs are found in brain areas considered to be involved in learning, cognition, and memory, such as the basal forebrain, hippocampus, cerebellum, and the temporal cortex [152]. nAChRs are found within lipid platforms in the plasma membrane. These cholesterol/sphingolipid-rich lipid domains determine nAChR nanocluster topography function and mobility on cell surfaces [153,154]. Numerous papers over the past 40 years have provided data on the multiplicity of effects of cholesterol on the peripheral nAChRs found in skeletal muscle, and in the electromotor synapse of electric fish.
nAChRs are influenced by cholesterol concentration at various levels of organization, within multiple time windows, and during ontogenetic development and adulthood [20,155]. Cholesterol homeostasis dysregulation produces alterations in the biophysical properties of the membrane bilayer that affect the mobility and protein–protein interactions of neurotransmitter receptors [22].
Cholesterol/sphingolipid/ceramide-rich lipid platforms are required for both muscle-type and neuronal nAChR trafficking to the plasma membrane [153,156,157,158,159]. Indeed, our group has provided evidence that disruption of these lipid platforms leads to both altered nAChR function and cell-surface expression [153,154].
Lowering cholesterol levels at the plasma membrane causes rapid nAChR (muscle type) internalization, and compensatory gain-of-function of the nAChRs remaining at the plasma membrane [160,161,162]. Studies using stimulated emission depletion (STED) [163] and single-molecule localization (SMLM) [164,165] superresolution microscopies further show contrasting changes in the distribution of nAChR nanoclusters and individual molecules upon cholesterol depletion or enrichment. In addition, changes in the cholesterol concentration of the membrane followed using fluorescence recovery after photobleaching and fluorescence correlation spectroscopy [153] and SMLM [164,165] have also been shown to modify the translational mobility of the receptor in the plane of the plasma membrane. Likewise, cholesterol content in the cell-surface membrane modulates neuronal nAChR subtypes. In a recent study by Báez-Pagán and coworkers, a reduction in the macroscopic response of the neuronal α7 nAChR subtype was described as cholesterol to phospholipid ratios increased [166]. The α7 nAChR subtype of nAChRs is predominantly located in cholesterol-rich Lo domains [157,159,167,168]. Other studies from our laboratory indicate that long-term inhibition of cholesterol biosynthesis can differentially augment cell-surface levels of both α4β2 and α7 nAChRs in the neurites and soma of rat hippocampal neurons [169].
Bearing in mind that neuronal nAChRs can be found on both the soma and synaptic terminals of excitatory and inhibitory neurons in different brain regions (Figure 1), the lipid microenvironment of the plasma membrane where they reside plays a crucial role in the modulation of nAChR responses. Additionally, alteration of cholesterol content at the plasma membrane will affect neuronal excitability through the modulation of the nAChR channels and by indirectly impacting on GABAergic and glutamatergic transmission, in turn possibly impinging on multivariable physiological or pathophysiological outcomes.
4.2. nAChR and EC Receptor Crosstalk
The overlapping distribution of CBRs and nAChRs is not fortuitous. Their distribution anatomically overlaps in brain areas such as the midbrain, the hippocampus, and the amygdala. Many papers support the notion that nicotinic and EC systems interact bidirectionally in the brain reward pathway [170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187]. Buczynski and coworkers, reported that nicotine self-administration in rats modified AN levels in the VTA [188]. Furthermore, fluctuations in AN but not 2-AG are associated with withdrawal from nicotine [189]. The lipophilic nature of ECs allows their incorporation into the plasma membrane, and their presence is shown to modulate the action of alcohol and volatile anesthetics on α7 nAChRs [190,191], suggesting that an alteration of the physicochemical properties of the plasma membrane is taking place. Using Xenopus oocytes, Oz and coworkers demonstrated that AN can reversibly inhibit nicotine-induced currents in a concentration-dependent manner without significantly affecting its half maximal effective concentration (EC50) value. These authors suggest that AN behaves as a noncompetitive antagonist of α7 nAChRs [192,193]. Likewise, AN was shown to inhibit the peak amplitudes of α4β2 nAChR-mediated currents in SH-EP1 cells [194] and in myenteric neurons [92,93]. In addition, synthetic cannabinoids have been reported to modulate cholinergic neurotransmission in the hippocampus [170,185,186,187]. In the latter, nAChRs and CB1Rs play a role in the cognition-impairing effects of the main psychoactive ingredient of cannabis, δ-9-tetrahydrocannabinol (THC) [195]. In the mesolimbic dopamine system, they are implicated in brain reward processes [195,196,197]. The blockade of α7 nAChR by methyllycaconitine at non-toxic concentrations reduces the behavioral and neurochemical effects of THC related to its abuse [198]. It has therefore been suggested that drugs that block α7 nAChR could be useful agents in the treatment of cannabis abuse in humans [198]. Contrarily, THC is reported to increase the response of the α7 nAChR to ACh by 128% [199]. Cannabidiol (CBD), the other most abundant component of cannabis, is reported to reduce cigarette consumption by 40% and to reduce the response of the α7 nAChR to ACh by 49% [199]. Understanding the complex interactions between the cholinergic and the endocannabinoid systems in the nervous system is critical to increasing our chances of using cannabinoids in the clinic as therapeutic tools.
Cohen and coworkers reported that SR141716 (rimonabant), a CB1R antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats [200], while other authors have also observed that rimonabant decreases the motivation to self-administer nicotine [201]. In support of the existence of a physiological interaction between the cannabinoid and cholinergic systems, genetic deletion of CB1R in knockout mice was demonstrated to inhibit nicotine-induced rewarding effects, evaluated by a conditioned place preference paradigm (CPP) [202,203]. In addition, a blockade of the CB1R in various areas of the brain, such as in the shell of the NAc, the basolateral amygdala, the prelimbic cortex, and in the bed nucleus of the stria terminalis, was reported to produce a reduction in nicotine-seeking behavior [204,205]. Genetic deletion or pharmacological inhibition of FAAH is shown to enhance the expression of nicotine CPP [203], thus suggesting that ECs play an important role in the rewarding properties of nicotine. Furthermore, CB1R stimulation in rats was reported to increase the motivation to self-administer nicotine and to enhance cue-induced reinstatement of nicotine-seeking behavior [206]. In contrast, neither the activation nor the inhibition of CB2R was shown to produce effects on the motivation to obtain nicotine or nicotine intake in rats [207]. However, studies performed in mice have documented the relevance of CB2R receptors on the rewarding/reinforcing properties of nicotine [208,209]. Thus, the current literature supports a distinct profile of CB1R and CB2R effects on behavior, and suggests that there may be important species differences mediating these effects.
The modulatory crossover effects taking place between CBRs and nAChRs at the plasma membrane point to EC and cholinergic systems as holding significant promise for the development of novel therapeutic strategies in behavior and addiction.
5. Concluding Remarks
Biological membranes are much more complex than double-layered lipid barriers acting simply as a boundary between the intracellular content and the extracellular medium. They are highly organized and multifaceted structures that provide an asymmetrical interface where many transport and enzymatic processes can simultaneously occur. The direct observation of nanoscopic lateral heterogeneities in the plane of the membrane accomplished in the last two decades has provided structural grounds that help explain how these dynamic platforms control and govern signal transduction, neurotransmitter synthesis, metabolism, and degradation. Here, we briefly reviewed the importance of these supramolecular structures and mechanisms using the CB1R and the nAChR as an example of two interacting cell-surface signaling systems. We also stressed the importance of the neutral lipid cholesterol in the phenomena involving these paradigmatic receptors. Cholesterol plays a direct role in orchestrating the membrane organization and endocytosis of the two receptor systems, contributes to the transport of the lipidic endocannabinoid AN to and from the CB1R, and participates in the regulation of the number, distribution, and functional states of nAChRs.
