
The Role of C2 Domains in Two Different Phosphatases: PTEN and SHIP2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction

2. Methods
2.1. Preparing Initial Structures: Addition of Missing Residues
2.2. Coarse-Grained MD Simulations
2.3. All-Atom MD Simulations
2.4. Analysis of Coarse-Grained Simulations
2.5. Analysis of All-Atom MD Simulations
2.6. Free Energy Calculations
2.7. Free Energy Surfaces, Visualization, and Plotting
2.8. Reproducibility and Data Availability
3. Results
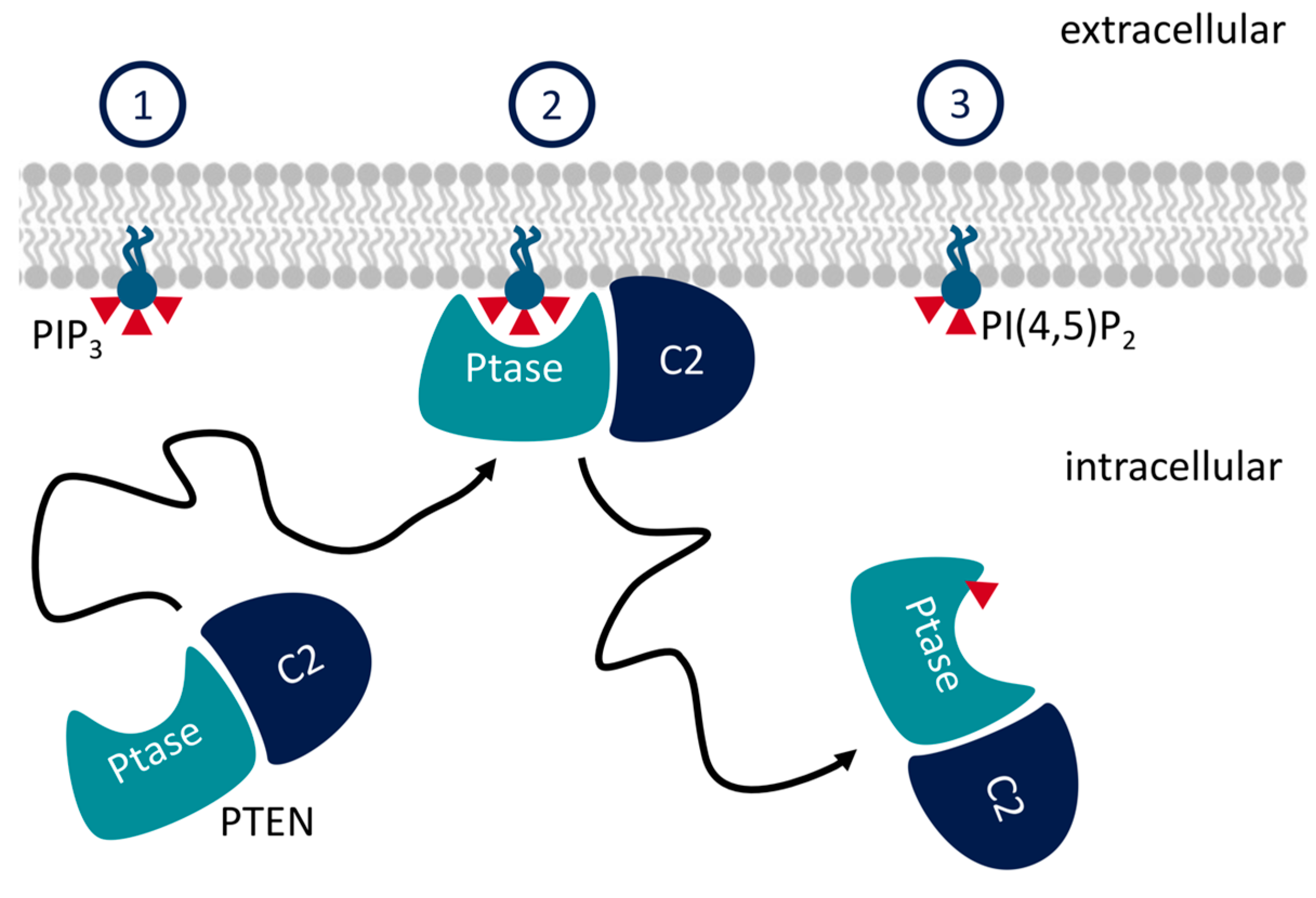
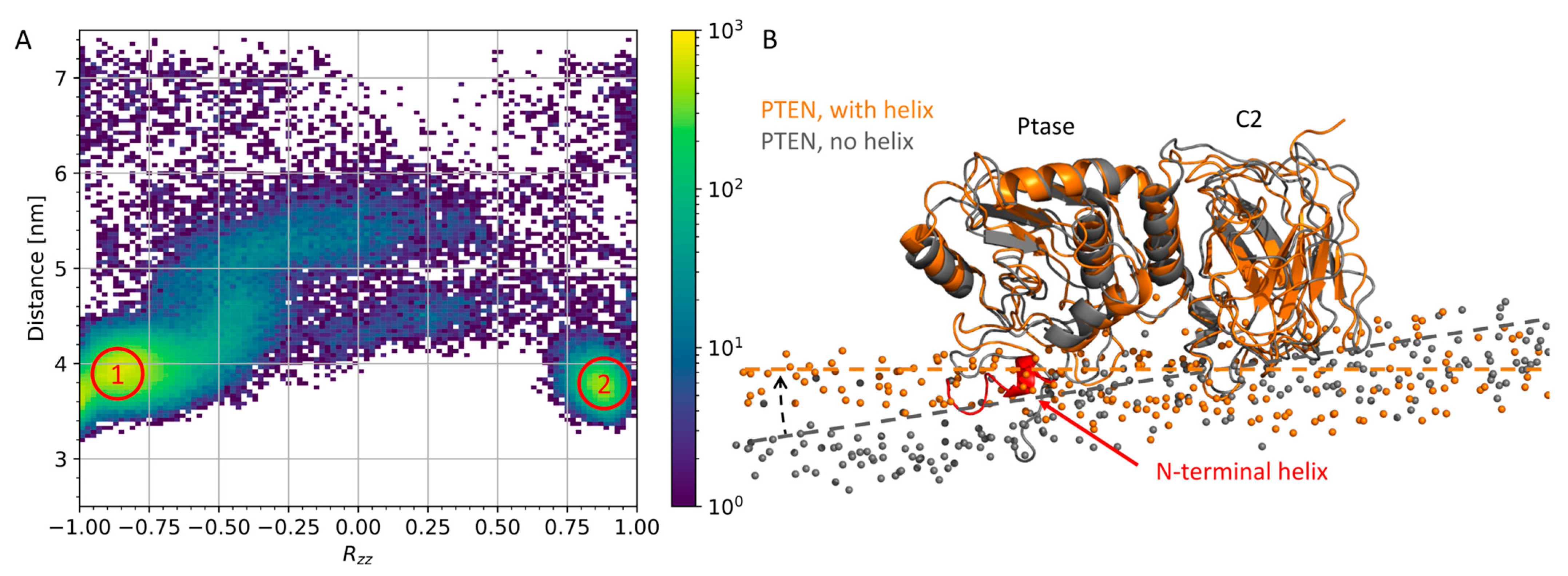
3.1. N-Terminal Helix in PTEN Participates in Lipid Binding
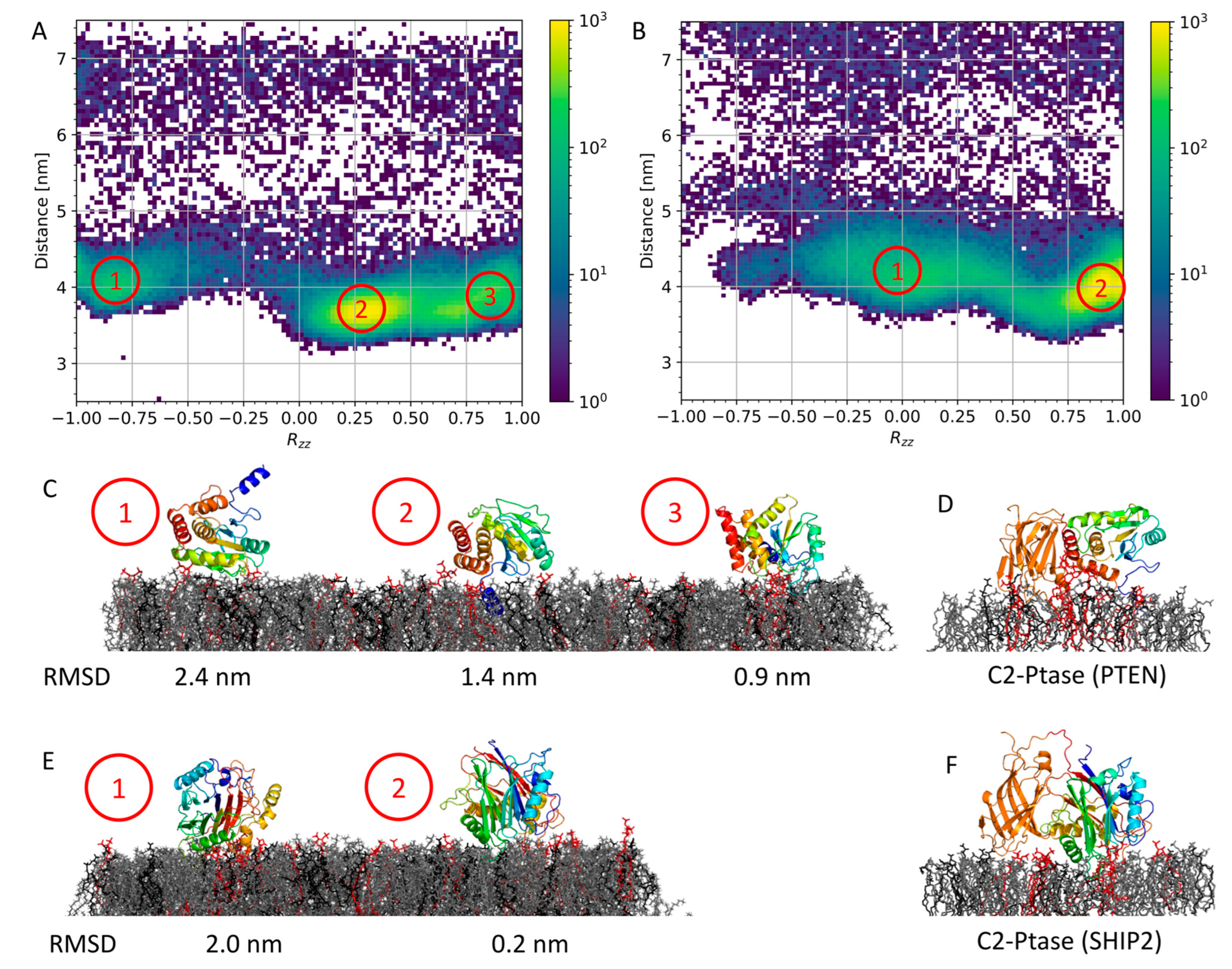
3.2. Membrane Binding of Isolated Ptase from PTEN and SHIP2
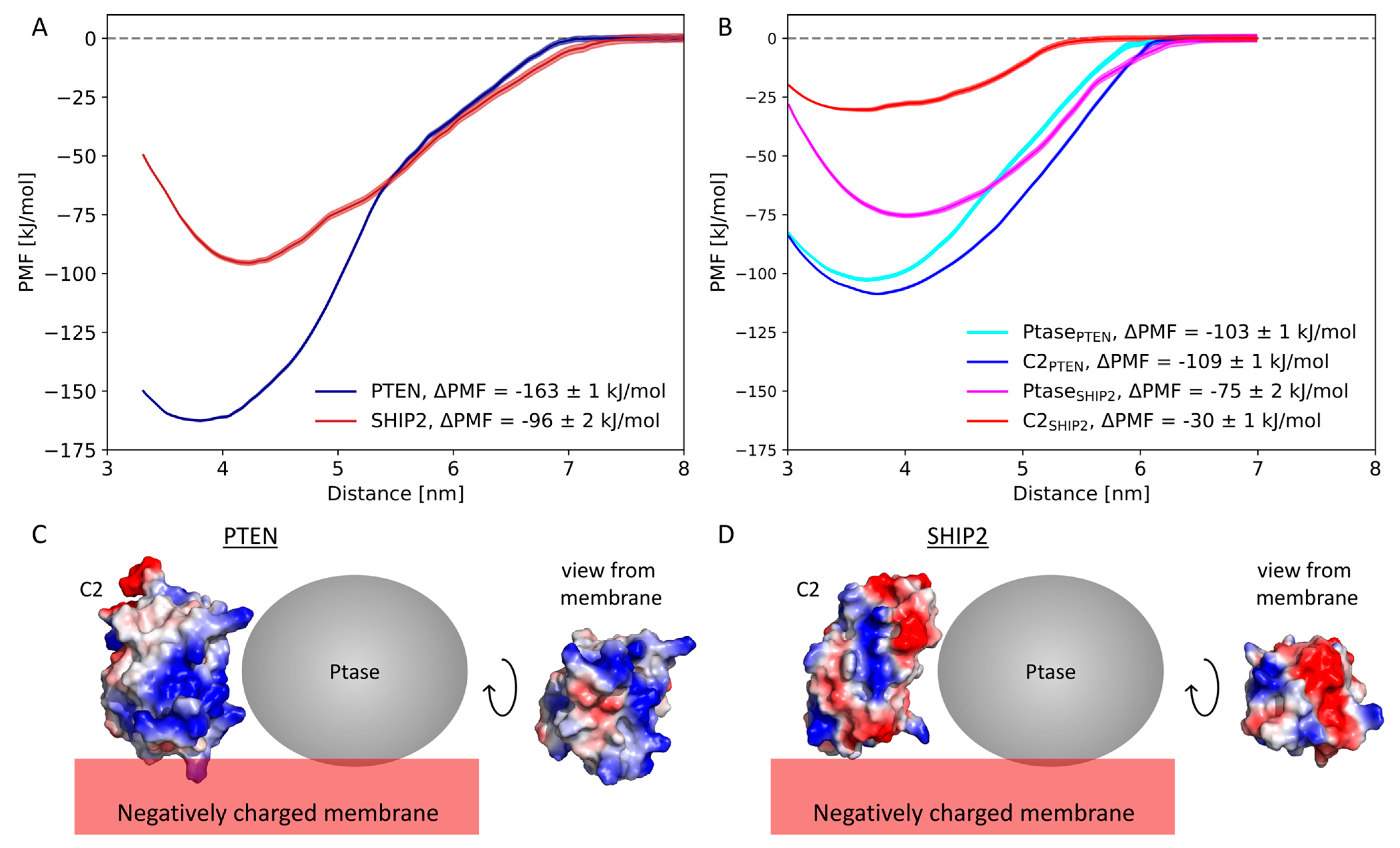
3.3. Free Energy Calculations
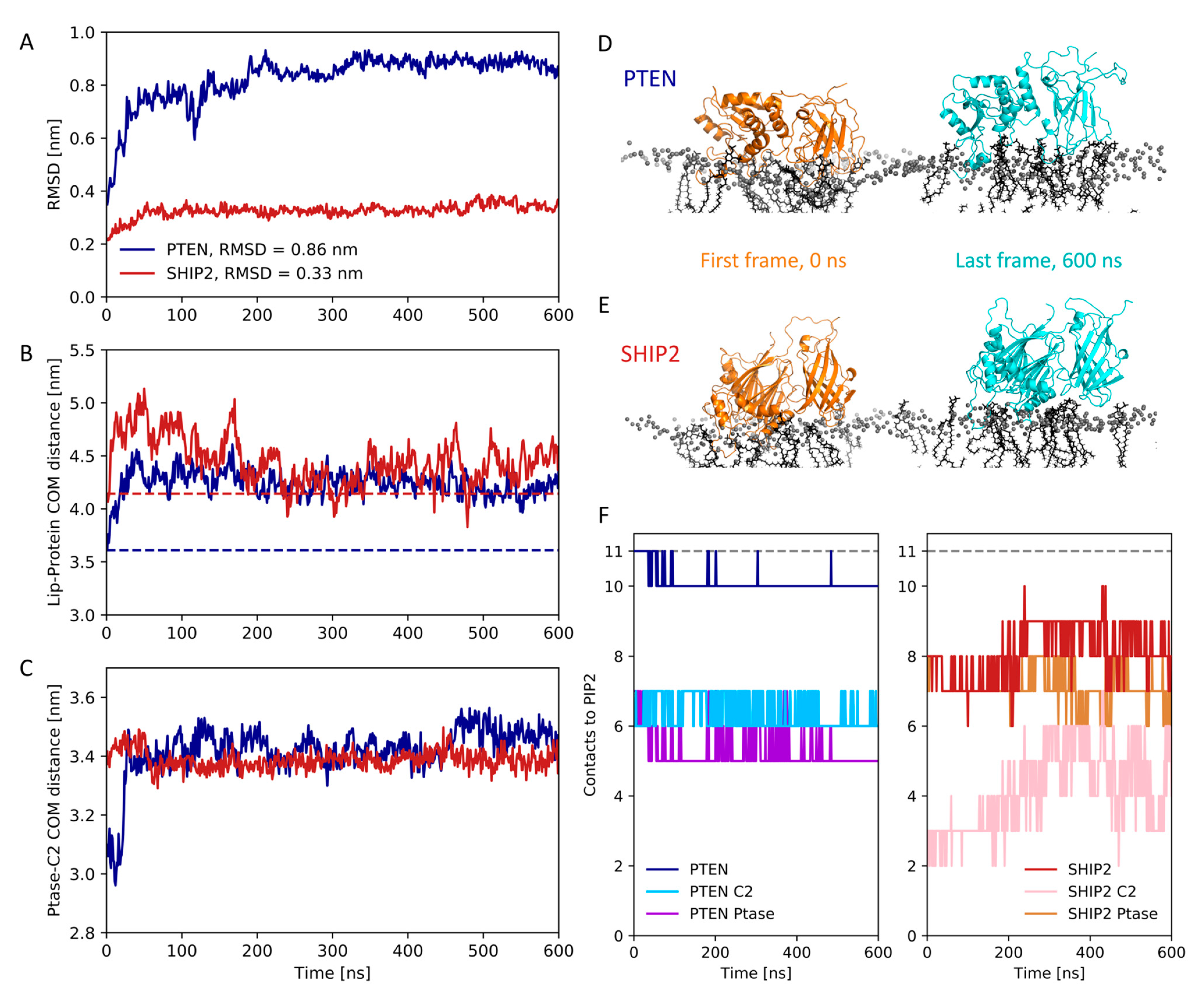
3.4. Atomistic Simulations of Full-Length Structures
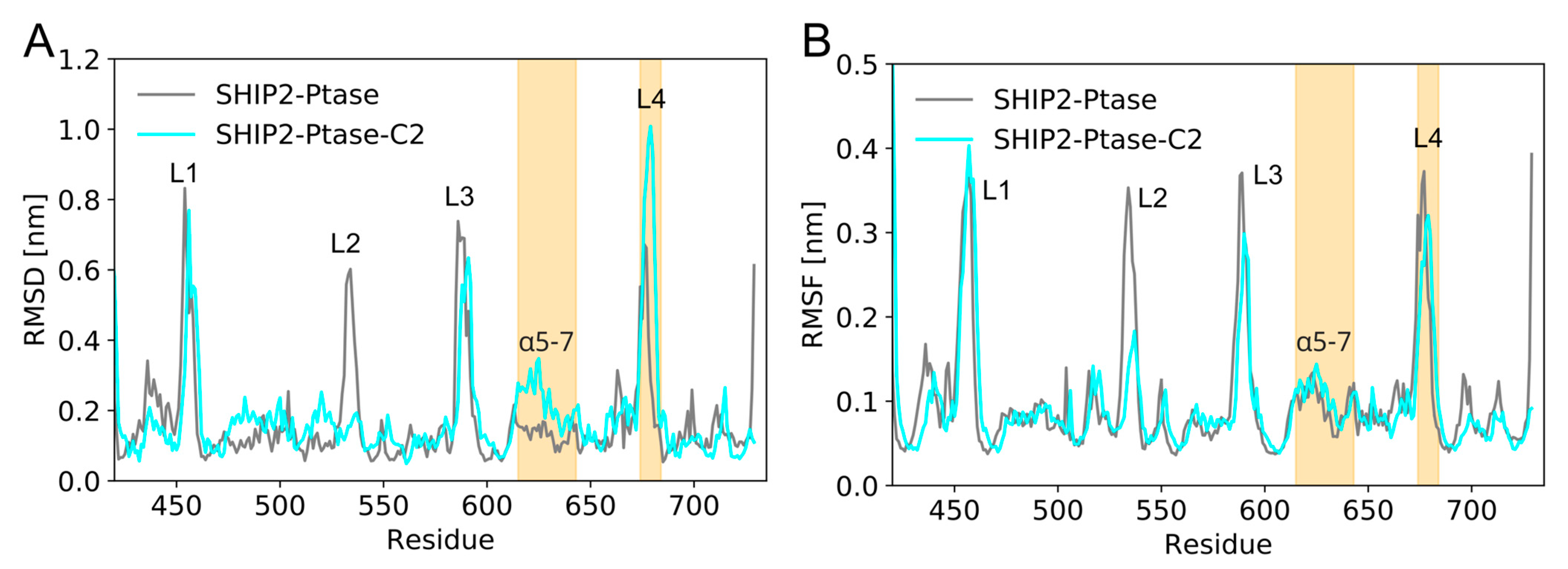
3.5. Dynamics of Full-Length SHIP2 vs PtaseSHIP2
4. Discussion
4.1. Negative Net Charge of the Membrane Is Critical for C2 Domain Binding
4.2. Comparison of Experimentally and Computationally Estimated Binding Energies
