
Current Methods for Identifying Plasma Membrane Proteins as Cancer Biomarkers
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
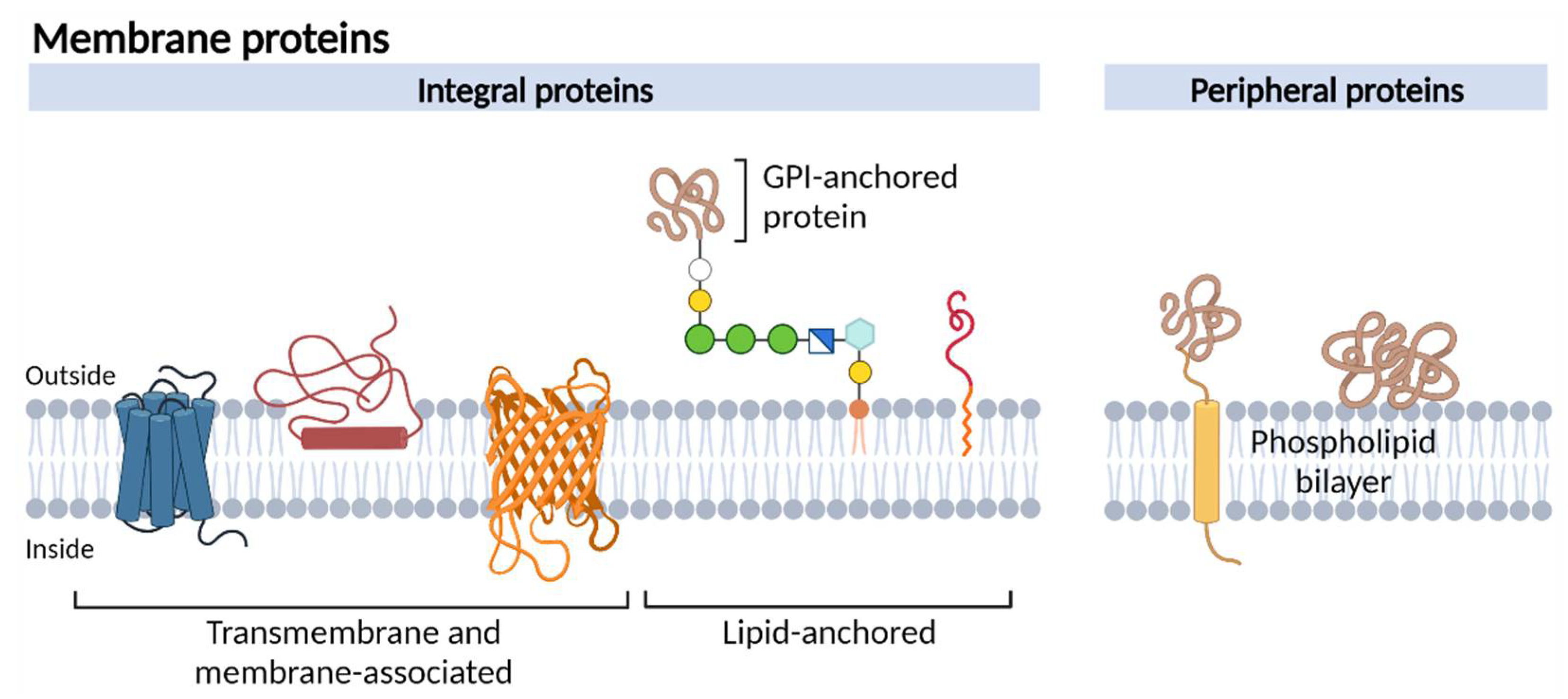
:1. Introduction
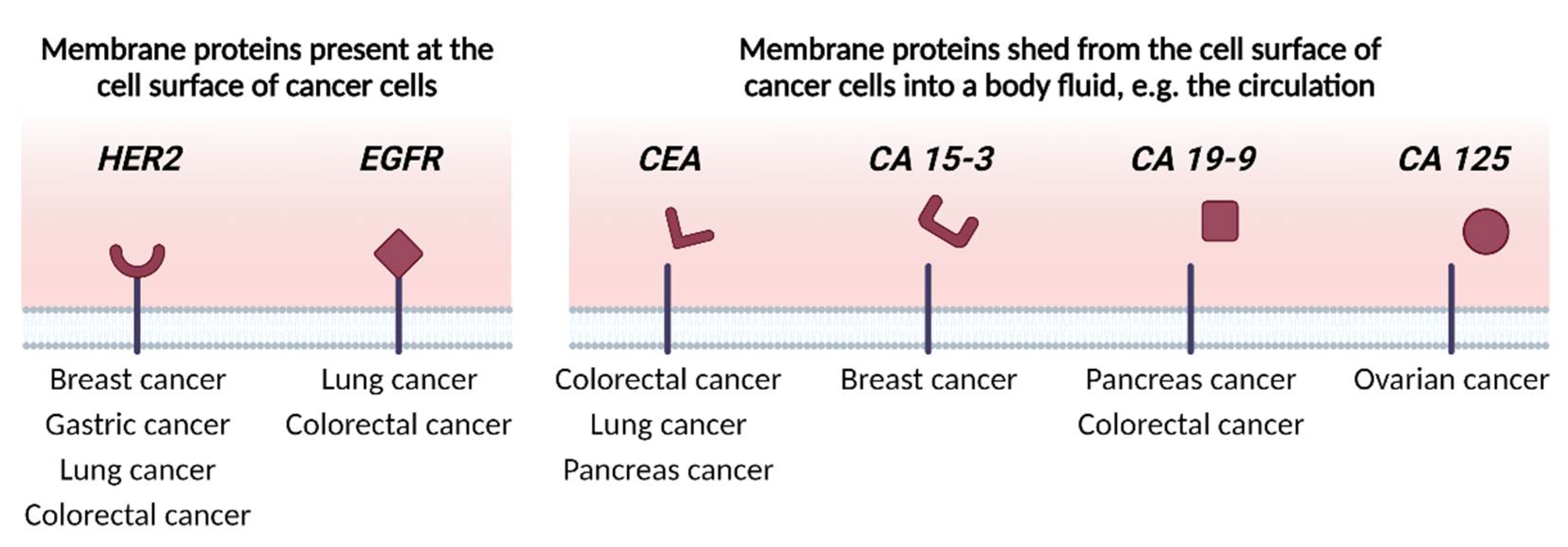
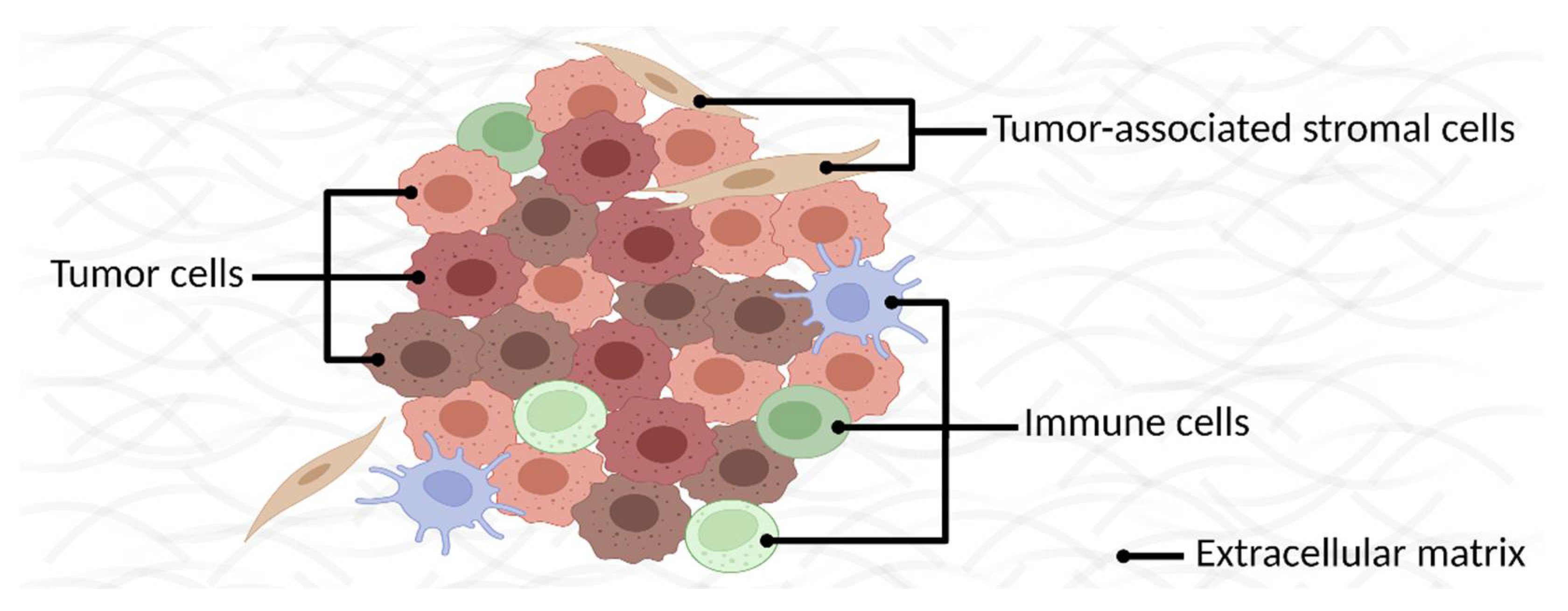
2. Identifying Membrane Proteins as Cancer Biomarkers
2.1. Indirect Discovery Methods
2.2. Direct, Biased Discovery Methods
2.2.1. Multiplexed Immunohistochemistry/Immunofluorescence
2.2.2. High-Throughput Flow Cytometry
2.3. Direct, Unbiased Discovery Methods
2.3.1. Bottom-Up Mass Spectrometry
Enrichment
Solubility
2.3.2. Top-Down Mass Spectrometry
2.4. Contemporary Methods
2.4.1. Mass Cytometry
2.4.2. Cell-SELEX
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hübner, C.A.; Jentsch, T.J. Ion Channel Diseases. Hum. Mol. Genet 2002, 11, 2435–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebhan, M.; Chalifa-Caspi, V.; Prilusky, J.; Lancet, D. GeneCards: Integrating Information about Genes, Proteins and Diseases. Trends Genet 1997, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, F.; Jurkat-Rott, K. Voltage-Gated Ion Channels and Hereditary Disease. Physiol. Rev. 1999, 79, 1317–1372. [Google Scholar] [CrossRef] [PubMed]
- Leth-Larsen, R.; Lund, R.R.; Ditzel, H.J. Plasma Membrane Proteomics and Its Application in Clinical Cancer Biomarker Discovery. Mol. Cell. Proteom. 2010, 9, 1369. [Google Scholar] [CrossRef] [Green Version]
- Dobson, L.; Reményi, I.; Tusnády, G.E. The Human Transmembrane Proteome. Biol. Direct 2015, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Jentsch, T.J.; Hübner, C.A.; Fuhrmann, J.C. Ion Channels: Function Unravelled by Dysfunction. Nat. Cell Biol. 2004, 6, 1039–1047. [Google Scholar] [CrossRef]
- Kampen, K.R. Membrane Proteins: The Key Players of a Cancer Cell. J. Membr. Biol. 2011, 242, 69–74. [Google Scholar] [CrossRef]
- Imai, K.; Takaoka, A. Comparing Antibody and Small-Molecule Therapies for Cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]
- Yildirim, M.A.; Goh, K.I.; Cusick, M.E.; Barabási, A.L.; Vidal, M. Drug—Target Network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef]
- Ye, X.; Kaczmarczyk, J.A.; Luke, B.; Saul, R.G.; Whiteley, G.R.; Nissley, D.V.; Blonder, J. Cell Surface Protein Enrichment for Biomarker and Drug Target Discovery Using Mass Spectrometry-Based Proteomics. In Proteomic and Metabolomic Approaches to Biomarker Discovery; Academic Press: Cambridge, MA, USA, 2020; pp. 409–420. [Google Scholar] [CrossRef]
- Davey, G.C.; Currie, G.A.; Alexander, P. Spontaneous Shedding and Antibody Induced Modulation of Histocompatibility Antigens on Murine Lymphomata: Correlation with Metastatic Capacity. Br. J. Cancer 1976, 33, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Lund, R.; Leth-Larsen, R.; Jensen, O.N.; Ditzel, H.J. Efficient Isolation and Quantitative Proteomic Analysis of Cancer Cell Plasma Membrane Proteins for Identification of Metastasis-Associated Cell Surface Markers. J. Proteome Res. 2009, 8, 3078–3090. [Google Scholar] [CrossRef]
- Kuhlmann, L.; Cummins, E.; Samudio, I.; Kislinger, T. Cell-Surface Proteomics for the Identification of Novel Therapeutic Targets in Cancer. Expert Rev. Proteom. 2018, 15, 259–275. [Google Scholar] [CrossRef]
- Cordwell, S.J.; Thingholm, T.E. Technologies for Plasma Membrane Proteomics. Proteomics 2010, 10, 611–627. [Google Scholar] [CrossRef]
