
Applications of Molecular Dynamics Simulation in Protein Study



Abstract
:1. Introduction
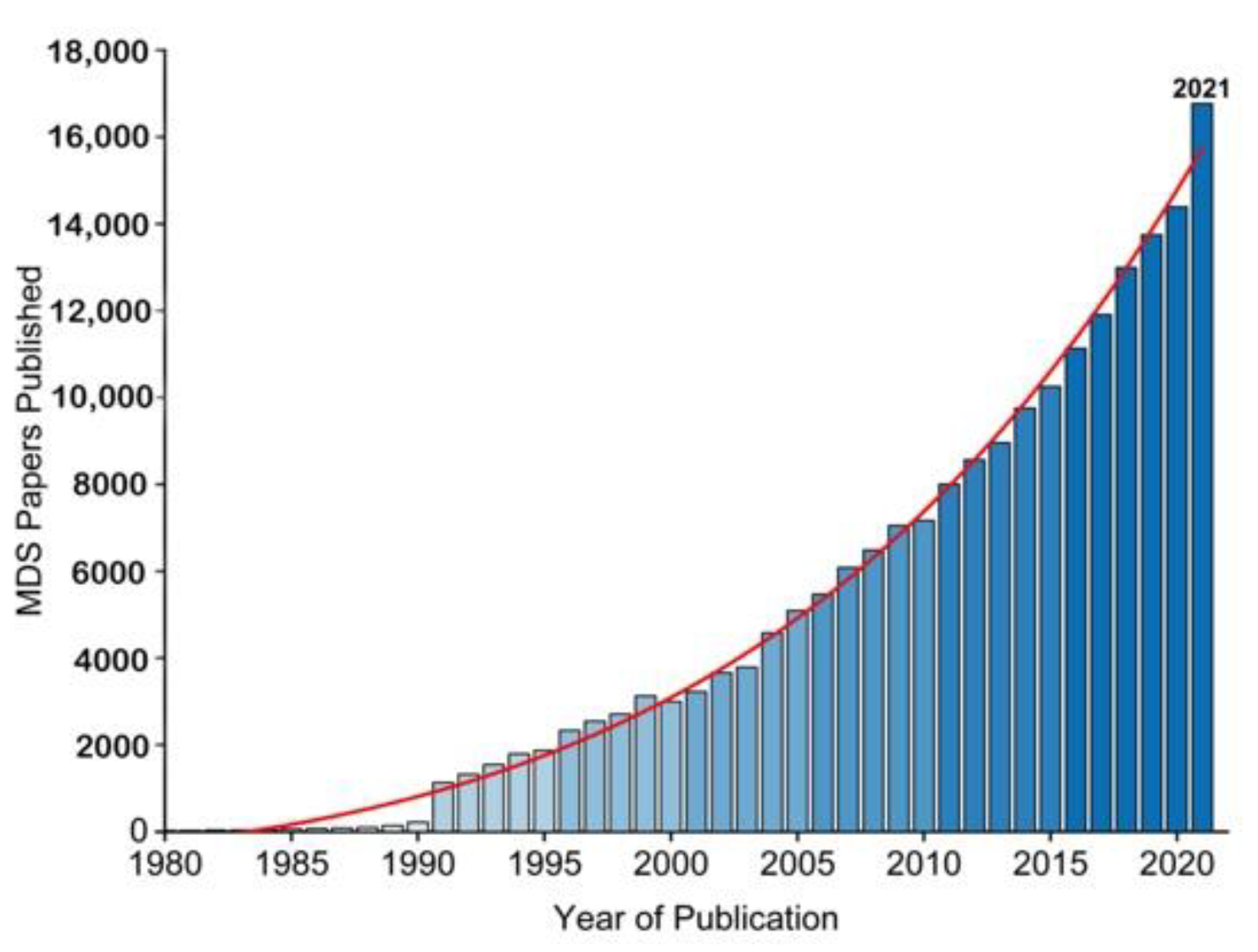
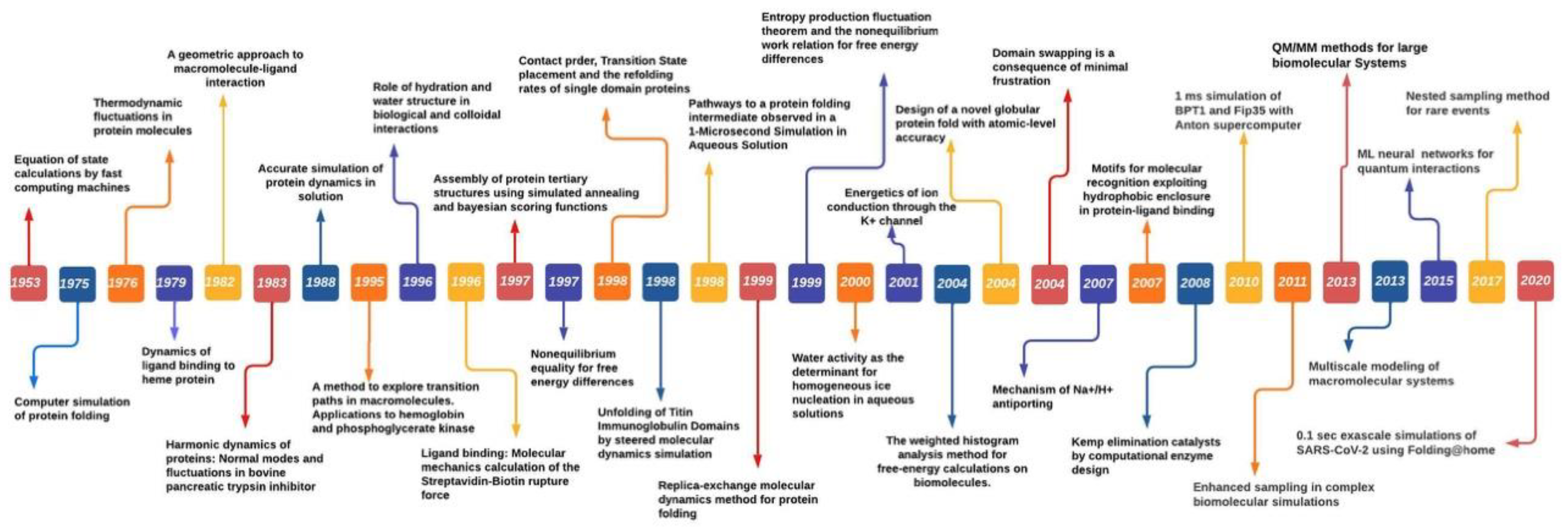
2. A Brief History of Molecular Simulations
3. Basic Concept of Force Field
4. Molecular Simulations in Protein Study
4.1. Applications of Molecular Simulations in Membrane Proteins
4.2. Simulations of Integral Membrane Protein (GPCRs)
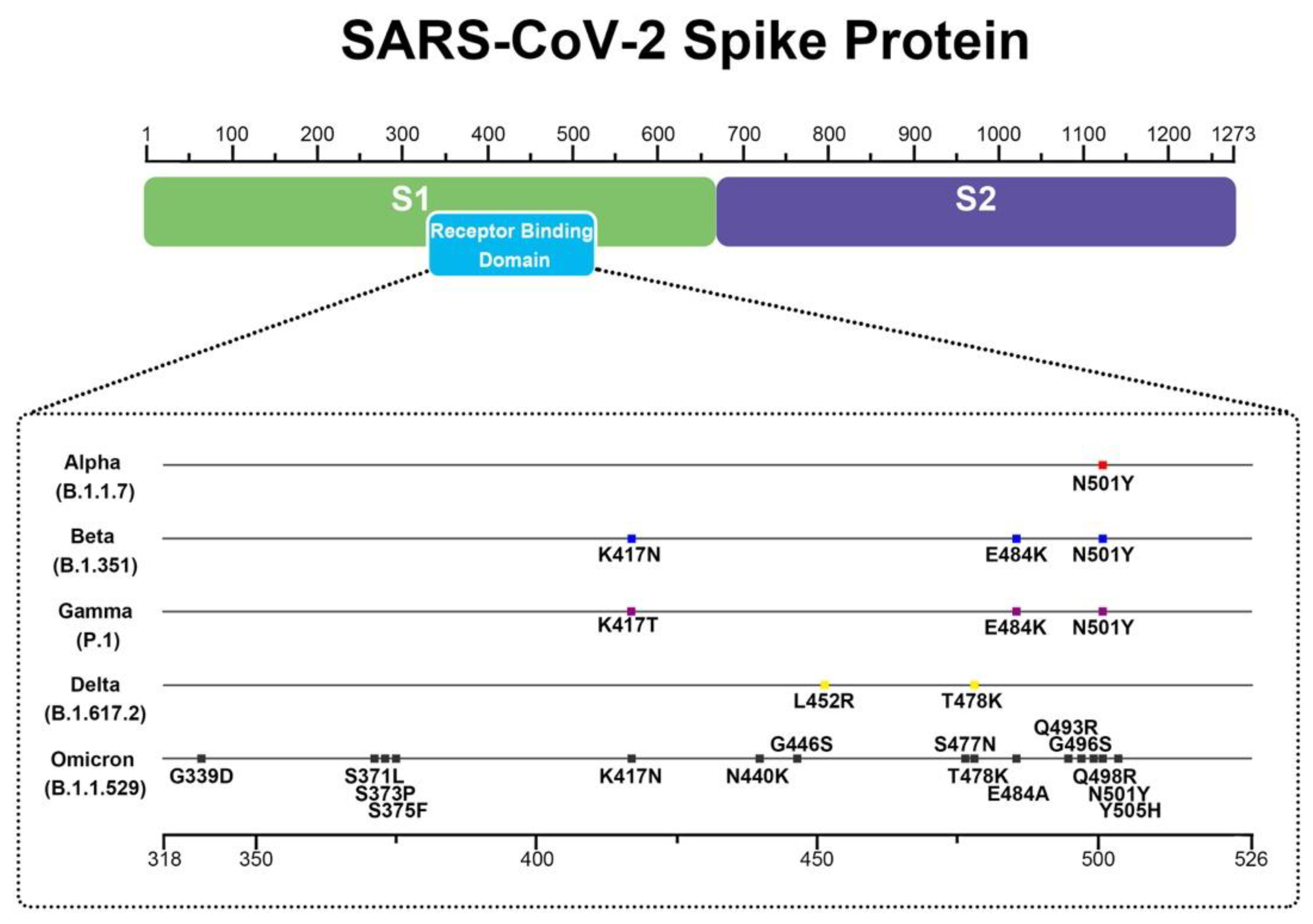
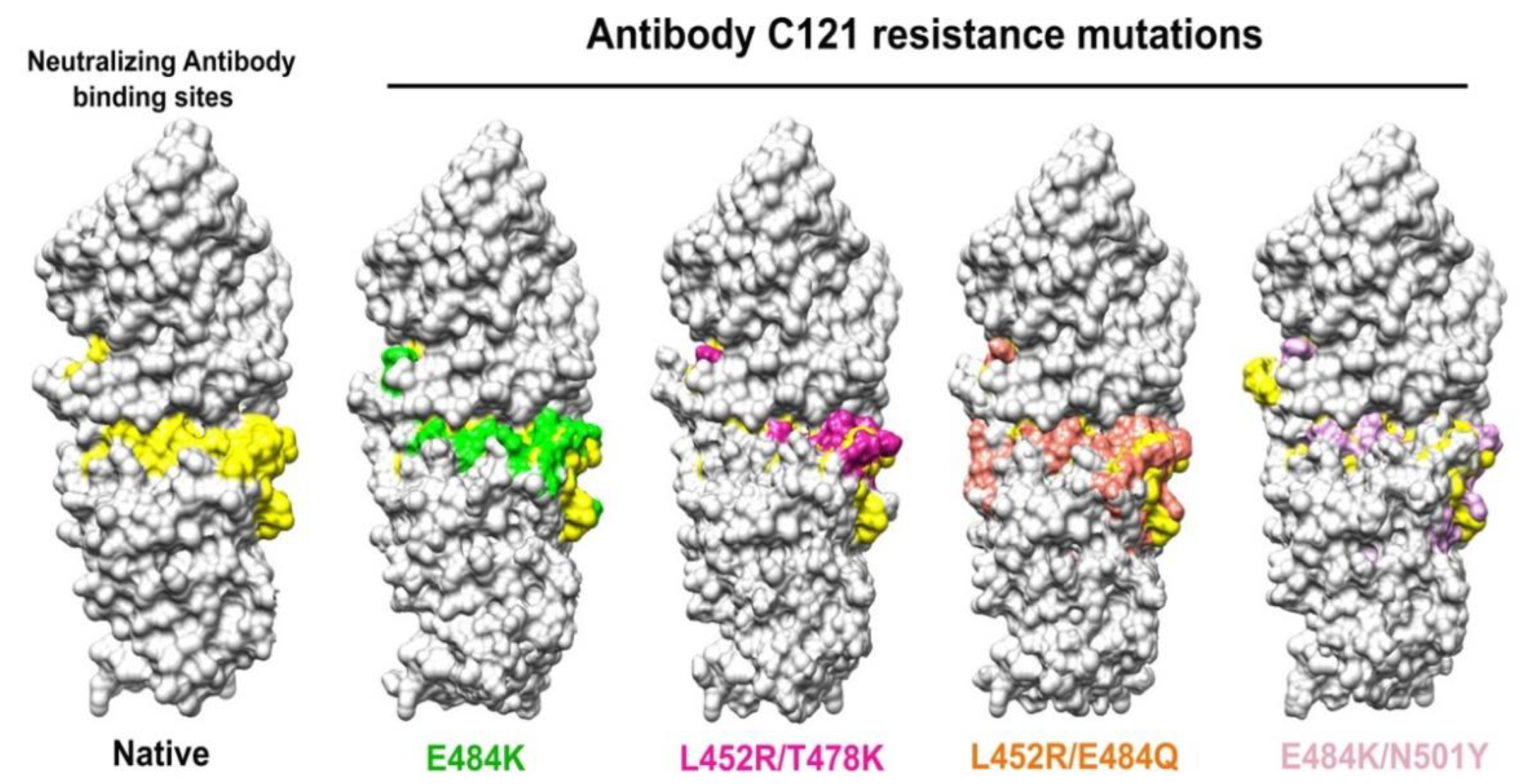
4.3. Simulations of Interaction between SARS-CoV-2 Spike and Membrane ACE2 Receptor
5. Challenges and Future Opportunities
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Minor, D.L., Jr. The neurobiologist’s guide to structural biology: A primer on why macromolecular structure matters and how to evaluate structural data. Neuron 2007, 54, 511–533. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Green, E.M.; Gouaux, E. X-ray structures and mechanism of the human serotonin transporter. Nature 2016, 532, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 6. [Google Scholar] [CrossRef]
- Alder, B.J.; Wainwright, T.E. Phase Transition for a Hard Sphere System. J. Chem. Phys. 1957, 27, 1208–1209. [Google Scholar] [CrossRef]
- Metropolis, N.; Ulam, S. The Monte Carlo Method. J. Am. Stat. Assoc. 1949, 44, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Alder, B.J.; Wainwright, T.E. Studies in Molecular Dynamics. I. General Method. J. Chem. Phys. 1959, 31, 459–466. [Google Scholar] [CrossRef]
- Rahman, A. Correlations in the Motion of Atoms in Liquid Argon. Phys. Rev. 1964, 136, A405–A411. [Google Scholar] [CrossRef]
- Rahman, A.; Stillinger, F.H. Molecular Dynamics Study of Liquid Water. J. Chem. Phys. 1971, 55, 3336–3359. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulos, A.Z. Direct determination of phase coexistence properties of fluids by Monte Carlo simulation in a new ensemble. Mol. Phys. 1987, 61, 813–826. [Google Scholar] [CrossRef]
- Carter, E.A.; Ciccotti, G.; Hynes, J.T.; Kapral, R. Constrained reaction coordinate dynamics for the simulation of rare events. Chem. Phys. Lett. 1989, 156, 472–477. [Google Scholar] [CrossRef]
- Voter, A.F. A method for accelerating the molecular dynamics simulation of infrequent events. J. Chem. Phys. 1997, 106, 4665–4677. [Google Scholar] [CrossRef]
- Mills, G.; Jacobsen, W. Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998. [Google Scholar]
- Weinan, E.; Ren, W.; Vanden-Eijnden, E. String method for the study of rare events. Phys. Rev. B 2002, 66, 052301. [Google Scholar]
- Ben-Nun, M.; Quenneville, J.; Martínez, T.J. Ab Initio Multiple Spawning: Photochemistry from First Principles Quantum Molecular Dynamics. J. Phys. Chem. A 2000, 104, 5161–5175. [Google Scholar] [CrossRef]
- Jones, C.M.; List, N.H.; Martínez, T.J. Steric and Electronic Origins of Fluorescence in GFP and GFP-like Proteins. J. Am. Chem. Soc. 2022, 144, 12732–12746. [Google Scholar] [CrossRef] [PubMed]
- Wasif Baig, M.; Pederzoli, M.; Kývala, M.; Cwiklik, L.; Pittner, J. Theoretical Investigation of the Effect of Alkylation and Bromination on Intersystem Crossing in BODIPY-Based Photosensitizers. J. Phys. Chem. B 2021, 125, 11617–11627. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Kaxiras, E. Real-time, local basis-set implementation of time-dependent density functional theory for excited state dynamics simulations. J. Chem. Phys. 2008, 129, 054110. [Google Scholar] [CrossRef]