Author Contributions
F.J.B. and A.S.V. conceived the work, searched the literature, and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
No funding is associated with this review.
Acknowledgments
Figures were created using Servier Medical Art Commons Attribution 3.0 Unported License. http://smart.servier.com (accessed on 1 July 2022). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
Conflicts of Interest
Authors declare no conflict of interest.
References
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.; Dietrich, C. Looking at lipid rafts? Trends Cell Biol. 1999, 9, 87–91. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Caldarone, B.J.; King, S.L.; Zachariou, V. Nicotinic Receptors in the Brain Links between Molecular Biology and Behavior. Neuropsychopharmacology 2000, 22, 451–465. [Google Scholar] [CrossRef]
- Schlicker, E.; Kathmann, M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol. Sci. 2001, 22, 565–572. [Google Scholar] [CrossRef]
- Le Foll, B.; Goldberg, S.R. Cannabinoid CB1 Receptor Antagonists as Promising New Medications for Drug Dependence. J. Pharmacol. Exp. Ther. 2005, 312, 875–883. [Google Scholar] [CrossRef]
- Lozada, A.F.; Wang, X.; Gounko, N.V.; Massey, K.A.; Duan, J.; Liu, Z.; Berg, D.K. Induction of Dendritic Spines by 2-Containing Nicotinic Receptors. J. Neurosci. 2012, 32, 8391–8400. [Google Scholar] [CrossRef]
- Halff, A.W.; Gómez-Varela, D.; John, D.; Berg, D.K. A novel mechanism for nicotinic potentiation of glutamatergic synapses. J. Neurosci. 2014, 34, 2051–2064. [Google Scholar] [CrossRef]
- Najafinobar, N.; Mellander, L.J.; Kurczy, M.E.; Dunevall, J.; Angerer, T.B.; Fletcher, J.S.; Cans, A.-S. Cholesterol Alters the Dynamics of Release in Protein Independent Cell Models for Exocytosis. Sci. Rep. 2016, 6, 33702. [Google Scholar] [CrossRef]
- Biswas, A.; Kashyap, P.; Datta, S.; Sengupta, T.; Sinha, B. Cholesterol Depletion by MβCD Enhances Cell Membrane Tension and Its Variations-Reducing Integrity. Biophys. J. 2019, 116, 1456–1468. [Google Scholar] [CrossRef]
- Karatekin, E.; Sandre, O.; Guitouni, H.; Borghi, N.; Puech, P.-H.; Brochard-Wyart, F. Cascades of Transient Pores in Giant Vesicles: Line Tension and Transport. Biophys. J. 2003, 84, 1734–1749. [Google Scholar] [CrossRef]
- Goñi, F.M.; Sot, J.; Alonso, A. Biophysical properties of sphingosine, ceramides and other simple sphingolipids. Biochem. Soc. Trans. 2014, 42, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.; Lin, H.; Fritch, M.R.; Tuan, R.S. Influence of cholesterol/caveolin-1/caveolae homeostasis on membrane properties and substrate adhesion characteristics of adult human mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Yano, Y. Effects of Membrane Cholesterol on Stability of Transmembrane Helix Associations. Chem. Pharm. Bull. 2022, 70, 514–518. [Google Scholar] [CrossRef]
- Bennett, W.F.D.; MacCallum, J.L.; Hinner, M.J.; Marrink, S.J.; Tieleman, D.P. Molecular View of Cholesterol Flip-Flop and Chemical Potential in Different Membrane Environments. J. Am. Chem. Soc. 2009, 131, 12714–12720. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska, A.; Draus, J.; Subczynski, W.K. Is a fluid-mosaic model of biological membranes fully relevant? Studies on lipid organization in model and biological membranes. Cell. Mol. Biol. Lett. 2003, 8, 147–159. [Google Scholar]
- Subczynski, W.K.; Kusumi, A. Dynamics of raft molecules in the cell and artificial membranes: Approaches by pulse EPR spin labeling and single molecule optical microscopy. Biochim. Biophys. Acta (BBA)-Biomembr. 2003, 1610, 231–243. [Google Scholar] [CrossRef]
- Aussenac, F.; Tavares, A.M.; Dufourc, E.J. Cholesterol Dynamics in Membranes of Raft Composition: A Molecular Point of View from 2H and 31P Solid-State NMR. Biochemistry 2003, 42, 1383–1390. [Google Scholar] [CrossRef]
- Baier, C.J.; Fantini, J.; Barrantes, F.J. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep. 2011, 1, 69. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F.J. Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 2345–2361. [Google Scholar] [CrossRef]
- Barrantes, F.J. Cholesterol effects on nicotinic acetylcholine receptor. J. Neurochem. 2007, 103, 72–80. [Google Scholar] [CrossRef]
- Levitan, I.; Barrantes, F.J. Cholesterol Regulation of Ion Channels and Receptors; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef]
- Oddi, S.; Dainese, E.; Fezza, F.; Lanuti, M.; Barcaroli, D.; De Laurenzi, V.; Centonze, D.; Maccarrone, M. Functional characterization of putative cholesterol binding sequence (CRAC) in human type-1 cannabinoid receptor. J. Neurochem. 2011, 116, 858–865. [Google Scholar] [CrossRef]
- Lozada, A.F.; Wang, X.; Gounko, N.; Massey, K.A.; Duan, J.; Liu, Z.; Berg, D.K. Glutamatergic Synapse Formation is Promoted by 7-Containing Nicotinic Acetylcholine Receptors. J. Neurosci. 2012, 32, 7651–7661. [Google Scholar] [CrossRef]
- Sodero, A.O.; Weissmann, C.; Ledesma, M.D.; Dotti, C.G. Cellular stress from excitatory neurotransmission contributes to cholesterol loss in hippocampal neurons aging in vitro. Neurobiol. Aging 2011, 32, 1043–1053. [Google Scholar] [CrossRef]
- Martin, M.G.; Ahmed, T.; Korovaichuk, A.; Venero, C.; Menchón, S.A.; Salas, I.; Munck, S.; Herreras, O.; Balschun, D.; Dotti, C.G. Constitutive hippocampal cholesterol loss underlies poor cognition in old rodents. EMBO Mol. Med. 2014, 6, 902–917. [Google Scholar] [CrossRef]
- Arroyo, A.I.; Camoletto, P.G.; Morando, L.; Sassoe-Pognetto, M.; Giustetto, M.; Van Veldhoven, P.P.; Schuchman, E.H.; Ledesma, M.D. Pharmacological reversion of sphingomyelin-induced dendritic spine anomalies in a Niemann Pick disease type A mouse model. EMBO Mol. Med. 2014, 6, 398–413. [Google Scholar] [CrossRef]
- Doria, M.; Maugest, L.; Moreau, T.; Lizard, G.; Vejux, A. Contribution of cholesterol and oxysterols to the pathophysiology of Parkinson’s disease. Free Radic. Biol. Med. 2016, 101, 393–400. [Google Scholar] [CrossRef]
- Karasinska, J.M.; Hayden, M.R. Cholesterol metabolism in Huntington disease. Nat. Rev. Neurol. 2011, 7, 561–572. [Google Scholar] [CrossRef]
- Bi, X.; Liao, G. Cholesterol in Niemann–Pick Type C disease. In Cholesterol Binding and Cholesterol Transport Proteins; Harris, J., Ed.; Springer: Dordrecht, The Netherlands, 2010; Volume 51, pp. 319–335. [Google Scholar] [CrossRef]