- Smolders, K.; Lombaert, N.; Valkenborg, D.; Baggerman, G.; Arckens, L. An Effective Plasma Membrane Proteomics Approach for Small Tissue Samples. Sci. Rep. 2015, 5, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Karhemo, P.R.; Hyvönen, M.; Laakkonen, P. Metastasis-Associated Cell Surface Oncoproteomics. Front. Pharmacol. 2012, 3, 192. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Griffin, A.; Qiang, Z.; Ren, J. Organelle-Targeted Therapies: A Comprehensive Review on System Design for Enabling Precision Oncology. Signal Transduct. Target. Ther. 2022, 7. [Google Scholar] [CrossRef]
- Díaz, P.; Sandoval-Bórquez, A.; Bravo-Sagua, R.; Quest, A.F.G.; Lavandero, S. Perspectives on Organelle Interaction, Protein Dysregulation, and Cancer Disease. Front. Cell Dev. Biol. 2021, 9, 1–10. [Google Scholar] [CrossRef]
- Gil-Hernández, A.; Arroyo-Campuzano, M.; Simoni-Nieves, A.; Zazueta, C.; Gomez-Quiroz, L.E.; Silva-Palacios, A. Relevance of Membrane Contact Sites in Cancer Progression. Front. Cell Dev. Biol. 2021, 8, 1–19. [Google Scholar] [CrossRef]
- Crick, F. Central Dogma of Molecular Biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Da Cunha, J.P.C.; Galante, P.A.F.; De Souza, J.E.; De Souza, R.F.; Carvalho, P.M.; Ohara, D.T.; Moura, R.P.; Oba-Shinja, S.M.; Marie, S.K.N.; Silva, W.A.; et al. Bioinformatics Construction of the Human Cell Surfaceome. Proc. Natl. Acad. Sci. USA 2009, 106, 16752–16757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almén, M.S.; Nordström, K.J.; Fredriksson, R.; Schiöth, H.B. Mapping the Human Membrane Proteome: A Majority of the Human Membrane Proteins Can Be Classified According to Function and Evolutionary Origin. BMC Biol. 2009, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Babcock, J.J.; Li, M. Deorphanizing the Human Transmembrane Genome: A Landscape of Uncharacterized Membrane Proteins. Acta Pharmacol. Sin. 2014, 35, 11–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Yu, X.; Xue, L.; Li, S.; Li, J.; Tong, D.; Du, Y. TP53-Associated Ion Channel Genes Serve as Prognostic Predictor and Therapeutic Targets in Head and Neck Squamous Cell Carcinoma. Technol. Cancer Res. Treat. 2020, 19, 1–13. [Google Scholar] [CrossRef]
- Zhao, Z.Y.; Liu, W. Pancreatic Cancer: A Review of Risk Factors, Diagnosis, and Treatment. Technol. Cancer Res. Treat. 2020, 19. [Google Scholar] [CrossRef]
- Diehn, M.; Eisen, M.B.; Botstein, D.; Brown, P.O. Large-Scale Identification of Secreted and Membrane-Associated Gene Products Using DNA Microarrays. Nat. Genet. 2000, 25, 58–62. [Google Scholar] [CrossRef]
- Diehn, M.; Bhattacharya, R.; Botstein, D.; Brown, P.O. Genome-Scale Identification of Membrane-Associated Human MRNAs. PLoS Genet 2006, 2, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chem. Int. Ed. Engl. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative Splicing and Cancer: A Systematic Review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L.; Linial, M.; Goodlett, D.; Langridge-Smith, P.; Goo, Y.A.; Safford, G.; Bonilla, L.; Kruppa, G.; Zubarev, R.; et al. Proteoform: A Single Term Describing Protein Complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of Multiplex Immunohistochemistry/Immunofluorescence Techniques in the Era of Cancer Immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [Green Version]
