

Modified Signaling of Membrane Formyl Peptide Receptors in NADPH-Oxidase Regulation in Obesity-Resistant Mice


Abstract
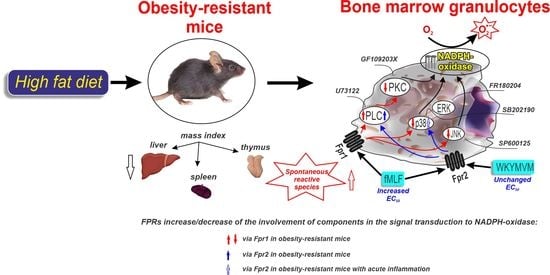
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Organ Isolation
2.3. Cell Isolation
2.4. Reagents
2.5. Generation of Reactive Oxygen Species
2.6. Data Processing
3. Results
3.1. Physiological and Biochemical Parameters
3.2. Ligands of Formyl Peptide Receptors (FPRs) and ROS Generation
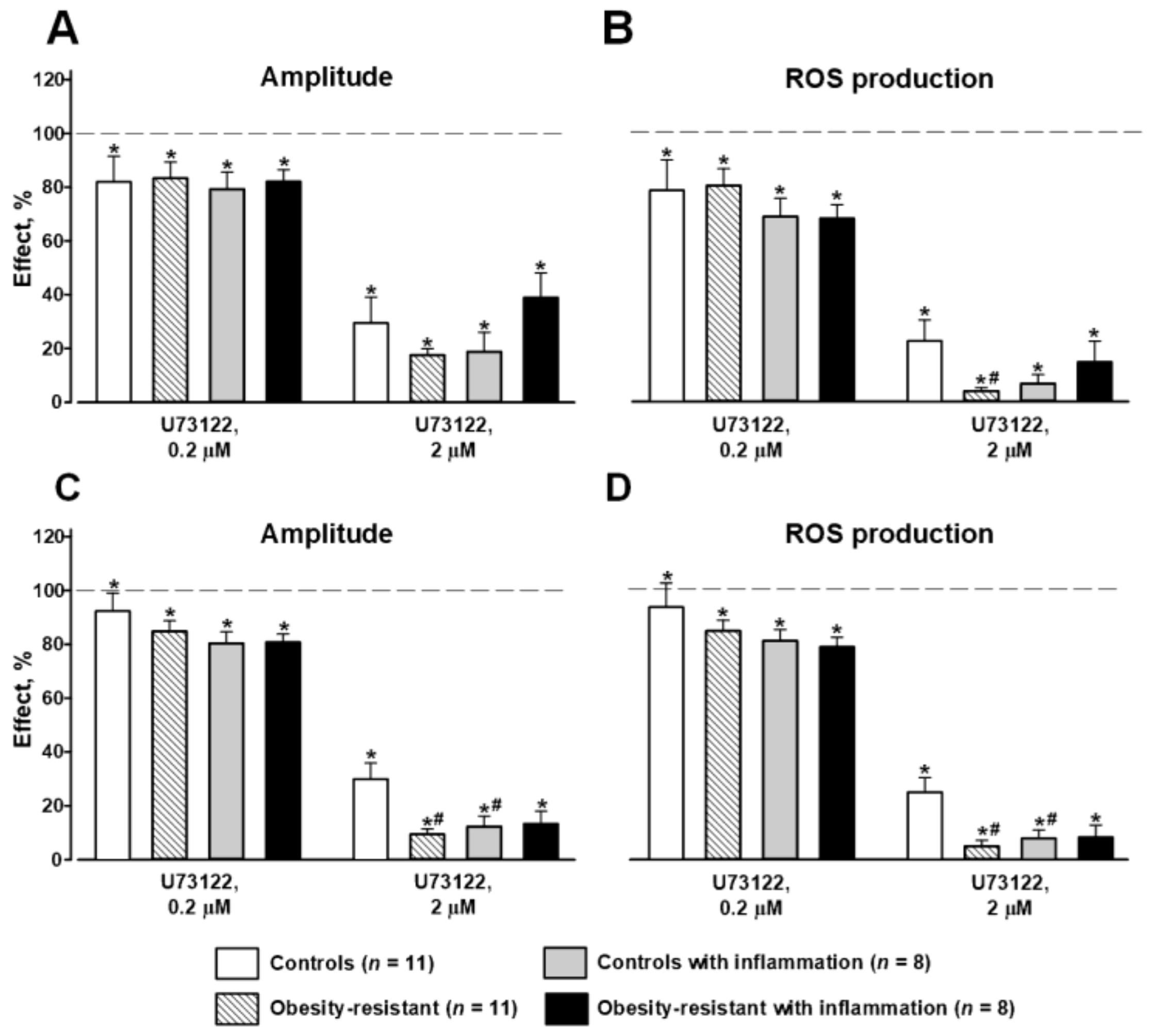
3.3. PLC and PKC Signaling in FPR-Mediated Respiratory Burst
3.4. Involvement of MAP Kinases in FPR-Mediated Respiratory Burst
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ibrahim, H.I.M. Epigenetic Regulation of Obesity-Associated Type 2 Diabetes. Medicina 2022, 58, 1366. [Google Scholar] [CrossRef] [PubMed]
- Belkina, A.C.; Denis, G.V. Obesity genes and insulin resistance. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef] [PubMed]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 2021, 22, 751–771. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, E.G. Genes and obesity: A cause and effect relationship. Endocrinol. Nutr. 2011, 58, 492–496. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Luo, Z.; Ji, Y.; Tang, K.; Jin, Z.; Ly, C.; Sears, D.D.; Mahata, S.; Ying, W. Accumulation of microbial DNAs promotes to islet inflammation and β cell abnormalities in obesity in mice. Nat. Commun. 2022, 13, 565. [Google Scholar] [CrossRef]
- Yang, W.S.; Wang, J.L.; Wu, W.; Wang, G.F.; Yan, J.; Liu, Q.; Wu, X.Y.; Zhou, Q.T.; Yang, D.H.; Wang, M.W.; et al. Formyl peptide receptor 2 as a potential therapeutic target for inflammatory bowel disease. Acta Pharmacol. Sin. 2023, 44, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Wollam, J.; Riopel, M.; Xu, Y.J.; Johnson, A.M.F.; Ofrecio, J.M.; Ying, W.; El Ouarrat, D.; Chan, L.S.; Han, A.W.; Mahmood, N.A.; et al. Microbiota-Produced N-Formyl Peptide fMLF Promotes Obesity-Induced Glucose Intolerance. Diabetes 2019, 68, 1415–1426. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef]
- Chen, G.; Wang, X.; Liao, Q.; Ge, Y.; Jiao, H.; Chen, Q.; Liu, Y.; Lyu, W.; Zhu, L.; van Zundert, G.C.P.; et al. Structural basis for recognition of N-formyl peptides as pathogen-associated molecular patterns. Nat. Commun. 2022, 13, 5232. [Google Scholar] [CrossRef] [PubMed]
- Marteau, J.B.; Samara, A.; Dedoussis, G.; Pfister, M.; Visvikis-Siest, S. Candidate gene microarray analysis in peripheral blood cells for studying hypertension/obesity. Per. Med. 2009, 6, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Tourki, B.; Kain, V.; Pullen, A.B.; Norris, P.C.; Patel, N.; Arora, P.; Leroy, X.; Serhan, C.N.; Halade, G.V. Lack of resolution sensor drives age-related cardiometabolic and cardiorenal defects and impedes inflammation-resolution in heart failure. Mol. Metab. 2020, 31, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhuo, S.; Zhu, T.; Yao, P.; Yang, M.; Mei, H.; Li, N.; Ma, F.; Wang, J.M.; Chen, S.; et al. Fpr2 Deficiency Alleviates Diet-Induced Insulin Resistance Through Reducing Body Weight Gain and Inhibiting Inflammation Mediated by Macrophage Chemotaxis and M1 Polarization. Diabetes 2019, 68, 1130–1142. [Google Scholar] [CrossRef]