- Huang, X.; Chen, H.; Miller, W.C.; Mailman, R.B.; Woodard, J.L.; Chen, P.C.; Xiang, D.; Murrow, R.W.; Wang, Y.-Z.; Poole, C. Lower low-density lipoprotein cholesterol levels are associated with Parkinson’s disease. Mov. Disord. 2007, 22, 377–381. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.-F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high β- and γ-secretase activities and Aβ production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef]
- Fecchio, C.; De Franceschi, G.; Relini, A.; Greggio, E.; Dalla Serra, M.; Bubacco, L.; De Laureto, P.P. α-Synuclein Oligomers Induced by Docosahexaenoic Acid Affect Membrane Integrity. PLoS ONE 2013, 8, e82732. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Solomon, T.; Malajczuk, C.; Mancera, R.; Howard, M.; Arrigan, D.; Newsholme, P.; Martins, R. Role of the cell membrane interface in modulating production and uptake of Alzheimer’s beta amyloid protein. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.-J.; Ho, D.H.; Park, E.; Jung, J.W.; Cho, K.; Hong, J.H.; Lee, H.-J.; Kim, K.P.; Lee, S.-J. Lipid Peroxidation Product 4-Hydroxy-2-Nonenal Promotes Seeding-Capable Oligomer Formation and Cell-to-Cell Transfer of α-Synuclein. Antioxid. Redox Signal. 2013, 18, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.; Aguayo, L.G.; Opazo, C. An extracellular mechanism that can explain the neurotoxic effects of α-synuclein aggregates in the brain. Front. Physiol. 2012, 3, 297. [Google Scholar] [CrossRef]
- Jang, H.; Connelly, L.; Arce, F.T.; Ramachandran, S.; Kagan, B.L.; Lal, R.; Nussinov, R. Mechanisms for the Insertion of Toxic, Fibril-like β-Amyloid Oligomers into the Membrane. J. Chem. Theory Comput. 2013, 9, 822–833. [Google Scholar] [CrossRef]
- Reynolds, N.P.; Soragni, A.; Rabe, M.; Verdes, D.; Liverani, E.; Handschin, S.; Riek, R.; Seeger, S. Mechanism of Membrane Interaction and Disruption by α-Synuclein. J. Am. Chem. Soc. 2011, 133, 19366–19375. [Google Scholar] [CrossRef]
- Dikiy, I.; Eliezer, D. Folding and misfolding of alpha-synuclein on membranes. Biochim. Biophys. Acta (BBA)-Biomembr. 2012, 1818, 1013–1018. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Aperia, A.; Melki, R.; Triller, A. Physico-Pathologic Mechanisms Involved in Neurodegeneration: Misfolded Protein-Plasma Membrane Interactions. Neuron 2017, 95, 33–50. [Google Scholar] [CrossRef]
- Sasahara, K.; Morigaki, K.; Shinya, K. Effects of membrane interaction and aggregation of amyloid β-peptide on lipid mobility and membrane domain structure. Phys. Chem. Chem. Phys. 2013, 15, 8929–8939. [Google Scholar] [CrossRef]
- Lee, J.; Gillman, A.; Jang, H.; Ramachandran, S.; Kagan, B.L.; Nussinov, R.; Arce, F.T. Role of the Fast Kinetics of Pyroglutamate-Modified Amyloid-β Oligomers in Membrane Binding and Membrane Permeability. Biochemistry 2014, 53, 4704–4714. [Google Scholar] [CrossRef]
- Ouberai, M.M.; Wang, J.; Swann, M.J.; Galvagnion, C.; Guilliams, T.; Dobson, C.M.; Welland, M.E. α-Synuclein Senses Lipid Packing Defects and Induces Lateral Expansion of Lipids Leading to Membrane Remodeling. J. Biol. Chem. 2013, 288, 20883–20895. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Sachs, J.N.; Rhoades, E.; Baumgart, T. Biophysics of α-synuclein induced membrane remodelling. Phys. Chem. Chem. Phys. 2015, 17, 15561–15568. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, U.; Ramalingam, N.; von Saucken, V.; Kim, T.-E.; Newman, A.J.; Terry-Kantor, E.; Nuber, S.; Ericsson, M.; Fanning, S.; Bartels, T.; et al. Loss of native α-synuclein multimerization by strategically mutating its amphipathic helix causes abnormal vesicle interactions in neuronal cells. Hum. Mol. Genet. 2017, 26, 3466–3481. [Google Scholar] [CrossRef] [PubMed]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane Curvature Induction and Tubulation Are Common Features of Synucleins and Apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.C.; Freeley, M.; Nield, J.; Palma, M.; Viles, J.H. Amyloid-β oligomers have a profound detergent-like effect on lipid membrane bilayers, imaged by atomic force and electron microscopy. J. Biol. Chem. 2019, 294, 7566–7572. [Google Scholar] [CrossRef]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different Species of α-Synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar α-Synuclein and Huntingtin Exon 1 Assemblies Are Toxic to the Cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef]
- Nakahata, Y.; Yasuda, R. Plasticity of Spine Structure: Local Signaling, Translation and Cytoskeletal Reorganization. Front. Synaptic Neurosci. 2018, 10, 29. [Google Scholar] [CrossRef]
- Penzes, P.; Rafalovich, I. Regulation of the Actin Cytoskeleton in Dendritic Spines. In Synaptic Plasticity. Advances in Experimental Medicine and Biology; Kreutz, M., Sala, C., Eds.; Springer: Vienna, Austria, 2012; Volume 970, pp. 81–95. [Google Scholar] [CrossRef]
- Runge, K.; Cardoso, C.; De Chevigny, A. Dendritic Spine Plasticity: Function and Mechanisms. Front. Synaptic Neurosci. 2020, 12, 36. [Google Scholar] [CrossRef]
- Spence, E.F.; Soderling, S.H. Actin Out: Regulation of the Synaptic Cytoskeleton. J. Biol. Chem. 2015, 290, 28613–28622. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, R. The actin cytoskeleton in memory formation. Prog. Neurobiol. 2014, 117, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Sala, C. PDZ Domains and the Organization of Supramolecular Complexes. Annu. Rev. Neurosci. 2001, 24, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Pak, D.T.S. Ligand-Gated Ion Channel Interactions with Cytoskeletal and Signaling Proteins. Annu. Rev. Physiol. 2000, 62, 755–778. [Google Scholar] [CrossRef]
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a Novel Family of Postsynaptic Density Proteins that Binds to the NMDA Receptor/PSD-95/GKAP Complex and Cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef]
- Sala, C.; Piëch, V.; Wilson, N.R.; Passafaro, M.; Liu, G.; Sheng, M. Regulation of Dendritic Spine Morphology and Synaptic Function by Shank and Homer. Neuron 2001, 31, 115–130. [Google Scholar] [CrossRef]
- Gorvel, J.-P.; Chavrier, P.; Zerial, M.; Gruenberg, J. rab5 controls early endosome fusion in vitro. Cell 1991, 64, 915–925. [Google Scholar] [CrossRef]
- Bucci, C.; Parton, R.G.; Mather, I.H.; Stunnenberg, H.; Simons, K.; Hoflack, B.; Zerial, M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 1992, 70, 715–728. [Google Scholar] [CrossRef]