- Solier, C.; Langen, H. Antibody-Based Proteomics and Biomarker Research-Current Status and Limitations. Proteomics 2014, 14, 774–783. [Google Scholar] [CrossRef]
- Lopes, N.; Bergsland, C.H.; Bjørnslett, M.; Pellinen, T.; Svindland, A.; Nesbakken, A.; Almeida, R.; Lothe, R.A.; David, L.; Bruun, J. Digital Image Analysis of Multiplex Fluorescence IHC in Colorectal Cancer Recognizes the Prognostic Value of CDX2 and Its Negative Correlation with SOX2. Lab. Investig. 2019, 100, 120–134. [Google Scholar] [CrossRef] [Green Version]
- Toh, J.; Hoppe, M.M.; Thakur, T.; Yang, H.; Tan, K.T.; Pang, B.; Ho, S.; Roy, R.; Ho, K.Y.; Yeoh, K.G.; et al. Profiling of Gastric Cancer Cell-Surface Markers to Achieve Tumour-Normal Discrimination. BMJ Open Gastroenterol. 2020, 7, e000452. [Google Scholar] [CrossRef]
- Adan, A.; Alizada, G.; Kiraz, Y.; Baran, Y.; Nalbant, A. Flow Cytometry: Basic Principles and Applications. Crit. Rev. Biotechnol. 2017, 37, 163–176. [Google Scholar] [CrossRef]
- Gedye, C.A.; Hussain, A.; Paterson, J.; Smrke, A.; Saini, H.; Sirskyj, D.; Pereira, K.; Lobo, N.; Stewart, J.; Go, C.; et al. Cell Surface Profiling Using High-Throughput Flow Cytometry: A Platform for Biomarker Discovery and Analysis of Cellular Heterogeneity. PLoS ONE 2014, 9, e105602. [Google Scholar] [CrossRef]
- Chen, K.; Ding, A.; Ding, Y.; Ghanekar, A. High-Throughput Flow Cytometry Screening of Human Hepatocellular Carcinoma Reveals CD146 to Be a Novel Marker of Tumor-Initiating Cells. Biochem. Biophys. Rep. 2016, 8, 107. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, N.L. Peer Reviewed: Top-Down Proteomics. Anal. Chem. 2004, 76, 196 A–203 A. [Google Scholar] [CrossRef] [Green Version]
- Cifani, P.; Kentsis, A. Towards Comprehensive and Quantitative Proteomics for Diagnosis and Therapy of Human Disease. Proteomics 2017, 17, 1600079. [Google Scholar] [CrossRef] [Green Version]
- Chait, B.T. Mass Spectrometry: Bottom-up or Top-Down? Science 2006, 314, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, X.; Fu, X.; Li, Z.; Huang, Y.; Liang, C. Advances in Aptamer-Based Biomarker Discovery. Front. Cell Dev. Biol. 2021, 9, 571. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qin, H.; Ye, M. An Overview on Enrichment Methods for Cell Surface Proteome Profiling. J. Sep. Sci. 2020, 43, 292–312. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, I.; Garrett, T.J. Mass Spectrometry Techniques in Emerging Pathogens Studies: COVID-19 Perspectives. J. Am. Soc. Mass Spectrom. 2020, 31, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Vit, O.; Petrak, J. Integral Membrane Proteins in Proteomics. How to Break Open the Black Box? J. Proteom. 2017, 153, 8–20. [Google Scholar] [CrossRef]
- Wollscheid, B.; Bausch-Fluck, D.; Henderson, C.; O’Brien, R.; Bibel, M.; Schiess, R.; Aebersold, R.; Watts, J.D. Mass-Spectrometric Identification and Relative Quantification of N-Linked Cell Surface Glycoproteins. Nat. Biotechnol. 2009, 27, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Elia, G. Biotinylation Reagents for the Study of Cell Surface Proteins. Proteomics 2008, 8, 4012–4024. [Google Scholar] [CrossRef]
- Gahmberg, C.G.; Andersson, L.C. Selective Radioactive Labeling of Cell Surface Sialoglycoproteins by Periodate-Tritiated Borohydride. J. Biol. Chem. 1977, 252, 5888–5894. [Google Scholar] [CrossRef]