- Ding, C.; Guo, J.; Su, Z. The status of research into resistance to diet-induced obesity. Horm. Metab. Res. 2015, 47, 404–410. [Google Scholar] [CrossRef]
- Barnes, M.J.; Holmes, G.; Primeaux, S.D.; York, D.A.; Bray, G.A. Increased expression of mu opioid receptors in animals susceptible to diet-induced obesity. Peptides 2006, 27, 3292–3298. [Google Scholar] [CrossRef]
- Kondo, H.; Minegishi, Y.; Komine, Y.; Mori, T.; Matsumoto, I.; Abe, K.; Tokimitsu, I.; Hase, T.; Murase, T. Differential regulation of intestinal lipid metabolism-related genes in obesity-resistant A/J vs. obesity-prone C57BL/6J mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1092–E1099. [Google Scholar] [CrossRef] [Green Version]
- Primeaux, S.D.; Braymer, H.D.; Bray, G.A. High fat diet differentially regulates the expression of olfactory receptors in the duodenum of obesity-prone and obesity-resistant rats. Dig. Dis. Sci. 2013, 58, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.B.; Luan, J.; Ye, J.; Chen, S.Y. RGC32 deficiency protects against high-fat diet-induced obesity and insulin resistance in mice. J. Endocrinol. 2015, 224, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, I.; Cho, W.; Oh, G.T.; Park, Y.M. Vimentin Deficiency Prevents High-Fat Diet-Induced Obesity and Insulin Resistance in Mice. Diabetes Metab. J. 2021, 45, 97–108. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duca, F.A.; Sakar, Y.; Lepage, P.; Devime, F.; Langelier, B.; Doré, J.; Covasa, M. Replication of obesity and associated signaling pathways through transfer of microbiota from obese-prone rats. Diabetes 2014, 63, 1624–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Ma, H.; Huang, Y.; Li, J.; Li, W. Ruminococcaceae_UCG-013 Promotes Obesity Resistance in Mice. Biomedicines 2022, 10, 3272. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Alcott, C.E.; Sullivan, E.L.; Takahashi, D.; McCurdy, C.E.; Comstock, S.; Baquero, K.; Blundell, P.; Frias, A.E.; Kahr, M.; et al. Genomic Variants Associated with Resistance to High Fat Diet Induced Obesity in a Primate Model. Sci. Rep. 2016, 6, 36123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielmann, J.; Naujoks, W.; Emde, M.; Allweyer, M.; Fänder, J.; Kielstein, H.; Quandt, D.; Bähr, I. The Impact of High-Fat Diet and Restrictive Feeding on Natural Killer Cells in Obese-Resistant BALB/c Mice. Front. Nutr. 2021, 8, 711824. [Google Scholar] [CrossRef]
- Boi, S.K.; Buchta, C.M.; Pearson, N.A.; Francis, M.B.; Meyerholz, D.K.; Grobe, J.L.; Norian, L.A. Obesity alters immune and metabolic profiles: New insight from obese-resistant mice on high-fat diet. Obesity 2016, 24, 2140–2149. [Google Scholar] [CrossRef]