- Fitch, B.G.; Rayshubskiy, A.; Eleftheriou, M.; Ward, T.J.C.; Giampapa, M.; Zhestkov, Y.; Pitman, M.C.; Suits, F.; Grossfield, A.; Pitera, J.; et al. Blue Matter: Strong Scaling of Molecular Dynamics on Blue Gene/L. Comp. Sci. 2006, 3992, 846–854. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Shaw, D.E.; Dror, R.O.; Salmon, J.K.; Grossman, J.P.; Mackenzie, K.M.; Bank, J.A.; Young, C.; Deneroff, M.M.; Batson, B.; Bowers, K.J.; et al. Millisecond-scale molecular dynamics simulations on Anton. In Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis, Portland, OR, USA, 14–20 November 2009; p. 65. [Google Scholar]
- Levitt, M.; Sharon, R. Accurate simulation of protein dynamics in solution. Proc. Natl. Acad. Sci. USA 1988, 85, 7557–7561. [Google Scholar] [CrossRef]
- Mackerell, A.D., Jr. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L., 3rd. Modern protein force fields behave comparably in molecular dynamics simulations. J. Comput. Chem. 2002, 23, 1045–1057. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, W.; Yong, C.W.; Rodger, P.M. DL_POLY: Application to molecular simulation. Mol. Simul. 2002, 28, 385–471. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Moore, P.B.; Lopez, C.F.; Klein, M.L. Dynamical properties of a hydrated lipid bilayer from a multinanosecond molecular dynamics simulation. Biophys. J. 2001, 81, 2484–2494. [Google Scholar] [CrossRef]
- Saiz, L.; Klein, M.L. Computer simulation studies of model biological membranes. Acc. Chem. Res. 2002, 35, 482–489. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef]
- Shi, Q.; Izvekov, S.; Voth, G.A. Mixed atomistic and coarse-grained molecular dynamics: Simulation of a membrane-bound ion channel. J. Phys. Chem. B 2006, 110, 15045–15048. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.-K.; Han, W.; Wu, Y.-D. Parameterization of PACE Force Field for Membrane Environment and Simulation of Helical Peptides and Helix–Helix Association. J. Chem. Theory Comput. 2012, 8, 300–313. [Google Scholar] [CrossRef]
- Kar, P.; Gopal, S.M.; Cheng, Y.-M.; Panahi, A.; Feig, M. Transferring the PRIMO Coarse-Grained Force Field to the Membrane Environment: Simulations of Membrane Proteins and Helix–Helix Association. J. Chem. Theory Comput. 2014, 10, 3459–3472. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Mark, A.E.; Van Gunsteren, W.F. Parametrization of aliphatic CHn united atoms of GROMOS96 force field. J. Comput. Chem. 1998, 19, 535–547. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Hart, K.; Foloppe, N.; Baker, C.M.; Denning, E.J.; Nilsson, L.; MacKerell, A.D. Optimization of the CHARMM Additive Force Field for DNA: Improved Treatment of the BI/BII Conformational Equilibrium. J. Chem. Theory Comput. 2012, 8, 348–362. [Google Scholar] [CrossRef]
- Denning, E.J.; Priyakumar, U.D.; Nilsson, L.; Mackerell, A.D., Jr. Impact of 2′-hydroxyl sampling on the conformational properties of RNA: Update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 2011, 32, 1929–1943. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Walker, R.C.; Gould, I.R. Lipid21: Complex Lipid Membrane Simulations with AMBER. J. Chem. Theory Comput. 2022, 18, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef]
- Zgarbová, M.; Otyepka, M.; Šponer, J.; Mládek, A.; Banáš, P.; Cheatham, T.E.; Jurečka, P. Refinement of the Cornell et al. Nucleic Acids Force Field Based on Reference Quantum Chemical Calculations of Glycosidic Torsion Profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Komáromi, I.; Owen, M.C.; Murphy, R.F.; Lovas, S. Development of glycyl radical parameters for the OPLS-AA/L force field. J. Comput. Chem. 2008, 29, 1999–2009. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef]