- Bayer, E.A.; Ben-Hur, H.; Wilchek, M. Biocytin Hydrazide--a Selective Label for Sialic Acids, Galactose, and Other Sugars in Glycoconjugates Using Avidin-Biotin Technology. Anal. Biochem. 1988, 170, 271–281. [Google Scholar] [CrossRef]
- Gundry, R.L.; Riordon, D.R.; Tarasova, Y.; Chuppa, S.; Bhattacharya, S.; Juhasz, O.; Wiedemeier, O.; Milanovich, S.; Noto, F.K.; Tchernyshyov, I.; et al. A Cell Surfaceome Map for Immunophenotyping and Sorting Pluripotent Stem Cells. Mol. Cell. Proteom. 2012, 11, 303. [Google Scholar] [CrossRef] [Green Version]
- Kailemia, M.J.; Park, D.; Lebrilla, C.B. Glycans and Glycoproteins as Specific Biomarkers for Cancer. Anal. Bioanal. Chem. 2017, 409, 395–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, J.; Sethi, A.; Li, Q.K.; Chen, L.; Collins, B.; Gillet, L.C.J.; Wollscheid, B.; Zhang, H.; Aebersold, R. Glycoproteomic Analysis of Prostate Cancer Tissues by SWATH Mass Spectrometry Discovers N-Acylethanolamine Acid Amidase and Protein Tyrosine Kinase 7 as Signatures for Tumor Aggressiveness. Mol. Cell. Proteom. 2014, 13, 1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Bova, G.S.; Zhang, H. Quantitative Glycoproteomic Analysis of Optimal Cutting Temperature-Embedded Frozen Tissues Identifying Glycoproteins Associated with Aggressive Prostate Cancer. Anal. Chem. 2011, 83, 7013–7019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Xi, J.; Tian, Y.; Bova, G.S.; Zhang, H. Identification, Prioritization and Evaluation of Glycoproteins for Aggressive Prostate Cancer Using Quantitative Glycoproteomics and Antibody-Based Assays on Tissue Specimens. Proteomics 2013, 13, 2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Oostrum, M.; Müller, M.; Klein, F.; Bruderer, R.; Zhang, H.; Pedrioli, P.G.A.; Reiter, L.; Tsapogas, P.; Rolink, A.; Wollscheid, B. Classification of Mouse B Cell Types Using Surfaceome Proteotype Maps. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Bausch-Fluck, D.; Hofmann, A.; Bock, T.; Frei, A.P.; Cerciello, F.; Jacobs, A.; Moest, H.; Omasits, U.; Gundry, R.L.; Yoon, C.; et al. A Mass Spectrometric-Derived Cell Surface Protein Atlas. PLoS ONE 2015, 10, e0121314. [Google Scholar] [CrossRef] [Green Version]
- Garbis, S.; Lubec, G.; Fountoulakis, M. Limitations of Current Proteomics Technologies. J. Chromatogr. A 2005, 1077, 1–18. [Google Scholar] [CrossRef]
- Rabilloud, T.; Chevallet, M.; Luche, S.; Lelong, C. Fully Denaturing Two-Dimensional Electrophoresis of Membrane Proteins: A Critical Update. Proteomics 2008, 8, 3965–3973. [Google Scholar] [CrossRef] [Green Version]
- Olaya-Abril, A.; Jiménez-Munguía, I.; Gómez-Gascón, L.; Rodríguez-Ortega, M.J. Surfomics: Shaving Live Organisms for a Fast Proteomic Identification of Surface Proteins. J. Proteom. 2014, 97, 164–176. [Google Scholar] [CrossRef]
- Zhang, X. Less Is More: Membrane Protein Digestion Beyond Urea-Trypsin Solution for Next-Level Proteomics. Mol. Cell. Proteom. 2015, 14, 2441–2453. [Google Scholar] [CrossRef] [Green Version]
- Manza, L.L.; Stamer, S.L.; Ham, A.J.L.; Codreanu, S.G.; Liebler, D.C. Sample Preparation and Digestion for Proteomic Analyses Using Spin Filters. Proteomics 2005, 5, 1742–1745. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Erde, J.; Loo, R.R.O.; Loo, J.A. Enhanced FASP (EFASP) to Increase Proteome Coverage and Sample Recovery for Quantitative Proteomic Experiments. J. Proteome Res. 2014, 13, 1885–1895. [Google Scholar] [CrossRef]