- Leterrier, C.; Bonnard, D.; Carrel, D.; Rossier, J.; Lenkei, Z. Constitutive Endocytic Cycle of the CB1 Cannabinoid Receptor. J. Biol. Chem. 2004, 279, 36013–36021. [Google Scholar] [CrossRef]
- van der Sluijs, P.; Hull, M.; Webster, P.; Mâle, P.; Goud, B.; Mellman, I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 1992, 70, 729–740. [Google Scholar] [CrossRef]
- Ullrich, O.; Reinsch, S.; Urbe, S.; Zerial, M.; Parton, R. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 1996, 135, 913–924. [Google Scholar] [CrossRef]
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455. [Google Scholar] [CrossRef]
- Correa, F.; Wolfson, M.L.; Valchi, P.; Aisemberg, J.; Franchi, A.M. Endocannabinoid system and pregnancy. Reproduction 2016, 152, R191–R200. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Rockwell, C.; Snider, N.T.; Thompson, J.T.; Heuvel, J.P.V.; Kaminski, N.E. Interleukin-2 Suppression by 2-Arachidonyl Glycerol Is Mediated through Peroxisome Proliferator-Activated Receptor γ Independently of Cannabinoid Receptors 1 and 2. Mol. Pharmacol. 2006, 70, 101–111. [Google Scholar] [CrossRef]
- Ghosh, M.; Wang, H.; Ai, Y.; Romeo, E.; Luyendyk, J.P.; Peters, J.; Mackman, N.; Dey, S.K.; Hla, T. COX-2 suppresses tissue factor expression via endocannabinoid-directed PPARδ activation. J. Exp. Med. 2007, 204, 2053–2061. [Google Scholar] [CrossRef]
- Jhaveri, M.D.; Richardson, D.; Robinson, I.; Garle, M.J.; Patel, A.; Sun, Y.; Sagar, D.R.; Bennett, A.J.; Alexander, S.P.; Kendall, D.A.; et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase-2 increases levels of endocannabinoid related molecules and produces analgesia via peroxisome proliferator-activated receptor-alpha in a model of inflammatory pain. Neuropharmacology 2008, 55, 85–93. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.-Y.; Lu, H.-C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Bodor, L.; Katona, I.; Nyiri, G.; Mackie, K.; Ledent, C.; Hajos, N.; Freund, T.F. Endocannabinoid Signaling in Rat Somatosensory Cortex: Laminar Differences and Involvement of Specific Interneuron Types. J. Neurosci. 2005, 25, 6845–6856. [Google Scholar] [CrossRef]
- Katona, I.; Urbán, G.M.; Wallace, M.; Ledent, C.; Jung, K.-M.; Piomelli, D.; Mackie, K.; Freund, T.F. Molecular Composition of the Endocannabinoid System at Glutamatergic Synapses. J. Neurosci. 2006, 26, 5628–5637. [Google Scholar] [CrossRef]
- Nyíri, G.; Cserép, C.; Szabadits, E.; MacKie, K.; Freund, T. CB1 cannabinoid receptors are enriched in the perisynaptic annulus and on preterminal segments of hippocampal GABAergic axons. Neuroscience 2005, 136, 811–822. [Google Scholar] [CrossRef]
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar] [CrossRef]
- Melis, M.; Perra, S.; Muntoni, A.L.; Pillolla, G.; Lutz, B.; Marsicano, G.; Di Marzo, V.; Gessa, G.L.; Pistis, M. Prefrontal Cortex Stimulation Induces 2-Arachidonoyl-Glycerol-Mediated Suppression of Excitation in Dopamine Neurons. J. Neurosci. 2004, 24, 10707–10715. [Google Scholar] [CrossRef]
- Melis, M.; Pistis, M.; Perra, S.; Muntoni, A.L.; Pillolla, G.; Gessa, G.L. Endocannabinoids Mediate Presynaptic Inhibition of Glutamatergic Transmission in Rat Ventral Tegmental Area Dopamine Neurons through Activation of CB1 Receptors. J. Neurosci. 2004, 24, 53–62. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-Mediated Control of Synaptic Transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid Signaling in the Brain. Science 2002, 296, 678–682. [Google Scholar] [CrossRef]
- Alger, B.E.; Kim, J. Supply and demand for endocannabinoids. Trends Neurosci. 2011, 34, 304–315. [Google Scholar] [CrossRef]
- Goonawardena, A.V.; Robinson, L.; Hampson, R.E.; Riedel, G. Cannabinoid and cholinergic systems interact during performance of a short-term memory task in the rat. Learn. Mem. 2010, 17, 502–511. [Google Scholar] [CrossRef]
- Nyíri, G.; Szabadits, E.; Cserép, C.; Mackie, K.; Shigemoto, R.; Freund, T.F. GABAB and CB1cannabinoid receptor expression identifies two types of septal cholinergic neurons. Eur. J. Neurosci. 2005, 21, 3034–3042. [Google Scholar] [CrossRef]
- Fortin, D.A.; Levine, E.S. Differential Effects of Endocannabinoids on Glutamatergic and GABAergic Inputs to Layer 5 Pyramidal Neurons. Cereb. Cortex 2007, 17, 163–174. [Google Scholar] [CrossRef]
- Lange, M.D.; Daldrup, T.; Remmers, F.; Szkudlarek, H.J.; Lesting, J.; Guggenhuber, S.; Ruehle, S.; Jüngling, K.; Seidenbecher, T.; Lutz, B.; et al. Cannabinoid CB1 receptors in distinct circuits of the extended amygdala determine fear responsiveness to unpredictable threat. Mol. Psychiatry 2017, 22, 1422–1430. [Google Scholar] [CrossRef]
- Ohno-Shosaku, T.; Tsubokawa, H.; Mizushima, I.; Yoneda, N.; Zimmer, A.; Kano, M. Presynaptic Cannabinoid Sensitivity Is a Major Determinant of Depolarization-Induced Retrograde Suppression at Hippocampal Synapses. J. Neurosci. 2002, 22, 3864–3872. [Google Scholar] [CrossRef]
- Covey, D.P.; Mateo, Y.; Sulzer, D.; Cheer, J.F.; Lovinger, D.M. Endocannabinoid modulation of dopamine neurotransmission. Neuropharmacology 2017, 124, 52–61. [Google Scholar] [CrossRef]
- Di Chiara, G.; Imperato, A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 1988, 85, 5274–5278. [Google Scholar] [CrossRef]
- De Biasi, M.; Dani, J.A. Reward, Addiction, Withdrawal to Nicotine. Annu. Rev. Neurosci. 2011, 34, 105–130. [Google Scholar] [CrossRef]
- Medeiros, D.; Silva-Gonçalves, L.D.C.; Da Silva, A.M.B.; Cabrera, M.P.D.S.; Arcisio-Miranda, M. Membrane-mediated action of the endocannabinoid anandamide on membrane proteins: Implications for understanding the receptor-independent mechanism. Sci. Rep. 2017, 7, 41362. [Google Scholar] [CrossRef]
- Chemin, J.; Monteil, A.; Perez-Reyes, E.; Nargeot, J.; Lory, P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 2001, 20, 7033–7040. [Google Scholar] [CrossRef]
- Oz, M. Receptor-Independent Effects of Endocannabinoids on Ion Channels. Curr. Pharm. Des. 2006, 12, 227–239. [Google Scholar] [CrossRef]
- Oz, M. Receptor-independent actions of cannabinoids on cell membranes: Focus on endocannabinoids. Pharmacol. Ther. 2006, 111, 114–144. [Google Scholar] [CrossRef]