- Reif, M.M.; Hünenberger, P.H.; Oostenbrink, C. New Interaction Parameters for Charged Amino Acid Side Chains in the GROMOS Force Field. J. Chem. Theory Comput. 2012, 8, 3705–3723. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, J.J.; Ingólfsson, H.I.; Akhshi, P.; Tieleman, D.P.; Marrink, S.J. Martini Coarse-Grained Force Field: Extension to DNA. J. Chem. Theory Comput. 2015, 11, 3932–3945. [Google Scholar] [CrossRef] [PubMed]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Doye, J.P.; Louis, A.A. A nucleotide-level coarse-grained model of RNA. J. Chem. Phys. 2014, 140, 235102. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.; Warshel, A. Computer simulation of protein folding. Nature 1975, 253, 694–698. [Google Scholar] [CrossRef]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Periole, X.; Cavalli, M.; Marrink, S.-J.; Ceruso, M.A. Combining an Elastic Network With a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. [Google Scholar] [CrossRef]
- Arnarez, C.; Uusitalo, J.J.; Masman, M.F.; Ingólfsson, H.I.; de Jong, D.H.; Melo, M.N.; Periole, X.; de Vries, A.H.; Marrink, S.J. Dry Martini, a Coarse-Grained Force Field for Lipid Membrane Simulations with Implicit Solvent. J. Chem. Theory Comput. 2015, 11, 260–275. [Google Scholar] [CrossRef]
- Bashford, D.; Case, D.A. Generalized born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 2000, 51, 129–152. [Google Scholar] [CrossRef]
- Im, W.; Chen, J.; Brooks, C.L. Peptide and Protein Folding and Conformational Equilibria: Theoretical Treatment of Electrostatics and Hydrogen Bonding with Implicit Solvent Models. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 2005; Volume 72, pp. 173–198. [Google Scholar]
- Zhang, J.; Zhang, H.; Wu, T.; Wang, Q.; van der Spoel, D. Comparison of Implicit and Explicit Solvent Models for the Calculation of Solvation Free Energy in Organic Solvents. J. Chem. Theory Comput. 2017, 13, 1034–1043. [Google Scholar] [CrossRef]
- Zhou, R. Free energy landscape of protein folding in water: Explicit vs. implicit solvent. Proteins Struct. Funct. Bioinform. 2003, 53, 148–161. [Google Scholar] [CrossRef]
- Chouard, T. Structural biology: Breaking the protein rules. Nature 2011, 471, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)--round x. Proteins 2014, 82 (Suppl. S2), 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liwo, A.; Baranowski, M.; Czaplewski, C.; Gołaś, E.; He, Y.; Jagieła, D.; Krupa, P.; Maciejczyk, M.; Makowski, M.; Mozolewska, M.A.; et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Modeling 2014, 20, 2306. [Google Scholar] [CrossRef]
- Davtyan, A.; Schafer, N.P.; Zheng, W.; Clementi, C.; Wolynes, P.G.; Papoian, G.A. AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinformatically Based Local Structure Biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [Google Scholar] [CrossRef]
- Sterpone, F.; Melchionna, S.; Tuffery, P.; Pasquali, S.; Mousseau, N.; Cragnolini, T.; Chebaro, Y.; St-Pierre, J.-F.; Kalimeri, M.; Barducci, A.; et al. The OPEP protein model: From single molecules, amyloid formation, crowding and hydrodynamics to DNA/RNA systems. Chem. Soc. Rev. 2014, 43, 4871–4893. [Google Scholar] [CrossRef] [PubMed]
- Dawid, A.E.; Gront, D.; Kolinski, A. SURPASS Low-Resolution Coarse-Grained Protein Modeling. J. Chem. Theory Comput. 2017, 13, 5766–5779. [Google Scholar] [CrossRef]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar] [CrossRef]