- Macklin, A.; Khan, S.; Kislinger, T. Recent Advances in Mass Spectrometry Based Clinical Proteomics: Applications to Cancer Research. Clin. Proteom. 2020, 17, 1–25. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, L.; Gunawardena, H.P.; Khatun, J.; Maier, C.; Spitzer, W.; Leerkes, M.; Giddings, M.C.; Chen, X. GOFAST: An Integrated Approach for Efficient and Comprehensive Membrane Proteome Analysis. Anal. Chem. 2012, 84, 9008–9014. [Google Scholar] [CrossRef]
- Raimondo, F.; Corbetta, S.; Savoia, A.; Chinello, C.; Cazzaniga, M.; Rocco, F.; Bosari, S.; Grasso, M.; Bovo, G.; Magni, F.; et al. Comparative Membrane Proteomics: A Technical Advancement in the Search of Renal Cell Carcinoma Biomarkers. Mol. Biosyst. 2015, 11, 1708–1716. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, D.P.; Rawlins, C.M.; DeHart, C.J.; Fornelli, L.; Schachner, L.F.; Lin, Z.; Lippens, J.L.; Aluri, K.C.; Sarin, R.; Chen, B.; et al. Best Practices and Benchmarks for Intact Protein Analysis for Top-down Mass Spectrometry. Nat. Methods 2019, 16, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Konijnenberg, A.; Bannwarth, L.; Yilmaz, D.; Koçer, A.; Venien-Bryan, C.; Sobott, F. Top-down Mass Spectrometry of Intact Membrane Protein Complexes Reveals Oligomeric State and Sequence Information in a Single Experiment. Protein Sci. 2015, 24, 1292–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konijnenberg, A.; Yilmaz, D.; Ingólfsson, H.I.; Dimitrova, A.; Marrink, S.J.; Li, Z.; Vénien-Bryan, C.; Sobott, F.; Kocer, A. Global Structural Changes of an Ion Channel during Its Gating Are Followed by Ion Mobility Mass Spectrometry. Proc. Natl. Acad. Sci. USA 2014, 111, 17170–17175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laganowsky, A.; Reading, E.; Hopper, J.T.S.; Robinson, C.V. Mass Spectrometry of Intact Membrane Protein Complexes. Nat. Protoc. 2013, 8, 639–651. [Google Scholar] [CrossRef]
- Delcourt, V.; Franck, J.; Leblanc, E.; Narducci, F.; Robin, Y.M.; Gimeno, J.P.; Quanico, J.; Wisztorski, M.; Kobeissy, F.; Jacques, J.F.; et al. Combined Mass Spectrometry Imaging and Top-down Microproteomics Reveals Evidence of a Hidden Proteome in Ovarian Cancer. EBioMedicine 2017, 21, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erady, C.; Boxall, A.; Puntambekar, S.; Suhas Jagannathan, N.; Chauhan, R.; Chong, D.; Meena, N.; Kulkarni, A.; Kasabe, B.; Prathivadi Bhayankaram, K.; et al. Pan-Cancer Analysis of Transcripts Encoding Novel Open-Reading Frames (NORFs) and Their Potential Biological Functions. npj Genom. Med. 2021, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Whitelegge, J.P.; Zhang, H.; Aguilera, R.; Taylor, R.M.; Cramer, W.A. Full Subunit Coverage Liquid Chromatography Electrospray Ionization Mass Spectrometry (LCMS+) of an Oligomeric Membrane Protein: Cytochrome B6f Complex From Spinach and the Cyanobacterium Mastigocladus Laminosus. Mol. Cell. Proteom. 2002, 1, 816–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, U.K.; Simonian, M.; Whitelegge, J.P. Integral Membrane Proteins: Bottom-up, Top-down and Structural Proteomics. Expert. Rev. Proteom. 2017, 14, 715. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Fearnley, I.M.; Walker, J.E. Definition of the Mitochondrial Proteome by Measurement of Molecular Masses of Membrane Proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 16170. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.A.; Tucholski, T.; Alpert, A.J.; Eken, C.; Wesemann, L.; Kyrvasilis, A.; Jin, S.; Ge, Y. Top-Down Proteomics of Endogenous Membrane Proteins Enabled by Cloud Point Enrichment and Multidimensional Liquid Chromatography-Mass Spectrometry. Anal. Chem. 2020, 92, 15726–15735. [Google Scholar] [CrossRef]
- Carroll, J.; Altman, M.C.; Fearnley, I.M.; Walker, J.E. Identification of Membrane Proteins by Tandem Mass Spectrometry of Protein Ions. Proc. Natl. Acad. Sci. USA 2007, 104, 14330–14335. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Brown, K.A.; Lin, Z.; Ge, Y. Top-Down Proteomics: Ready for Prime Time? Anal. Chem. 2018, 90, 110–127. [Google Scholar] [CrossRef]
- Jeong, K.; Kim, J.; Gaikwad, M.; Hidayah, S.N.; Heikaus, L.; Schlüter, H.; Kohlbacher, O. FLASHDeconv: Ultrafast, High-Quality Feature Deconvolution for Top-Down Proteomics. Cell. Syst. 2020, 10, 213–218.e6. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.D.; Liu, X.; Payne, S.H.; et al. Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, e1800361. [Google Scholar] [CrossRef]
- Wu, Z.; Roberts, D.S.; Melby, J.A.; Wenger, K.; Wetzel, M.; Gu, Y.; Ramanathan, S.G.; Bayne, E.F.; Liu, X.; Sun, R.; et al. MASH Explorer: A Universal Software Environment for Top-Down Proteomics. J. Proteome Res. 2020, 19, 3867–3876. [Google Scholar] [CrossRef]
- Brodin, P. The Biology of the Cell - Insights from Mass Cytometry. FEBS J. 2019, 286, 1514–1522. [Google Scholar] [CrossRef]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass Cytometry: Technique for Real Time Single Cell Multitarget Immunoassay Based on Inductively Coupled Plasma Time-of-Flight Mass Spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef]
- Zhang, T.; Warden, A.R.; Li, Y.; Ding, X. Progress and Applications of Mass Cytometry in Sketching Immune Landscapes. Clin. Transl. Med. 2020, 10. [Google Scholar] [CrossRef]
- Gadalla, R.; Noamani, B.; MacLeod, B.L.; Dickson, R.J.; Guo, M.; Xu, W.; Lukhele, S.; Elsaesser, H.J.; Razak, A.R.A.; Hirano, N.; et al. Validation of CyTOF against Flow Cytometry for Immunological Studies and Monitoring of Human Cancer Clinical Trials. Front. Oncol. 2019, 9, 415. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yan, S.; Lin, B.; Shi, Q.; Lu, Y. Single-Cell Proteomics for Cancer Immunotherapy. Adv. Cancer Res. 2018, 139, 185–207. [Google Scholar] [CrossRef]
- Iyer, A.; Hamers, A.A.J.; Pillai, A.B. CyTOF® for the Masses. Front. Immunol. 2022, 13, 1636. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780. [Google Scholar] [CrossRef] [Green Version]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA Aptamers Using Cell-SELEX. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef]
- Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z.C.; Chen, H.W.; Mallikaratchy, P.; Sefah, K.; Yang, C.J.; Tan, W. Aptamers Evolved from Live Cells as Effective Molecular Probes for Cancer Study. Proc. Natl. Acad. Sci. USA 2006, 103, 11838–11843. [Google Scholar] [CrossRef] [Green Version]