4.3. C2 Facilitates Membrane Binding in PTEN
4.4. C2 May Allosterically Affect Catalytic Activity in SHIP2
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Corbalan-Garcia, S.; Gómez-Fernández, J.C. Signaling through C2 Domains: More than One Lipid Target. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1536–1547. [Google Scholar] [CrossRef]
- Edimo, E.; Schurmans, S.; Roger, P.P.; Erneux, C. Advances in Biological Regulation SHIP2 Signaling in Normal and Pathological Situations: Its Impact on Cell Proliferation. Adv. Biol. Regul. 2014, 54, 142–151. [Google Scholar] [CrossRef]
- Clément, S.; Krause, U.; Desmedt, F.; Tanti, J.F.; Behrends, J.; Pesesse, X.; Sasaki, T.; Penninger, J.; Doherty, M.; Malaisse, W.; et al. The Lipid Phosphatase SHIP2 Controls Insulin Sensitivity. Nature 2001, 409, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ross, A.H. Why Is PTEN an Important Tumor Suppressor? J. Cell. Biochem. 2007, 102, 1368–1374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Aravind, L. Identification of Novel Families and Classification of the C2 Domain Superfamily Elucidate the Origin and Evolution of Membrane Targeting Activities in Eukaryotes. Gene 2010, 469, 18–30. [Google Scholar] [CrossRef]