- Barana, A.; Amorós, I.; Caballero, R.; Gómez, R.; Osuna, L.; Lillo, M.P.; Blázquez, C.; Guzmán, M.; Delpón, E.; Tamargo, J. Endocannabinoids and cannabinoid analogues block cardiac hKv1.5 channels in a cannabinoid receptor-independent manner. Cardiovasc. Res. 2009, 85, 56–67. [Google Scholar] [CrossRef]
- Prather, P.L.; Martin, N.A.; Breivogel, C.S.; Childers, S.R. Activation of cannabinoid receptors in rat brain by WIN 55212-2 produces coupling to multiple G protein alpha-subunits with different potencies. Mol. Pharmacol. 2000, 57, 1000–1010. [Google Scholar]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Fisyunov, A.; Tsintsadze, V.; Min, R.; Burnashev, N.; Lozovaya, N. Cannabinoids Modulate the P-Type High-Voltage-Activated Calcium Currents in Purkinje Neurons. J. Neurophysiol. 2006, 96, 1267–1277. [Google Scholar] [CrossRef]
- Gebremedhin, D.; Lange, A.R.; Campbell, W.B.; Hillard, C.J.; Harder, D.R. Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am. J. Physiol. Circ. Physiol. 1999, 276, H2085–H2093. [Google Scholar] [CrossRef]
- Mackie, K.; Lai, Y.; Westenbroek, R.; Mitchell, R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J. Neurosci. 1995, 15, 6552–6561. [Google Scholar] [CrossRef]
- Deadwyler, S.A.; Hampson, R.E.; Mu, J.; Whyte, A.; Childers, S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J. Pharmacol. Exp. Ther. 1995, 273, 734–743. [Google Scholar]
- Alger, B.E. Endocannabinoid Signaling in Neural Plasticity. In Behavioral Neurobiology of the Endocannabinoid System; Kendall, D., Alexander, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 1, pp. 141–172. [Google Scholar] [CrossRef]
- Chevaleyre, V.; Takahashi, K.A.; Castillo, P.E. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 2006, 29, 37–76. [Google Scholar] [CrossRef]
- Dainese, E.; Oddi, S.; Bari, M.; Maccarrone, M. Modulation of the Endocannabinoid System by Lipid Rafts. Curr. Med. Chem. 2007, 14, 2702–2715. [Google Scholar] [CrossRef]
- Siegmund, S.V.; Uchinami, H.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. Anandamide induces necrosis in primary hepatic stellate cells. Hepatology 2005, 41, 1085–1095. [Google Scholar] [CrossRef]
- Bari, M.; Spagnuolo, P.; Fezza, F.; Oddi, S.; Pasquariello, N.; Finazzi-Agrò, A.; Maccarrone, M. Effect of Lipid Rafts on Cb2 Receptor Signaling and 2-Arachidonoyl-Glycerol Metabolism in Human Immune Cells. J. Immunol. 2006, 177, 4971–4980. [Google Scholar] [CrossRef]
- Bari, M.; Paradisi, A.; Pasquariello, N.; Maccarrone, M. Cholesterol-dependent modulation of type 1 cannabinoid receptors in nerve cells. J. Neurosci. Res. 2005, 81, 275–283. [Google Scholar] [CrossRef]
- Sarnataro, D.; Grimaldi, C.; Pisanti, S.; Gazzerro, P.; Laezza, C.; Zurzolo, C.; Bifulco, M. Plasma membrane and lysosomal localization of CB1 cannabinoid receptor are dependent on lipid rafts and regulated by anandamide in human breast cancer cells. FEBS Lett. 2005, 579, 6343–6349. [Google Scholar] [CrossRef]
- Moffett, S.; Brown, D.A.; Linder, M.E. Lipid-dependent Targeting of G Proteins into Rafts. J. Biol. Chem. 2000, 275, 2191–2198. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. G Protein-coupled Receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J. Biol. Chem. 1998, 273, 18677–18680. [Google Scholar] [CrossRef]
- Garcia, D.E.; Brown, S.; Hille, B.; Mackie, K. Protein Kinase C Disrupts Cannabinoid Actions by Phosphorylation of the CB1 Cannabinoid Receptor. J. Neurosci. 1998, 18, 2834–2841. [Google Scholar] [CrossRef]
- Jin, W.; Brown, S.; Roche, J.P.; Hsieh, C.; Celver, J.P.; Kovoor, A.; Chavkin, C.; Mackie, K. Distinct Domains of the CB1 Cannabinoid Receptor Mediate Desensitization and Internalization. J. Neurosci. 1999, 19, 3773–3780. [Google Scholar] [CrossRef]
- Ostrom, R.; Insel, P.A. The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: Implications for molecular pharmacology. Br. J. Pharmacol. 2004, 143, 235–245. [Google Scholar] [CrossRef]
- Hsieh, C.; Brown, S.; Derleth, C.; Mackie, K. Internalization and Recycling of the CB1 Cannabinoid Receptor. J. Neurochem. 1999, 73, 493–501. [Google Scholar] [CrossRef]
- Coutts, A.A.; Anavi-Goffer, S.; Ross, R.A.; MacEwan, D.J.; Mackie, K.; Pertwee, R.G.; Irving, A.J. Agonist-Induced Internalization and Trafficking of Cannabinoid CB1 Receptors in Hippocampal Neurons. J. Neurosci. 2001, 21, 2425–2433. [Google Scholar] [CrossRef]
- Sánchez, C.; Rueda, D.; Ségui, B.; Galve-Roperh, I.; Levade, T.; Guzmán, M. The CB1 Cannabinoid Receptor of Astrocytes Is Coupled to Sphingomyelin Hydrolysis through the Adaptor Protein Fan. Mol. Pharmacol. 2001, 59, 955–959. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Sánchez, C.; Cortés, M.L.; Del Pulgar, T.G.; Izquierdo, M.; Guzmán, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef]
- Tian, X.; Guo, J.; Yao, F.; Yang, D.-P.; Makriyannis, A. The Conformation, Location, and Dynamic Properties of the Endocannabinoid Ligand Anandamide in a Membrane Bilayer. J. Biol. Chem. 2005, 280, 29788–29795. [Google Scholar] [CrossRef]
- Barnett-Norris, J.; Hurst, D.P.; Lynch, D.L.; Guarnieri, F.; Makriyannis, A.; Reggio, P.H. Conformational Memories and the Endocannabinoid Binding Site at the Cannabinoid CB1 Receptor. J. Med. Chem. 2002, 45, 3649–3659. [Google Scholar] [CrossRef]
- Howlett, A.C.; Reggio, P.H.; Childers, S.R.; Hampson, R.E.; Ulloa, N.M.; Deutsch, D.G. Endocannabinoid tone versus constitutive activity of cannabinoid receptors. Br. J. Pharmacol. 2011, 163, 1329–1343. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Chahinian, H.; Sanchez, P.; Fantini, J. The Insertion and Transport of Anandamide in Synthetic Lipid Membranes Are Both Cholesterol-Dependent. PLoS ONE 2009, 4, e4989. [Google Scholar] [CrossRef]
- Di Scala, C.; Fantini, J.; Yahi, N.; Barrantes, F.J.; Chahinian, H. Anandamide Revisited: How Cholesterol and Ceramides Control Receptor-Dependent and Receptor-Independent Signal Transmission Pathways of a Lipid Neurotransmitter. Biomolecules 2018, 8, 31. [Google Scholar] [CrossRef]
- Metcalf, R.; Pandit, S.A. Mixing Properties of Sphingomyelin Ceramide Bilayers: A Simulation Study. J. Phys. Chem. B 2012, 116, 4500–4509. [Google Scholar] [CrossRef]
- Lönnfors, M.; Långvik, O.; Björkbom, A.; Slotte, J. Cholesteryl Phosphocholine—A Study on Its Interactions with Ceramides and Other Membrane Lipids. Langmuir 2013, 29, 2319–2329. [Google Scholar] [CrossRef]