- Boxio, R.; Bossenmeyer-Pourié, C.; Steinckwich, N.; Dournon, C.; Nüsse, O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J. Leukoc. Biol. 2004, 75, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Filina, Y.; Gabdoulkhakova, A.; Rizvanov, A.; Safronova, V. MAP kinases in regulation of NOX activity stimulated through two types of formyl peptide receptors in murine bone marrow granulocytes. Cell Signal. 2022, 90, 110205. [Google Scholar] [CrossRef]
- Ohori, M.; Kinoshita, T.; Okubo, M.; Sato, K.; Yamazaki, A.; Arakawa, H.; Nishimura, S.; Inamura, N.; Nakajima, H.; Neya, M.; et al. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem. Biophys. Res. Commun. 2005, 336, 357–363. [Google Scholar] [CrossRef]
- Lai, K.H.; Chen, P.J.; Chen, C.C.; Yang, S.H.; El-Shazly, M.; Chang, Y.C.; Wu, Y.H.; Wu, Y.H.; Wang, Y.H.; Hsieh, H.L.; et al. Lophatherum gracile Brongn. attenuates neutrophilic inflammation through inhibition of JNK and calcium. J. Ethnopharmacol. 2021, 264, 113224. [Google Scholar] [CrossRef]
- Zhang, K.; Jiang, N.; Sang, X.; Feng, Y.; Chen, R.; Chen, Q. Trypanosoma brucei Lipophosphoglycan Induces the Formation of Neutrophil Extracellular Traps and Reactive Oxygen Species Burst via Toll-Like Receptor 2, Toll-Like Receptor 4, and c-Jun N-Terminal Kinase Activation. Front. Microbiol. 2021, 12, 713531. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Liu, Y.; Rundberg Nilsson, A.; Gatchalian, R.; Crother, T.R.; Tourtellotte, W.G.; Zhang, Y.; Aleman-Muench, G.R.; Lewis, G.; Chen, W.; et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 2021, 54, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, I.V.; Grinevich, A.A.; Guseva, I.E.; Safronova, V.G. Modified kinetics of generation of reactive species in peripheral blood of patients with type 2 diabetes. Free Radic. Biol. Med. 2020, 159, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Karlsson, A.; Bylund, J. Measurement of respiratory burst products generated by professional phagocytes. Methods Mol. Biol. 2007, 412, 349–363. [Google Scholar] [CrossRef] [Green Version]
- Vladimirov, Y.A.; Proskurnina, E.V. Free radicals and cell chemiluminescence. Biochemistry 2009, 74, 1545–1566. [Google Scholar] [CrossRef]
- Monteseirín, J.; Vega, A. Different methods to analyze reactive oxygen species production. Clin. Immunol. 2011, 138, 331–332. [Google Scholar] [CrossRef]
- Foyouzi-Youssefi, R.; Petersson, F.; Lew, D.P.; Krause, K.H.; Nüsse, O. Chemoattractant-induced respiratory burst: Increases in cytosolic Ca2+ concentrations are essential and synergize with a kinetically distinct second signal. Biochem. J. 1997, 322, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Futosi, K.; Fodor, S.; Mócsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rees, D.J.; Szilagyi, K.; Kuijpers, T.W.; Matlung, H.L.; van den Berg, T.K. Immunoreceptors on neutrophils. Semin. Immunol. 2016, 28, 94–108. [Google Scholar] [CrossRef]
- Loh, W.; Vermeren, S. Anti-Inflammatory Neutrophil Functions in the Resolution of Inflammation and Tissue Repair. Cells 2022, 11, 4076. [Google Scholar] [CrossRef] [PubMed]