- Larsen, A.H.; Sansom, M.S.P. Binding of Ca2+-Independent C2 Domains to Lipid Membranes: A Multi-Scale Molecular Dynamics Study. Structure 2021, 29, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.H.; John, L.H.; Sansom, M.S.P.; Corey, R.A. Specific Interactions of Peripheral Membrane Proteins with Lipids: What Can Molecular Simulations Show Us? Biosci. Rep. 2022, 42, BSR20211406. [Google Scholar] [CrossRef]
- Lee, J.O.; Yang, H.; Georgescu, M.M.; Cristofano, A.D.; Maehama, T.; Shi, Y.; Dixon, J.E.; Pandolfi, P.; Pavletich, N.P. Crystal Structure of the PTEN Tumor Suppressor: Implications for Its Phosphoinositide Phosphatase Activity and Membrane Association. Cell 1999, 99, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.; Sali, A.; Fiser, A.; Kinh, R.; Do, G.; Andrej, S. Modeling of Loops in Protein Structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Le Coq, J.; Camacho-Artacho, M.; Velázquez, J.; Santiveri, C.M.; Gallego, L.H.; Campos-Olivas, R.; Dölker, N.; Lietha, D. Structural Basis for Interdomain Communication in SHIP2 Providing High Phosphatase Activity. eLife 2017, 6, e26640. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Ingólfsson, H.I.; Böckmann, R.A.; Tieleman, D.P.; Marrink, S.J. Computational Lipidomics with Insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- López, C.A.; Sovova, Z.; Van Eerden, F.J.; De Vries, A.H.; Marrink, S.J. Martini Force Field Parameters for Glycolipids. J. Chem. Theory Comput. 2013, 9, 1694–1708. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Vickery, O.N.; Stansfeld, P.J. CG2AT2: An Enhanced Fragment-Based Approach for Serial Multi-Scale Molecular Dynamics Simulations. J. Chem. Theory Comput. 2021, 17, 6472–6482. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Hub, J.S.; De Groot, B.L.; Van Der Spoel, D. G_wham—A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 2010, 6, 3713–3720. [Google Scholar] [CrossRef]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Song, W.; Corey, R.A.; Ansell, T.B.; Cassidy, C.K.; Horrell, M.R.; Duncan, A.L.; Stansfeld, P.J.; Sansom, M.S.P. PyLipID: A Python Package for Analysis of Protein-Lipid Interactions from Molecular Dynamics Simulations. J. Chem. Theory Comput. 2022, 18, 1188–1201. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.R.V.; Balla, T. Polyphosphoinositide Binding Domains: Key to Inositol Lipid Biology. Biochim. Biophys. Acta 2015, 1851, 746–758. [Google Scholar] [CrossRef]
- Das, S.; Dixon, J.E.; Cho, W. Membrane-Binding and Activation Mechanism of PTEN. Proc. Natl. Acad. Sci. USA 2003, 100, 7491–7496. [Google Scholar] [CrossRef]
- Le Coq, J.; López Navajas, P.; Rodrigo Martin, B.; Alfonso, C.; Lietha, D. A New Layer of Phosphoinositide-Mediated Allosteric Regulation Uncovered for SHIP2. FASEB J. 2021, 35, e21815. [Google Scholar] [CrossRef]
- Shenoy, S.S.; Nanda, H.; Lösche, M. Membrane Association of the PTEN Tumor Suppressor: Electrostatic Interaction with Phosphatidylserine-Containing Bilayers and Regulatory Role of the C-Terminal Tail. J. Struct. Biol. 2012, 180, 394–408. [Google Scholar] [CrossRef]
- Shenoy, S.; Shekhar, P.; Heinrich, F.; Daou, M.C.; Gericke, A.; Ross, A.H.; Lösche, M. Membrane Association of the PTEN Tumor Suppressor: Molecular Details of the Protein-Membrane Complex from SPR Binding Studies and Neutron Reflection. PLoS ONE 2012, 7, e32591. [Google Scholar] [CrossRef]