- Kar, P.; Gopal, S.M.; Cheng, Y.-M.; Predeus, A.; Feig, M. PRIMO: A Transferable Coarse-Grained Force Field for Proteins. J. Chem. Theory Comput. 2013, 9, 3769–3788. [Google Scholar] [CrossRef]
- Basdevant, N.; Borgis, D.; Ha-Duong, T. Modeling Protein–Protein Recognition in Solution Using the Coarse-Grained Force Field SCORPION. J. Chem. Theory Comput. 2013, 9, 803–813. [Google Scholar] [CrossRef]
- Rohl, C.A.; Strauss, C.E.; Misura, K.M.; Baker, D. Protein structure prediction using Rosetta. Methods Enzym. 2004, 383, 66–93. [Google Scholar] [CrossRef]
- Cao, S.; Chen, S.J. Predicting structures and stabilities for H-type pseudoknots with interhelix loops. RNA 2009, 15, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setny, P.; Zacharias, M. Elastic Network Models of Nucleic Acids Flexibility. J. Chem. Theory Comput. 2013, 9, 5460–5470. [Google Scholar] [CrossRef] [PubMed]
- Boniecki, M.J.; Lach, G.; Dawson, W.K.; Tomala, K.; Lukasz, P.; Soltysinski, T.; Rother, K.M.; Bujnicki, J.M. SimRNA: A coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 2016, 44, e63. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Sharma, S.; Chalasani, P.; Demidov, V.V.; Broude, N.E.; Dokholyan, N.V. Ab initio RNA folding by discrete molecular dynamics: From structure prediction to folding mechanisms. RNA 2008, 14, 1164–1173. [Google Scholar] [CrossRef]
- Jonikas, M.A.; Radmer, R.J.; Laederach, A.; Das, R.; Pearlman, S.; Herschlag, D.; Altman, R.B. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA 2009, 15, 189–199. [Google Scholar] [CrossRef]
- Orsi, M.; Essex, J.W. The ELBA Force Field for Coarse-Grain Modeling of Lipid Membranes. PLoS ONE 2011, 6, e28637. [Google Scholar] [CrossRef]
- Shih, A.Y.; Arkhipov, A.; Freddolino, P.L.; Schulten, K. Coarse grained protein-lipid model with application to lipoprotein particles. J. Phys. Chem. B 2006, 110, 3674–3684. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H.J.C. Computer Simulation of Molecular Dynamics: Methodology, Applications, and Perspectives in Chemistry. Angew. Chem. Int. Ed. Engl. 1990, 29, 992–1023. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Arcon, J.P.; Defelipe, L.A.; Modenutti, C.P.; López, E.D.; Alvarez-Garcia, D.; Barril, X.; Turjanski, A.G.; Martí, M.A. Molecular Dynamics in Mixed Solvents Reveals Protein-Ligand Interactions, Improves Docking, and Allows Accurate Binding Free Energy Predictions. J. Chem. Inf. Modeling 2017, 57, 846–863. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.C.; Miners, J.O. Molecular dynamics simulations: From structure function relationships to drug discovery. Silico Pharmacol. 2014, 2, 4. [Google Scholar] [CrossRef]
- Fernandez-Leiro, R.; Scheres, S.H. Unravelling biological macromolecules with cryo-electron microscopy. Nature 2016, 537, 339–346. [Google Scholar] [CrossRef]
- Xu, D.; Li, D. Molecular Dynamics Simulation Method. In Encyclopedia of Microfluidics and Nanofluidics; Li, D., Ed.; Springer: Boston, MA, USA, 2008; pp. 1391–1398. [Google Scholar]
- Mahdavi, A.; Jahandideh, S. Application of density similarities to predict membrane protein types based on pseudo-amino acid composition. J. Theor. Biol. 2011, 276, 132–137. [Google Scholar] [CrossRef]
- Nagle, J.F.; Tristram-Nagle, S. Structure of lipid bilayers. Biochim. Et Biophys. Acta 2000, 1469, 159–195. [Google Scholar] [CrossRef]