- Nosadini, R.; Tonolo, G. Role of oxidized low density lipoproteins and free fatty acids in the pathogenesis of glomerulopathy and tubulointerstitial lesions in type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 79–85. [Google Scholar] [CrossRef]
- Palvinskaya, T.; Antkowiak, M.; Burg, E.; Lenox, C.C.; Ubags, N.; Cramer, A.; Rincón, M.; Dixon, A.E.; Fessler, M.B.; Poynter, M.E.; et al. Effects of acute and chronic low density lipoprotein exposure on neutrophil function. Pulm. Pharmacol. Ther. 2013, 26, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef] [PubMed]
- Stott, L.A.; Hall, D.A.; Holliday, N.D. Unravelling intrinsic efficacy and ligand bias at G protein coupled receptors: A practical guide to assessing functional data. Biochem. Pharmacol. 2016, 101, 1–12. [Google Scholar] [CrossRef]
- Gabl, M.; Holdfeldt, A.; Sundqvist, M.; Lomei, J.; Dahlgren, C.; Forsman, H. FPR2 signaling without β-arrestin recruitment alters the functional repertoire of neutrophils. Biochem. Pharmacol. 2017, 145, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Xiong, M.; Zong, X.; Ge, Y.; Zhang, H.; Wang, M.; Won Han, G.; Yi, C.; Ma, L.; Ye, R.D.; et al. Structural basis of ligand binding modes at the human formyl peptide receptor 2. Nat. Commun. 2020, 11, 1208. [Google Scholar] [CrossRef] [Green Version]
- Lind, S.; Dahlgren, C.; Holmdahl, R.; Olofsson, P.; Forsman, H. Functional selective FPR1 signaling in favor of an activation of the neutrophil superoxide generating NOX2 complex. J. Leukoc. Biol. 2021, 109, 1105–1120. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wang, L.; Guo, J.; Sun, D.; Wang, Y.; Liu, W.; Xu, H.E.; Zhang, C. Molecular recognition of formylpeptides and diverse agonists by the formylpeptide receptors FPR1 and FPR2. Nat. Commun. 2022, 13, 1054. [Google Scholar] [CrossRef]
- Bhalla, P.; Su, D.M.; van Oers, N.S.C. Thymus Functionality Needs More Than a Few TECs. Front. Immunol. 2022, 13, 864777. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, T.; Liao, L.; Fan, X.; Chang, L.; Hashimoto, K. Brain-spleen axis in health and diseases: A review and future perspective. Brain Res. Bull. 2022, 182, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Tomaipitinca, L.; Mandatori, S.; Mancinelli, R.; Giulitti, F.; Petrungaro, S.; Moresi, V.; Facchiano, A.; Ziparo, E.; Gaudio, E.; Giampietri, C. The Role of Autophagy in Liver Epithelial Cells and Its Impact on Systemic Homeostasis. Nutrients 2019, 11, 827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, M.; Choi, M.S.; Jung, S.; Jung, Y.; Choi, J.Y.; Ryu, D.H.; Hwang, G.S. Lipidomic Profiling of Liver Tissue from Obesity-Prone and Obesity-Resistant Mice Fed a High Fat Diet. Sci. Rep. 2015, 5, 16984. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C.; Yu, H.P.; Liao, C.C.; Chou, A.H.; Liu, F.C. Escin protects against acetaminophen-induced liver injury in mice via attenuating inflammatory response and inhibiting ERK signaling pathway. Am. J. Transl. Res. 2019, 11, 5170–5182. [Google Scholar]
- Fiorentino, T.V.; Prioletta, A.; Zuo, P.; Folli, F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Curr. Pharm. Des. 2013, 19, 5695–5703. [Google Scholar] [CrossRef]