- Harishchandra, R.K.; Neumann, B.M.; Gericke, A.; Ross, A.H. Biophysical Methods for the Characterization of PTEN/Lipid Bilayer Interactions. Methods 2015, 77, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Van Hilten, N.; Stroh, K.S.; Risselada, H.J. Efficient Quantification of Lipid Packing Defect Sensing by Amphipathic Peptides: Comparing Martini 2 and 3 with CHARMM36. J. Chem. Theory Comput. 2022, 18, 4503–4514. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M.; Kirsch, K.H.; Kaloudis, P.; Yang, H.; Pavletich, N.P.; Hanafusa, H. Stabilization and Productive Positioning Roles of the C2 Domain of PTEN Tumor Suppressor. Cancer Res. 2000, 60, 7033–7038. [Google Scholar] [PubMed]
- Smith, I.N.; Dawson, J.E.; Eng, C. Comparative Protein Structural Network Analysis Reveals C-Terminal Tail Phosphorylation Structural Communication Fingerprint in PTEN-Associated Mutations in Autism and Cancer. J. Phys. Chem. B 2023, 127, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Sanlidag, S.; Jensen, S.A.; Burnap, S.A.; Struwe, W.B.; Larsen, A.H.; Feng, X.; Mittal, S.; Sansom, M.S.P.; Sahlgren, C.; et al. An N-Glycan on the C2 Domain of JAGGED1 Is Important for Notch Activation. Sci. Signal. 2022, 15, eabo3507. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
John, L.H.; Naughton, F.B.; Sansom, M.S.P.; Larsen, A.H. The Role of C2 Domains in Two Different Phosphatases: PTEN and SHIP2. Membranes 2023, 13, 408. https://doi.org/10.3390/membranes13040408
John LH, Naughton FB, Sansom MSP, Larsen AH. The Role of C2 Domains in Two Different Phosphatases: PTEN and SHIP2. Membranes. 2023; 13(4):408. https://doi.org/10.3390/membranes13040408
Chicago/Turabian StyleJohn, Laura H., Fiona B. Naughton, Mark S. P. Sansom, and Andreas Haahr Larsen. 2023. "The Role of C2 Domains in Two Different Phosphatases: PTEN and SHIP2" Membranes 13, no. 4: 408. https://doi.org/10.3390/membranes13040408
APA StyleJohn, L. H., Naughton, F. B., Sansom, M. S. P., & Larsen, A. H. (2023). The Role of C2 Domains in Two Different Phosphatases: PTEN and SHIP2. Membranes, 13(4), 408. https://doi.org/10.3390/membranes13040408