- Bari, M.; Battista, N.; Fezza, F.; Finazzi-Agrò, A.; Maccarrone, M. Lipid Rafts Control Signaling of Type-1 Cannabinoid Receptors in Neuronal Cells: Implications for anandamide-induced apoptosis. J. Biol. Chem. 2005, 280, 12212–12220. [Google Scholar] [CrossRef]
- Di Scala, C.; Mazzarino, M.; Yahi, N.; Varini, K.; Garmy, N.; Fantini, J.; Chahinian, H. Anandamide-ceramide interactions in a membrane environment: Molecular dynamic simulations data. Data Brief 2017, 14, 163–167. [Google Scholar] [CrossRef]
- Gotti, C.; Zoli, M.; Clementi, F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef]
- Jones, S.; Yakel, J.L. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J. Physiol. 1997, 504, 603–610. [Google Scholar] [CrossRef]
- Colombo, S.F.; Mazzo, F.; Pistillo, F.; Gotti, C. Biogenesis, trafficking and up-regulation of nicotinic ACh receptors. Biochem. Pharmacol. 2013, 86, 1063–1073. [Google Scholar] [CrossRef]
- Wonnacott, S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997, 20, 92–98. [Google Scholar] [CrossRef]
- Jones, S.; Sudweeks, S.; Yakel, J.L. Nicotinic receptors in the brain: Correlating physiology with function. Trends Neurosci. 1999, 22, 555–561. [Google Scholar] [CrossRef]
- Cooper, E.; Couturier, S.; Ballivet, M. Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature 1991, 350, 235–238. [Google Scholar] [CrossRef]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human α4β2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef]
- Jones, A.K.; Sattelle, D.B. Diversity of Insect Nicotinic Acetylcholine Receptor Subunits. In Insect Nicotinic Acetylcholine Receptors; Thany, S.H., Ed.; Springer: New York, NY, USA, 2010; Volume 683, pp. 25–43. [Google Scholar] [CrossRef]
- Quick, M.W.; Lester, R.A. Desensitization of neuronal nicotinic receptors. J. Neurobiol. 2002, 53, 457–478. [Google Scholar] [CrossRef]
- Wooltorton, J.; Pidoplichko, V.I.; Broide, R.S.; Dani, J.A. Differential Desensitization and Distribution of Nicotinic Acetylcholine Receptor Subtypes in Midbrain Dopamine Areas. J. Neurosci. 2003, 23, 3176–3185. [Google Scholar] [CrossRef]
- Mansvelder, H.; McGehee, D.S. Cellular and synaptic mechanisms of nicotine addiction. J. Neurobiol. 2002, 53, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Giniatullin, R.; Nistri, A.; Yakel, J.L. Desensitization of nicotinic ACh receptors: Shaping cholinergic signaling. Trends Neurosci. 2005, 28, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Addy, N.A.; Mineur, Y.S.; Brunzell, D.H. It is not “either/or”: Activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog. Neurobiol. 2008, 84, 329–342. [Google Scholar] [CrossRef]
- Wang, J.; Lindstrom, J. Orthosteric and allosteric potentiation of heteromeric neuronal nicotinic acetylcholine receptors. Br. J. Pharmacol. 2018, 175, 1805–1821. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Bertrand, D. Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef]
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat. Rev. Drug Discov. 2009, 8, 733–750. [Google Scholar] [CrossRef]
- Ji, D.; Lape, R.; Dani, J.A. Timing and Location of Nicotinic Activity Enhances or Depresses Hippocampal Synaptic Plasticity. Neuron 2001, 31, 131–141. [Google Scholar] [CrossRef]
- McQuiston, A.R. Acetylcholine release and inhibitory interneuron activity in hippocampal CA1. Front. Synaptic Neurosci. 2014, 6, 20. [Google Scholar] [CrossRef]
- Oda, A.; Yamagata, K.; Nakagomi, S.; Uejima, H.; Wiriyasermkul, P.; Ohgaki, R.; Nagamori, S.; Kanai, Y.; Tanaka, H. Nicotine induces dendritic spine remodeling in cultured hippocampal neurons. J. Neurochem. 2014, 128, 246–255. [Google Scholar] [CrossRef]
- Tang, A.-H.; Karson, M.A.; Nagode, D.A.; McIntosh, J.M.; Uebele, V.N.; Renger, J.J.; Klugmann, M.; Milner, T.A.; Alger, B.E. Nerve Terminal Nicotinic Acetylcholine Receptors Initiate Quantal GABA Release from Perisomatic Interneurons by Activating Axonal T-Type (Cav3) Ca2+ Channels and Ca2+ Release from Stores. J. Neurosci. 2011, 31, 13546–13561. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.-J.; Lozada, A.F.; Gou, C.-Y.; Xu, J.; Chen, Y.; Berg, D.K. Nicotine recruits glutamate receptors to postsynaptic sites. Mol. Cell. Neurosci. 2015, 68, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Alkondon, M.; Pereira, E.F.; Albuquerque, E.X. α-Bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res. 1998, 810, 257–263. [Google Scholar] [CrossRef]
- Gu, Z.; Yakel, J.L. Timing-Dependent Septal Cholinergic Induction of Dynamic Hippocampal Synaptic Plasticity. Neuron 2011, 71, 155–165. [Google Scholar] [CrossRef]
- Pidoplichko, V.I.; Prager, E.M.; Aroniadou-Anderjaska, V.; Braga, M.F.M. α7-Containing nicotinic acetylcholine receptors on interneurons of the basolateral amygdala and their role in the regulation of the network excitability. J. Neurophysiol. 2013, 110, 2358–2369. [Google Scholar] [CrossRef]
- Udakis, M.; Wright, V.L.; Wonnacott, S.; Bailey, C.P. Integration of inhibitory and excitatory effects of α7 nicotinic acetylcholine receptor activation in the prelimbic cortex regulates network activity and plasticity. Neuropharmacology 2016, 105, 618–629. [Google Scholar] [CrossRef]
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef]
- Baier, C.J.; Gallegos, C.E.; Levi, V.; Barrantes, F.J. Cholesterol modulation of nicotinic acetylcholine receptor surface mobility. Eur. Biophys. J. 2009, 39, 213–227. [Google Scholar] [CrossRef]
- Borroni, V.; Barrantes, F.J. Cholesterol Modulates the Rate and Mechanism of Acetylcholine Receptor Internalization. J. Biol. Chem. 2011, 286, 17122–17132. [Google Scholar] [CrossRef]
- Barrantes, F. Structural basis for lipid modulation of nicotinic acetylcholine receptor function. Brain Res. Rev. 2004, 47, 71–95. [Google Scholar] [CrossRef]
- Baier, C.J.; Barrantes, F.J. Sphingolipids are necessary for nicotinic acetylcholine receptor export in the early secretory pathway. J. Neurochem. 2007, 101, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Brusés, J.L.; Chauvet, N.; Rutishauser, U. Membrane Lipid Rafts Are Necessary for the Maintenance of the α7 Nicotinic Acetylcholine Receptor in Somatic Spines of Ciliary Neurons. J. Neurosci. 2001, 21, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, C.; Pediconi, M.; Barrantes, F. Ceramides modulate cell-surface acetylcholine receptor levels. Biochim. Biophys. Acta (BBA)-Biomembr. 2008, 1778, 917–930. [Google Scholar] [CrossRef]