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. J. Physiol. Pharmacol. 2019, 70, 809–824. [Google Scholar] [CrossRef]
- Johnson, J.; Jaggers, R.M.; Gopalkrishna, S.; Dahdah, A.; Murphy, A.J.; Hanssen, N.M.J.; Nagareddy, P.R. Oxidative Stress in Neutrophils: Implications for Diabetic Cardiovascular Complications. Antioxid. Redox. Signal. 2022, 36, 652–666. [Google Scholar] [CrossRef]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Invest. 2018, 48, 12951. [Google Scholar] [CrossRef] [Green Version]
- Immler, R.; Simon, S.I.; Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Invest. 2018, 48, e12964. [Google Scholar] [CrossRef] [PubMed]
- Talior, I.; Tennenbaum, T.; Kuroki, T.; Eldar-Finkelman, H. PKC-delta-dependent activation of oxidative stress in adipocytes of obese and insulin-resistant mice: Role for NADPH oxidase. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E405–E411. [Google Scholar] [CrossRef] [Green Version]
- Eshima, H.; Tamura, Y.; Kakehi, S.; Kakigi, R.; Hashimoto, R.; Funai, K.; Kawamori, R.; Watada, H. A chronic high-fat diet exacerbates contractile dysfunction with impaired intracellular Ca2+ release capacity in the skeletal muscle of aged mice. J. Appl. Physiol. 2020, 128, 1153–1162. [Google Scholar] [CrossRef]
- Lien, C.-F.; Chen, S.-J.; Tsai, M.-C.; Lin, C.-S. Potential Role of Protein Kinase C in the Pathophysiology of Diabetes-Associated Atherosclerosis. Front. Pharmacol. 2021, 12, 716332. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westenberger, G.; Sellers, J.; Fernando, S.; Junkins, S.; Han, S.M.; Min, K.; Lawan, A. Function of Mitogen-Activated Protein Kinases in Hepatic Inflammation. J. Cell Signal. 2021, 2, 172–180. [Google Scholar]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O′Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Sumara, G.; Formentini, I.; Collins, S.; Sumara, I.; Windak, R.; Bodenmiller, B.; Ramracheya, R.; Caille, D.; Jiang, H.; Platt, K.A.; et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 2009, 136, 235–248. [Google Scholar] [CrossRef]
- Lee, J.; Sun, C.; Zhou, Y.; Lee, J.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fan, L.; Wu, J.; Xu, H.; Leung, W.Y.; Fu, K.; Wu, J.; Liu, K.; Man, K.; Yang, X.; et al. Macrophage p38α promotes nutritional steatohepatitis through M1 polarization. J. Hepatol. 2019, 71, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, S.H.; Hirsova, P.; Malhi, H.; Gores, G.J. Nonalcoholic Steatohepatitis Promoting Kinases. Semin. Liver Dis. 2020, 40, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Hemi, R.; Yochananov, Y.; Barhod, E.; Kasher-Meron, M.; Karasik, A.; Tirosh, A.; Kanety, H. p38 mitogen-activated protein kinase-dependent transactivation of ErbB receptor family: A novel common mechanism for stress-induced IRS-1 serine phosphorylation and insulin resistance. Diabetes 2011, 60, 1134–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, S.; Yu, W.Q.; Moore, J.; Mori, Y.; Tsiani, E.; Giacca, A. Effect of a p38 