- Cuello, L.G.; Jogini, V.; Cortes, D.M.; Pan, A.C.; Gagnon, D.G.; Dalmas, O.; Cordero-Morales, J.F.; Chakrapani, S.; Roux, B.; Perozo, E. Structural basis for the coupling between activation and inactivation gates in K(+) channels. Nature 2010, 466, 272–275. [Google Scholar] [CrossRef]
- Shenoy, S.K.; Lefkowitz, R.J. β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Xu, Q.; Dunbrack, R.L., Jr. The protein common interface database (ProtCID)—A comprehensive database of interactions of homologous proteins in multiple crystal forms. Nucleic Acids Res. 2011, 39, D761–D770. [Google Scholar] [CrossRef] [PubMed]
- White, S.H. The progress of membrane protein structure determination. Protein Sci. 2004, 13, 1948–1949. [Google Scholar] [CrossRef]
- Kozma, D.; Simon, I.; Tusnády, G.E. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2013, 41, D524–D529. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.E.; Hallock, M.J.; Phillips, J.C.; Peterson, J.R.; Luthey-Schulten, Z.; Schulten, K. Evaluation of Emerging Energy-Efficient Heterogeneous Computing Platforms for Biomolecular and Cellular Simulation Workloads. IEEE Int. Parallel Distrib. Processing Symp. Workshops 2016, 2016, 89–100. [Google Scholar] [CrossRef]
- Stone, J.E.; Sener, M.; Vandivort, K.L.; Barragan, A.; Singharoy, A.; Teo, I.; Ribeiro, J.V.; Isralewitz, B.; Liu, B.; Goh, B.C.; et al. Atomic detail visualization of photosynthetic membranes with GPU-accelerated ray tracing. Parallel Comput. 2016, 55, 17–27. [Google Scholar] [CrossRef]
- Marrink, S.-J.; Berendsen, H.J.C. Simulation of water transport through a lipid membrane. J. Phys. Chem. 1994, 98, 4155–4168. [Google Scholar] [CrossRef]
- Feller, S.E.; Venable, R.M.; Pastor, R.W. Computer Simulation of a DPPC Phospholipid Bilayer: Structural Changes as a Function of Molecular Surface Area. Langmuir 1997, 13, 6555–6561. [Google Scholar] [CrossRef]
- Hansmann, U.H.E.; Okamoto, Y. Generalized-ensemble Monte Carlo method for systems with rough energy landscape. Phys. Rev. E 1997, 56, 2228–2233. [Google Scholar] [CrossRef]
- Lou, H.; Cukier, R.I. Molecular dynamics of apo-adenylate kinase: A distance replica exchange method for the free energy of conformational fluctuations. J. Phys. Chem. B 2006, 110, 24121–24137. [Google Scholar] [CrossRef]
- Im, W.; Brooks, C.L., 3rd. De novo folding of membrane proteins: An exploration of the structure and NMR properties of the fd coat protein. J. Mol. Biol. 2004, 337, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Im, W.; Brooks, C.L. Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc. Natl. Acad. Sci. 2005, 102, 6771–6776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nymeyer, H.; Woolf, T.B.; Garcia, A.E. Folding is not required for bilayer insertion: Replica exchange simulations of an alpha-helical peptide with an explicit lipid bilayer. Proteins 2005, 59, 783–790. [Google Scholar] [CrossRef]
- Mori, T.; Miyashita, N.; Im, W.; Feig, M.; Sugita, Y. Molecular dynamics simulations of biological membranes and membrane proteins using enhanced conformational sampling algorithms. Biochim. Et Biophys. Acta (BBA) Biomembr. 2016, 1858, 1635–1651. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174 (Suppl. S1), S17–S129. [Google Scholar] [CrossRef]
- Lundstrom, K. Latest development in drug discovery on G protein-coupled receptors. Curr. Protein Pept. Sci. 2006, 7, 465–470. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Heilker, R.; Wolff, M.; Tautermann, C.S.; Bieler, M. G-protein-coupled receptor-focused drug discovery using a target class platform approach. Drug Discov. Today 2009, 14, 231–240. [Google Scholar] [CrossRef]