- Marchand, S.; Devillers-Thiéry, A.; Pons, S.; Changeux, J.-P.; Cartaud, J. Rapsyn Escorts the Nicotinic Acetylcholine Receptor Along the Exocytic Pathway via Association with Lipid Rafts. J. Neurosci. 2002, 22, 8891–8901. [Google Scholar] [CrossRef]
- Borroni, V.; Baier, C.J.; Lang, T.; Bonini, I.; White, M.M.; Garbus, I.; Barrantes, F.J. Cholesterol depletion activates rapid internalization of submicron-sized acetylcholine receptor domains at the cell membrane. Mol. Membr. Biol. 2007, 24, 1–15. [Google Scholar] [CrossRef]
- Santiago, J.; Guzmán, G.R.; Rojas, L.V.; Marti, R.; Asmar-Rovira, G.A.; Santana, L.F.; McNamee, M.; Lasalde-Dominicci, J.A. Probing the Effects of Membrane Cholesterol in the Torpedo californica Acetylcholine Receptor and the Novel Lipid-exposed Mutation αC418W in XenopusOocytes. J. Biol. Chem. 2001, 276, 46523–46532. [Google Scholar] [CrossRef]
- Pediconi, M.; Gallegos, C.; Santos, E.D.L.; Barrantes, F. Metabolic cholesterol depletion hinders cell-surface trafficking of the nicotinic acetylcholine receptor. Neuroscience 2004, 128, 239–249. [Google Scholar] [CrossRef]
- Kellner, R.; Baier, C.; Willig, K.; Hell, S.; Barrantes, F. Nanoscale organization of nicotinic acetylcholine receptors revealed by stimulated emission depletion microscopy. Neuroscience 2007, 144, 135–143. [Google Scholar] [CrossRef]
- Mosqueira, A.; Camino, P.A.; Barrantes, F.J. Cholesterol modulates acetylcholine receptor diffusion by tuning confinement sojourns and nanocluster stability. Sci. Rep. 2018, 8, 11974. [Google Scholar] [CrossRef]
- Mosqueira, A.; Camino, P.A.; Barrantes, F.J. Antibody-induced crosslinking and cholesterol-sensitive, anomalous diffusion of nicotinic acetylcholine receptors. J. Neurochem. 2020, 152, 663–674. [Google Scholar] [CrossRef]
- Báez-Pagán, C.A.; Del Hoyo-Rivera, N.; Quesada, O.; Otero-Cruz, J.D.; Lasalde-Dominicci, J.A. Heterogeneous Inhibition in Macroscopic Current Responses of Four Nicotinic Acetylcholine Receptor Subtypes by Cholesterol Enrichment. J. Membr. Biol. 2016, 249, 539–549. [Google Scholar] [CrossRef]
- Oshikawa, J.; Toya, Y.; Fujita, T.; Egawa, M.; Kawabe, J.; Umemura, S.; Ishikawa, Y. Nicotinic acetylcholine receptor α7regulates cAMP signal within lipid rafts. Am. J. Physiol. Cell Physiol. 2003, 285, C567–C574. [Google Scholar] [CrossRef]
- Zhu, D.; Xiong, W.C.; Mei, L. Lipid Rafts Serve as a Signaling Platform for Nicotinic Acetylcholine Receptor Clustering. J. Neurosci. 2006, 26, 4841–4851. [Google Scholar] [CrossRef]
- Borroni, V.; Kamerbeek, C.; Pediconi, M.F.; Barrantes, F.J. Lovastatin Differentially Regulates α7 and α4 Neuronal Nicotinic Acetylcholine Receptor Levels in Rat Hippocampal Neurons. Molecules 2020, 25, 4838. [Google Scholar] [CrossRef]
- Scherma, M.; Muntoni, A.L.; Melis, M.; Fattore, L.; Fadda, P.; Fratta, W.; Pistis, M. Interactions between the endocannabinoid and nicotinic cholinergic systems: Preclinical evidence and therapeutic perspectives. Psychopharmacology 2016, 233, 1765–1777. [Google Scholar] [CrossRef]
- Jackson, K.J.; Marks, M.J.; Vann, R.E.; Chen, X.; Gamage, T.F.; Warner, J.A.; Damaj, M.I. Role of α5 Nicotinic Acetylcholine Receptors in Pharmacological and Behavioral Effects of Nicotine in Mice. J. Pharmacol. Exp. Ther. 2010, 334, 137–146. [Google Scholar] [CrossRef]
- Howlett, A.; Bidaut-Russell, M.; Devane, W.A.; Melvin, L.S.; Johnson, M.; Herkenham, M. The cannabinoid receptor: Biochemical, anatomical and behavioral characterization. Trends Neurosci. 1990, 13, 420–423. [Google Scholar] [CrossRef]
- Lichtman, A.H.; Cook, S.A.; Martin, B.R. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: Evidence supporting periaqueductal gray involvement. J. Pharmacol. Exp. Ther. 1996, 276, 585–593. [Google Scholar]
- Sañudo-Peña, M.; Romero, J.; Seale, G.E.; Fernandez-Ruiz, J.; Walker, J. Activational role of cannabinoids on movement. Eur. J. Pharmacol. 2000, 391, 269–274. [Google Scholar] [CrossRef]
- Justinova, Z.; Goldberg, S.R.; Heishman, S.J.; Tanda, G. Self-administration of cannabinoids by experimental animals and human marijuana smokers. Pharmacol. Biochem. Behav. 2005, 81, 285–299. [Google Scholar] [CrossRef]
- Ahsan, H.M.; De La Peña, J.B.I.; Botanas, C.J.; Kim, H.J.; Yu, G.Y.; Cheong, J.H. Conditioned Place Preference and Self-Administration Induced by Nicotine in Adolescent and Adult Rats. Biomol. Ther. 2014, 22, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Scherma, M.; Justinová, Z.; Zanettini, C.; Panlilio, L.V.; Mascia, P.; Fadda, P.; Fratta, W.; Makriyannis, A.; Vadivel, S.K.; Gamaleddin, I.; et al. The anandamide transport inhibitor AM404 reduces the rewarding effects of nicotine and nicotine-induced dopamine elevations in the nucleus accumbens shell in rats. Br. J. Pharmacol. 2011, 165, 2539–2548. [Google Scholar] [CrossRef]
- Balerio, G.N.; Aso, E.; Berrendero, F.; Murtra, P.; Maldonado, R. Delta9-tetrahydrocannabinol decreases somatic and motivational manifestations of nicotine withdrawal in mice. Eur. J. Neurosci. 2004, 20, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Balerio, G.N.; Aso, E.; Maldonado, R. Role of the cannabinoid system in the effects induced by nicotine on anxiety-like behaviour in mice. Psychopharmacology 2006, 184, 504–513. [Google Scholar] [CrossRef]
- Valjent, E.; Mitchell, J.M.; Besson, M.-J.; Caboche, J.; Maldonado, R. Behavioural and biochemical evidence for interactions between Δ9-tetrahydrocannabinol and nicotine. Br. J. Pharmacol. 2002, 135, 564–578. [Google Scholar] [CrossRef]
- Xi, Z.-X.; Spiller, K.; Gardner, E.L. Mechanism-based medication development for the treatment of nicotine dependence. Acta Pharmacol. Sin. 2009, 30, 723–739. [Google Scholar] [CrossRef]
- Cooper, S.Y.; Henderson, B.J. The Impact of Electronic Nicotine Delivery System (ENDS) Flavors on Nicotinic Acetylcholine Receptors and Nicotine Addiction-Related Behaviors. Molecules 2020, 25, 4223. [Google Scholar] [CrossRef]
- Engle, S.E.; Shih, P.-Y.; McIntosh, J.M.; Drenan, R.M. α4α6β2* Nicotinic Acetylcholine Receptor Activation on Ventral Tegmental Area Dopamine Neurons Is Sufficient to Stimulate a Depolarizing Conductance and Enhance Surface AMPA Receptor Function. Mol. Pharmacol. 2013, 84, 393–406. [Google Scholar] [CrossRef]