MAPK inhibitor on FFA-induced hepatic insulin resistance in vivo. Nutr. Diabetes 2016, 6, e210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Terán, B.; Matesanz, N.; Nikolic, I.; Verdugo, M.A.; Sreeramkumar, V.; Hernández-Cosido, L.; Mora, A.; Crainiciuc, G.; Sáiz, M.L.; Bernardo, E.; et al. p38γ and p38δ reprogram liver metabolism by modulating neutrophil infiltration. EMBO J. 2016, 35, 536–552. [Google Scholar] [CrossRef] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Sabio, G.; Davis, R.J. cJun NH2-terminal kinase 1 (JNK1): Roles in metabolic regulation of insulin resistance. Trends Biochem. Sci. 2010, 35, 490–496. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls, n = 9 | Obesity-Resistant, n = 12 | |
|---|---|---|
| Physiological characteristics | ||
| Body weight, g | 27.7 ± 0.7 | 29.1 ± 0.6 |
| Thymus mass index, % | 0.16 ± 0.03 | 0.14 ± 0.01 |
| Spleen mass index, % | 0.30 ± 0.03 | 0.37 ± 0.03 |
| Liver mass index, % | 6.62 ± 0.35 | 3.63 ± 0.22 * |
| Biochemical blood analysis | ||
| Glucose, mmol/L | 7.43 ± 0.52 | 10.42 ± 0.54 * |
| Total cholesterol, mmol/L | 1.80 ± 0.01 | 2.17 ± 0.12 |
| HDL, mmol/L | 0.95 ± 0.05 | 1.08 ± 0.07 |
| LDL, mmol/L | 0.56 ± 0.05 | 0.13 ± 0.01 * |
| Triglycerides, mmol/L | 0.41 ± 0.06 | 0.54 ± 0.03 |
| ALT, U/L | 21.7 ± 3.4 | 89.2 ± 30.8 * |
| AST, U/L | 41.7 ± 8.6 | 141.5 ± 0.2 * |
| Total bilirubin, µmol/L | 3.2 ± 1.3 | 1.70 ± 0.30 |
| Creatinine, µmol/L | 54.0 ± 2.6 | 32.4 ± 4.0 * |
| Urea, mmol/L | 11.5 ± 1.0 | 8.4 ± 1.1 |
| EC50, µM fMLF-Induced Response | EC50, µM WKYMVM-Induced Response | |||
|---|---|---|---|---|
| Amplitude | ROS Production | Amplitude | ROS Production | |
| Controls | 2.44 ± 0.25 | 2.39 ± 0.13 | 0.99 ± 0.19 | 0.98 ± 0.18 |
| ORM | 6.76 ± 0.78 * | 6.44 ± 0.82 * | 0.81 ± 0.04 | 0.76 ± 0.05 |
| Controls with inflammation | 1.65 ± 0.38 | 1.67 ±0.34 | 0.56 ± 0.10 | 0.56 ± 0.09 |
| ORM with inflammation | 8.94 ± 0.23 # | 9.19 ±0.28 # | 0.07 ± 0.69 | 0.06 ± 0.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tikhonova, I.; Dyukina, A.; Shaykhutdinova, E.; Safronova, V. Modified Signaling of Membrane Formyl Peptide Receptors in NADPH-Oxidase Regulation in Obesity-Resistant Mice. Membranes 2023, 13, 306. https://doi.org/10.3390/membranes13030306
Tikhonova I, Dyukina A, Shaykhutdinova E, Safronova V. Modified Signaling of Membrane Formyl Peptide Receptors in NADPH-Oxidase Regulation in Obesity-Resistant Mice. Membranes. 2023; 13(3):306. https://doi.org/10.3390/membranes13030306
Chicago/Turabian StyleTikhonova, Irina, Alsu Dyukina, Elvira Shaykhutdinova, and Valentina Safronova. 2023. "Modified Signaling of Membrane Formyl Peptide Receptors in NADPH-Oxidase Regulation in Obesity-Resistant Mice" Membranes 13, no. 3: 306. https://doi.org/10.3390/membranes13030306
APA StyleTikhonova, I., Dyukina, A., Shaykhutdinova, E., & Safronova, V. (2023). Modified Signaling of Membrane Formyl Peptide Receptors in NADPH-Oxidase Regulation in Obesity-Resistant Mice. Membranes, 13(3), 306. https://doi.org/10.3390/membranes13030306