- Mizuno, H.; Kihara, Y. Druggable Lipid GPCRs: Past, Present, and Prospects. In Druggable Lipid Signaling Pathways; Kihara, Y., Ed.; Springer International Publishing: Cham, Germany, 2020; pp. 223–258. [Google Scholar]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Marino, K.A.; Filizola, M. Investigating Small-Molecule Ligand Binding to G Protein-Coupled Receptors with Biased or Unbiased Molecular Dynamics Simulations. Methods Mol. Biol. 2018, 1705, 351–364. [Google Scholar] [CrossRef]
- Miao, Y.; McCammon, J.A. G-protein coupled receptors: Advances in simulation and drug discovery. Curr. Opin. Struct. Biol. 2016, 41, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.; Menon, S.; Sakmar, T.P. Structural basis for ligand binding and specificity in adrenergic receptors: Implications for GPCR-targeted drug discovery. Biochemistry 2008, 47, 11013–11023. [Google Scholar] [CrossRef] [PubMed]
- Cang, X.; Du, Y.; Mao, Y.; Wang, Y.; Yang, H.; Jiang, H. Mapping the functional binding sites of cholesterol in β2-adrenergic receptor by long-time molecular dynamics simulations. J. Phys. Chem. B 2013, 117, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef]
- Shaw, D.E.; Deneroff, M.M.; Dror, R.O.; Kuskin, J.S.; Larson, R.H.; Salmon, J.K.; Young, C.; Batson, B.; Bowers, K.J.; Chao, J.C.; et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM 2008, 51, 91–97. [Google Scholar] [CrossRef]
- Schneider, S.; Provasi, D.; Filizola, M. How Oliceridine (TRV-130) Binds and Stabilizes a μ-Opioid Receptor Conformational State That Selectively Triggers G Protein Signaling Pathways. Biochemistry 2016, 55, 6456–6466. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- Jones, A.J.Y.; Gabriel, F.; Tandale, A.; Nietlispach, D. Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules 2020, 25, 4729. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Stanley, N.; Pardo, L.; Fabritiis, G.D. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016, 6, 22639. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Modeling 2014, 54, 372–376. [Google Scholar] [CrossRef]
- Dickson, C.J.; Hornak, V.; Velez-Vega, C.; McKay, D.J.; Reilly, J.; Sandham, D.A.; Shaw, D.; Fairhurst, R.A.; Charlton, S.J.; Sykes, D.A.; et al. Uncoupling the Structure-Activity Relationships of β2 Adrenergic Receptor Ligands from Membrane Binding. J. Med. Chem. 2016, 59, 5780–5789. [Google Scholar] [CrossRef]
- Hedger, G.; Sansom, M.S.P. Lipid interaction sites on channels, transporters and receptors: Recent insights from molecular dynamics simulations. Biochim. Et Biophys. Acta 2016, 1858, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lyman, E. Predictions for cholesterol interaction sites on the A2A adenosine receptor. J. Am. Chem. Soc. 2012, 134, 16512–16515. [Google Scholar] [CrossRef] [PubMed]
- Neale, C.; Herce, H.D.; Pomès, R.; García, A.E. Can Specific Protein-Lipid Interactions Stabilize an Active State of the Beta 2 Adrenergic Receptor? Biophys. J. 2015, 109, 1652–1662. [Google Scholar] [CrossRef]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Masureel, M.; Van Antwerpen, P.; Kobilka, B.K.; Govaerts, C. Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol. 2016, 12, 35–39. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2020, 49, D335–D343. [Google Scholar] [CrossRef]
- GPCRs: G Protein Coupled Receptors Database. Available online: https://gproteindb.org (accessed on 13 August 2022).