- Liu, L.; Zhao-Shea, R.; McIntosh, J.M.; Gardner, P.D.; Tapper, A.R. Nicotine Persistently Activates Ventral Tegmental Area Dopaminergic Neurons via Nicotinic Acetylcholine Receptors Containing α4 and α6 Subunits. Mol. Pharmacol. 2012, 81, 541–548. [Google Scholar] [CrossRef]
- Acquas, E.; Pisanu, A.; Marrocu, P.; Goldberg, S.R.; Di Chiara, G. Δ9-tetrahydrocannabinol enhances cortical and hippocampal acetylcholine release in vivo: A microdialysis study. Eur. J. Pharmacol. 2001, 419, 155–161. [Google Scholar] [CrossRef]
- Pisanu, A.; Acquas, E.; Fenu, S.; Di Chiara, G. Modulation of Δ9-THC-induced increase of cortical and hippocampal acetylcholine release by μ opioid and D1 dopamine receptors. Neuropharmacology 2006, 50, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Tzavara, E.T.; Wade, M.; Nomikos, G.G. Biphasic Effects of Cannabinoids on Acetylcholine Release in the Hippocampus: Site and Mechanism of Action. J. Neurosci. 2003, 23, 9374–9384. [Google Scholar] [CrossRef] [PubMed]
- Buczynski, M.W.; Polis, I.Y.; Parsons, L.H. The Volitional Nature of Nicotine Exposure Alters Anandamide and Oleoylethanolamide Levels in the Ventral Tegmental Area. Neuropsychopharmacology 2012, 38, 574–584. [Google Scholar] [CrossRef]
- Cippitelli, A.; Astarita, G.; Duranti, A.; Caprioli, G.; Ubaldi, M.; Stopponi, S.; Kallupi, M.; Sagratini, G.; de Fonseca, F.R.; Piomelli, D.; et al. Endocannabinoid Regulation of Acute and Protracted Nicotine Withdrawal: Effect of FAAH Inhibition. PLoS ONE 2011, 6, e28142. [Google Scholar] [CrossRef] [PubMed]
- Oz, M.; Jackson, S.N.; Woods, A.S.; Morales, M.; Zhang, L. Additive Effects of Endogenous Cannabinoid Anandamide and Ethanol on α7-Nicotinic Acetylcholine Receptor-Mediated Responses in Xenopus Oocytes. J. Pharmacol. Exp. Ther. 2005, 313, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.N.; Singhal, S.K.; Woods, A.S.; Morales, M.; Shippenberg, T.; Zhang, L.; Oz, M. Volatile anesthetics and endogenous cannabinoid anandamide have additive and independent inhibitory effects on α7-nicotinic acetylcholine receptor-mediated responses in Xenopus oocytes. Eur. J. Pharmacol. 2008, 582, 42–51. [Google Scholar] [CrossRef][Green Version]
- Oz, M.; Ravindran, A.; Diaz-Ruiz, O.; Zhang, L.; Morales, M. The Endogenous Cannabinoid Anandamide Inhibits α7Nicotinic Acetylcholine Receptor-Mediated Responses in Xenopus Oocytes. J. Pharmacol. Exp. Ther. 2003, 306, 1003–1010. [Google Scholar] [CrossRef]
- Oz, M.; Zhang, L.; Ravindran, A.; Morales, M.; Lupica, C.R. Differential Effects of Endogenous and Synthetic Cannabinoids on α7-Nicotinic Acetylcholine Receptor-Mediated Responses in Xenopus Oocytes. J. Pharmacol. Exp. Ther. 2004, 310, 1152–1160. [Google Scholar] [CrossRef]
- Spivak, C.E.; Lupica, C.R.; Oz, M. The Endocannabinoid Anandamide Inhibits the Function of α4β2 Nicotinic Acetylcholine Receptors. Mol. Pharmacol. 2007, 72, 1024–1032. [Google Scholar] [CrossRef]
- Lichtman, A.; Varvel, S.; Martin, B. Endocannabinoids in cognition and dependence. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 269–285. [Google Scholar] [CrossRef]
- Fattore, L.; Spano, M.S.; Deiana, S.; Melis, V.; Cossu, G.; Fadda, P.; Fratta, W. An endocannabinoid mechanism in relapse to drug seeking: A review of animal studies and clinical perspectives. Brain Res. Rev. 2007, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Ji, D.; Zhou, F.-M. Synaptic Plasticity and Nicotine Addiction. Neuron 2001, 31, 349–352. [Google Scholar] [CrossRef]
- Solinas, M.; Scherma, M.; Fattore, L.; Stroik, J.; Wertheim, C.; Tanda, G.; Fratta, W.; Goldberg, S.R. Nicotinic 7 Receptors as a New Target for Treatment of Cannabis Abuse. J. Neurosci. 2007, 27, 5615–5620. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Tirado, C.S.; Colon-Saez, J.O.; Lasalde-Dominicci, J. Modulation of Nicotinic Receptors by Cannabinoids. FASEB J. 2018, 32, 533–578. [Google Scholar] [CrossRef]
- Cohen, C.; Perrault, G.; Voltz, C.; Steinberg, R.; Soubrié, P. SR141716, a central cannabinoid (CB1) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav. Pharmacol. 2002, 13, 451–463. [Google Scholar] [CrossRef]
- Forget, B.; Coen, K.M.; Le Foll, B. Inhibition of fatty acid amide hydrolase reduces reinstatement of nicotine seeking but not break point for nicotine self-administration—Comparison with CB1 receptor blockade. Psychopharmacology 2009, 205, 613–624. [Google Scholar] [CrossRef]
- Castañé, A.; Valjent, E.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Lack of CB1 cannabinoid receptors modifies nicotine behavioural responses, but not nicotine abstinence. Neuropharmacology 2002, 43, 857–867. [Google Scholar] [CrossRef]
- Merritt, L.L.; Martin, B.R.; Walters, C.; Lichtman, A.H.; Damaj, M.I. The Endogenous Cannabinoid System Modulates Nicotine Reward and Dependence. J. Pharmacol. Exp. Ther. 2008, 326, 483–492. [Google Scholar] [CrossRef]
- Reisiger, A.-R.; Kaufling, J.; Manzoni, O.; Cador, M.; Georges, F.; Caillé, S. Nicotine Self-Administration Induces CB1-Dependent LTP in the Bed Nucleus of the Stria Terminalis. J. Neurosci. 2014, 34, 4285–4292. [Google Scholar] [CrossRef]
- Kodas, E.; Cohen, C.; Louis, C.; Griebel, G. Cortico-limbic circuitry for conditioned nicotine-seeking behavior in rats involves endocannabinoid signaling. Psychopharmacology 2007, 194, 161–171. [Google Scholar] [CrossRef]
- Gamaleddin, I.; Wertheim, C.; Zhu, A.Z.; Coen, K.M.; Vemuri, K.; Makryannis, A.; Goldberg, S.R.; Le Foll, B. Cannabinoid receptor stimulation increases motivation for nicotine and nicotine seeking. Addict. Biol. 2011, 17, 47–61. [Google Scholar] [CrossRef]
- Gamaleddin, I.; Zvonok, A.; Makriyannis, A.; Goldberg, S.R.; Le Foll, B. Effects of a Selective Cannabinoid CB2 Agonist and Antagonist on Intravenous Nicotine Self Administration and Reinstatement of Nicotine Seeking. PLoS ONE 2012, 7, e29900. [Google Scholar] [CrossRef]
- Ignatowska-Jankowska, B.M.; Muldoon, P.P.; Lichtman, A.H.; Damaj, M.I. The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology 2013, 229, 591–601. [Google Scholar] [CrossRef]
- Navarrete, F.; Rodriguez-Arias, M.; Martin-García, E.; Navarro, D.; García-Gutiérrez, M.S.; Aguilar, M.A.; Aracil-Fernández, A.; Berbel, P.; Miñarro, J.; Maldonado, R.; et al. Role of CB2 Cannabinoid Receptors in the Rewarding, Reinforcing, and Physical Effects of Nicotine. Neuropsychopharmacology 2013, 38, 2515–2524. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).