- Da Costa, C.H.S.; de Freitas, C.A.B.; Alves, C.N.; Lameira, J. Assessment of mutations on RBD in the Spike protein of SARS-CoV-2 Alpha, Delta and Omicron variants. Sci. Rep. 2022, 12, 8540. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Rath, S.L.; Tripathi, T. Accelerating COVID-19 Research Using Molecular Dynamics Simulation. J. Phys. Chem. B 2021, 125, 9078–9091. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Tam, B.; Wang, S.M. RBD Double Mutations of SARS-CoV-2 Strains Increase Transmissibility through Enhanced Interaction between RBD and ACE2 Receptor. Viruses 2021, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Mulholland, A.J. Biomolecular Simulations in the Time of COVID-19, and After. Comput. Sci. Eng. 2020, 22, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Komatsu, T.S.; Koyama, Y.; Okimoto, N.; Morimoto, G.; Ohno, Y.; Taiji, M. COVID-19 related trajectory data of 10 microseconds all atom molecular dynamics simulation of SARS-CoV-2 dimeric main protease. Mendeley Data 2020. [Google Scholar] [CrossRef]
- Acharya, A.; Agarwal, R.; Baker, M.B.; Baudry, J.; Bhowmik, D.; Boehm, S.; Byler, K.G.; Chen, S.Y.; Coates, L.; Cooper, C.J.; et al. Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19. J. Chem. Inf. Modeling 2020, 60, 5832–5852. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat. Chem. 2021, 13, 651–659. [Google Scholar] [CrossRef]
- Li, B.; Deng, A.; Li, K.; Hu, Y.; Li, Z.; Shi, Y.; Xiong, Q.; Liu, Z.; Guo, Q.; Zou, L.; et al. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. Nat. Commun. 2022, 13, 460. [Google Scholar] [CrossRef]
- Silva, S.; Pena, L. Collapse of the public health system and the emergence of new variants during the second wave of the COVID-19 pandemic in Brazil. One Health 2021, 13, 100287. [Google Scholar] [CrossRef]
- Ho, D.; Wang, P.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.; et al. Increased Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7 to Antibody Neutralization. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Demoliner, M.; da Silva, M.S.; Gularte, J.S.; Hansen, A.W.; de Almeida, P.R.; Weber, M.N.; Heldt, F.H.; Silveira, F.; Filippi, M.; de Abreu Góes Pereira, V.M.; et al. Predominance of SARS-CoV-2 P.1 (Gamma) lineage inducing the recent COVID-19 wave in southern Brazil and the finding of an additional S: D614A mutation. Infect. Genet. Evol. 2021, 96, 105134. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Teruel, N.; Mailhot, O.; Najmanovich, R.J. Modelling conformational state dynamics and its role on infection for SARS-CoV-2 Spike protein variants. PLOS Comput. Biol. 2021, 17, e1009286. [Google Scholar] [CrossRef]
- Ali, F.; Kasry, A.; Amin, M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov. 2021, 10, 100086. [Google Scholar] [CrossRef]
- Chakraborty, S. E484K and N501Y SARS-CoV 2 spike mutants Increase ACE2 recognition but reduce affinity for neutralizing antibody. Int. Immunopharmacol. 2022, 102, 108424. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Li, Q.; Nie, J.; Wu, J.; Zhang, L.; Ding, R.; Wang, H.; Zhang, Y.; Li, T.; Liu, S.; Zhang, M.; et al. SARS-CoV-2 501Y.V2 variants lack higher infectivity but do have immune escape. Cell 2021, 184, 2362–2371.e2369. [Google Scholar] [CrossRef]
- Zhao, S.; Lou, J.; Chong, M.K.C.; Cao, L.; Zheng, H.; Chen, Z.; Chan, R.W.Y.; Zee, B.C.Y.; Chan, P.K.S.; Wang, M.H. Inferring the Association between the Risk of COVID-19 Case Fatality and N501Y Substitution in SARS-CoV-2. Viruses 2021, 13, 638. [Google Scholar] [CrossRef]
- Istifli, E.S.; Netz, P.A.; Sihoglu Tepe, A.; Sarikurkcu, C.; Tepe, B. Understanding the molecular interaction of SARS-CoV-2 spike mutants with ACE2 (angiotensin converting enzyme 2). J. Biomol. Struct. Dyn. 2021, 1–12. [Google Scholar] [CrossRef]
- Collier, D.A.; De Marco, A.; Ferreira, I.A.T.M.; Meng, B.; Datir, R.P.; Walls, A.C.; Kemp, S.A.; Bassi, J.; Pinto, D.; Silacci-Fregni, C.; et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 2021, 593, 136–141. [Google Scholar] [CrossRef]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; García-Sastre, A.; Schotsaert, M. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe 2021, 2, e283–e284. [Google Scholar] [CrossRef]
- Hospital, A.; Andrio, P.; Cugnasco, C.; Codo, L.; Becerra, Y.; Dans, P.D.; Battistini, F.; Torres, J.; Goñi, R.; Orozco, M.; et al. BIGNASim: A NoSQL database structure and analysis portal for nucleic acids simulation data. Nucleic Acids Res. 2016, 44, D272–D278. [Google Scholar] [CrossRef] [PubMed]
- Thibault, J.C.; Cheatham, T.E.; Facelli, J.C. iBIOMES Lite: Summarizing Biomolecular Simulation Data in Limited Settings. J. Chem. Inf. Modeling 2014, 54, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.; Murdock, S.; Wu, B.; Ng, M.H.; Johnston, S.; Fangohr, H.; Cox, S.J.; Jeffreys, P.; Essex, J.W.; Sansom, M.S. BioSimGrid: Towards a worldwide repository for biomolecular simulations. Org. Biomol. Chem. 2004, 2, 3219–3221. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; D’Abramo, M.; Hospital, A.; Rueda, M.; Ferrer-Costa, C.; Pérez, A.; Carrillo, O.; Camps, J.; Fenollosa, C.; Repchevsky, D.; et al. MoDEL (Molecular Dynamics Extended Library): A database of atomistic molecular dynamics trajectories. Structure 2010, 18, 1399–1409. [Google Scholar] [CrossRef]
- Feig, M.; Nawrocki, G.; Yu, I.; Wang, P.-h.; Sugita, Y. Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J. Phys. Conf. Ser. 2018, 1036, 012010. [Google Scholar] [CrossRef]
- Petrov, D.; Zagrovic, B. Are Current Atomistic Force Fields Accurate Enough to Study Proteins in Crowded Environments? PLOS Comput. Biol. 2014, 10, e1003638. [Google Scholar] [CrossRef]
- Lin, H.; Truhlar, D.G. QM/MM: What have we learned, where are we, and where do we go from here? Theor. Chem. Acc. 2007, 117, 185–199. [Google Scholar] [CrossRef]
- Botu, V.; Ramprasad, R. Adaptive machine learning framework to accelerate ab initio molecular dynamics. Int. J. Quantum Chem. 2015, 115, 1074–1083. [Google Scholar] [CrossRef]
- Wang, J.; Olsson, S.; Wehmeyer, C.; Pérez, A.; Charron, N.E.; de Fabritiis, G.; Noé, F.; Clementi, C. Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. ACS Cent. Sci. 2019, 5, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Kermode, J.R.; De Vita, A. Molecular Dynamics with On-the-Fly Machine Learning of Quantum-Mechanical Forces. Phys. Rev. Lett. 2015, 114, 096405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermayr, J.; Gastegger, M.; Menger, M.F.S.J.; Mai, S.; González, L.; Marquetand, P. Machine learning enables long time scale molecular photodynamics simulations. Chem. Sci. 2019, 10, 8100–8107. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ding, J.; Wang, Z.; Cao, D.; Ding, X.; Hou, T. From machine learning to deep learning: Advances in scoring functions for protein–ligand docking. WIREs Comput. Mol. Sci. 2020, 10, e1429. [Google Scholar] [CrossRef]
- Casadio, R.; Martelli, P.L.; Savojardo, C. Machine learning solutions for predicting protein–protein interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, e1618. [Google Scholar] [CrossRef]
- Arantes, P.R.; Polêto, M.D.; Pedebos, C.; Ligabue-Braun, R. Making it Rain: Cloud-Based Molecular Simulations for Everyone. J. Chem. Inf. Modeling 2021, 61, 4852–4856. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Forcefield | Drugs | Lipid | DNA & RNA | Protein |
|---|---|---|---|---|---|
| 1 | GROMOS | GROMOS 43A1, GROMOS 45A3/4, GROMOS53A5/6, GROMOS54A7, GROMOS54B7, GROMOS54A8 | |||
| 2 | OPLS | OPLS-AA | OPLS-AA | OPLS-AA/M | OPLS-AA, OPLS-AA/L |
| 3 | CHARMM | CHARMM general force field (CGenFF) | CHARMM27 lipids, CHARMM36 lipids | CHARMM27 DNA, CHARMM27 RNA/DNA, CHARMM 36 RNA, CHARMM 36 DNA | CHARMM22/CMAP, CHARM27, CHARMM36, CHARMM36m |
| 4 | AMBER | General AMBER force field (GAFF) | LIPID14, LIPID21 | AMBER99 OL3, AMBER99bsc, AMBER OL15 | AMBER94, AMBER96, AMBER99, AMBER99sb, AMBER03, AMBER14sb, AMBER15ipq, AMBER19sb |
| 5 | MARTINI | MARTINI 2, MARTINI22, MARTINI22p, MARTINI 3, MARTINI dry, MARTINI ELNEDYN22, MARTINI ELNEDYNP22 | MARTINI 2, MARTINI22, MARTINI22p, MARTINI 3, MARTINI-Dry, MARTINI ELNEDYN22, MARTINI ELNEDYNP22 | MARTINI 2015 | MARTINI 2, MARTINI22, MARTINI22p, MARTINI 3, MARTINI dry, MARTINI ELNEDYN22, MARTINI ELNEDYNP22 |
| 6 | Coarse-grained forcefield models (additional) | - | Electrostatics-based model (ELBA) [90] protein-lipid CG model [91] | PRIMONA, DMD, NAST, ENMs, oxRNA, SimRNA, SPQR | Rosetta centroid (CEN), UNRES, CABS, PRIMO, AWSEM, SURPASS, Scorpion, OPEP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, S.; Tam, B.; Wang, S.M. Applications of Molecular Dynamics Simulation in Protein Study. Membranes 2022, 12, 844. https://doi.org/10.3390/membranes12090844
Sinha S, Tam B, Wang SM. Applications of Molecular Dynamics Simulation in Protein Study. Membranes. 2022; 12(9):844. https://doi.org/10.3390/membranes12090844
Chicago/Turabian StyleSinha, Siddharth, Benjamin Tam, and San Ming Wang. 2022. "Applications of Molecular Dynamics Simulation in Protein Study" Membranes 12, no. 9: 844. https://doi.org/10.3390/membranes12090844
APA StyleSinha, S., Tam, B., & Wang, S. M. (2022). Applications of Molecular Dynamics Simulation in Protein Study. Membranes, 12(9), 844. https://doi.org/10.3390/membranes12090844