- Shigdar, S.; Agnello, L.; Fedele, M.; Camorani, S.; Cerchia, L. Profiling Cancer Cells by Cell-SELEX: Use of Aptamers for Discovery of Actionable Biomarkers and Therapeutic Applications Thereof. Pharmaceutics 2021, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, D.; Cao, Z.C.; Li, Y.; Tan, W. Aptamers Evolved from Cultured Cancer Cells Reveal Molecular Differences of Cancer Cells in Patient Samples. Clin. Chem. 2007, 53, 1153–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shangguan, D.; Cao, Z.; Meng, L.; Mallikaratchy, P.; Sefah, K.; Wang, H.; Li, Y.; Tan, W. Cell-Specific Aptamer Probes for Membrane Protein Elucidation in Cancer Cells. J. Proteome Res. 2008, 7, 2133–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicco, E.; Baez, J.; Ibarra, M.; Fernández, M.; Cabral, P.; Moreno, M.; Cerecetto, H.; Calzada, V. Sgc8-c Aptamer as a Potential Theranostic Agent for Hemato-Oncological Malignancies. Cancer Biother Radiopharm 2020, 35, 262–270. [Google Scholar] [CrossRef]
- Jia, W.; Ren, C.; Wang, L.; Zhu, B.; Jia, W.; Gao, M.; Zeng, F.; Zeng, L.; Xia, X.; Zhang, X.; et al. CD109 Is Identified as a Potential Nasopharyngeal Carcinoma Biomarker Using Aptamer Selected by Cell-SELEX. Oncotarget 2016, 7, 55328–55342. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Jiang, X.; Chen, Y.; Guo, Q.; Wang, K.; Meng, X.; Huang, Z.; Wen, X. Metastatic Cancer Cell and Tissue-Specific Fluorescence Imaging Using a New DNA Aptamer Developed by Cell-SELEX. Talanta 2017, 170, 56–62. [Google Scholar] [CrossRef]
- Li, W.M.; Zhou, L.L.; Zheng, M.; Fang, J. Selection of Metastatic Breast Cancer Cell-Specific Aptamers for the Capture of CTCs with a Metastatic Phenotype by Cell-SELEX. Mol. Ther. Nucleic Acids 2018, 12, 707. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Liu, L.; Zhu, Z.; Ouyang, G.; An, Y.; Zhao, C.; Yang, C.J. In Vitro Selection of DNA Aptamers for Metastatic Breast Cancer Cell Recognition and Tissue Imaging. Anal. Chem. 2014, 86, 6596–6603. [Google Scholar] [CrossRef]
- Wang, L.; Li, P.; Xiao, X.; Li, J.; Li, J.; Yang, H.H.; Tan, W. Generating Lung-Metastatic Osteosarcoma Targeting Aptamers for in Vivo and Clinical Tissue Imaging. Talanta 2018, 188, 66–73. [Google Scholar] [CrossRef]
- Duan, M.; Long, Y.; Yang, C.; Wu, X.; Sun, Y.; Li, J.; Hu, X.; Lin, W.; Han, D.; Zhao, Y.; et al. Selection and Characterization of DNA Aptamer for Metastatic Prostate Cancer Recognition and Tissue Imaging. Oncotarget 2016, 7, 36436–36446. [Google Scholar] [CrossRef]
- Speransky, S.; Serafini, P.; Caroli, J.; Bicciato, S.; Lippman, M.E.; Bishopric, N.H. A Novel RNA Aptamer Identifies Plasma Membrane ATP Synthase Beta Subunit as an Early Marker and Therapeutic Target in Aggressive Cancer. Breast Cancer Res. Treat. 2019, 176, 271–289. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.B.; Rong, Y.; Fang, M.; Yuan, J.P.; Peng, C.W.; Liu, S.P.; Li, Y. Recognition and Capture of Metastatic Hepatocellular Carcinoma Cells Using Aptamer-Conjugated Quantum Dots and Magnetic Particles. Biomaterials 2013, 34, 3816–3827. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yuan, C.H.; Yang, Y.F.; Yin, C.Q.; Guan, Q.; Wang, F.B.; Tu, J.C. Subtractive Cell-SELEX Selection of DNA Aptamers Binding Specifically and Selectively to Hepatocellular Carcinoma Cells with High Metastatic Potential. Biomed Res. Int. 2016, 2016, 5735869. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; Chen, H.; Zhou, X.F.; Yin, C.Q.; Wang, B.C.; Peng, C.W.; Liu, S.P.; Wang, F.B. Identification of an Aptamer through Whole Cell-SELEX for Targeting High Metastatic Liver Cancers. Oncotarget 2016, 7, 8282–8294. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; An, Y.; Jin, J.; Zhu, Z.; Hao, L.; Liu, L.; Shi, Y.; Fan, D.; Ji, T.; Yang, C.J. Evolution of DNA Aptamers through in Vitro Metastatic-Cell-Based Systematic Evolution of Ligands by Exponential Enrichment for Metastatic Cancer Recognition and Imaging. Anal. Chem. 2015, 87, 4941–4948. [Google Scholar] [CrossRef]
- Li, W.M.; Bing, T.; Wei, J.Y.; Chen, Z.Z.; Shangguan, D.H.; Fang, J. Cell-SELEX-Based Selection of Aptamers That Recognize Distinct Targets on Metastatic Colorectal Cancer Cells. Biomaterials 2014, 35, 6998–7007. [Google Scholar] [CrossRef]
- Ding, M.; Clark, R.; Bardelle, C.; Backmark, A.; Norris, T.; Williams, W.; Wigglesworth, M.; Howes, R. Application of High-Throughput Flow Cytometry in Early Drug Discovery: An AstraZeneca Perspective. SLAS Discov. 2018, 23, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Herold, D.A. Evolving Platforms for Clinical Mass Spectrometry. In Mass Spectrometry for the Clinical Laboratory; Academic Press: Cambridge, MA, USA, 2017; pp. 261–276. [Google Scholar] [CrossRef]
- Bendall, S.C.; Simonds, E.F.; Qiu, P.; Amir, E.A.D.; Krutzik, P.O.; Finck, R.; Bruggner, R.V.; Melamed, R.; Trejo, A.; Ornatsky, O.I.; et al. Single-Cell Mass Cytometry of Differential Immune and Drug Responses Across a Human Hematopoietic Continuum. Science 2011, 332, 687. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.M.; Fu, T.; Zhang, X.B.; Tan, W. Cell-SELEX-Based Aptamer-Conjugated Nanomaterials for Cancer Diagnosis and Therapy. Natl. Sci. Rev. 2015, 2, 71–84. [Google Scholar] [CrossRef]
- Chen, M.; Yu, Y.; Jiang, F.; Zhou, J.; Li, Y.; Liang, C.; Dang, L.; Lu, A.; Zhang, G. Development of Cell-SELEX Technology and Its Application in Cancer Diagnosis and Therapy. Int. J. Mol. Sci. 2016, 17, 2079. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Crosby, D.; Bhatia, S.; Brindle, K.M.; Coussens, L.M.; Dive, C.; Emberton, M.; Esener, S.; Fitzgerald, R.C.; Gambhir, S.S.; Kuhn, P.; et al. Early Detection of Cancer. Science 2022, 375. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sáez, O.; Prat, A. Current and Future Management of HER2-Positive Metastatic Breast Cancer. JCO Oncol. Pract. 2021, 17, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Dormann, C. Metastatic Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer: Current Treatment Standards and Future Perspectives. Breast Care 2020, 15, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Klempner, S.J.; Chao, J. Progress and Challenges in HER2-Positive Gastroesophageal Adenocarcinoma. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Bang, Y.J.; Feng-yi, F.; Xu, J.M.; Lee, K.W.; Jiao, S.C.; Chong, J.L.; López-Sanchez, R.I.; Price, T.; Gladkov, O.; et al. HER2 Screening Data from ToGA: Targeting HER2 in Gastric and Gastroesophageal Junction Cancer. Gastric Cancer 2015, 18, 476–484. [Google Scholar] [CrossRef]
- Riudavets, M.; Sullivan, I.; Abdayem, P.; Planchard, D. Targeting HER2 in Non-Small-Cell Lung Cancer (NSCLC): A Glimpse of Hope? An Updated Review on Therapeutic Strategies in NSCLC Harbouring HER2 Alterations. ESMO Open 2021, 6. [Google Scholar] [CrossRef]
- Zhao, J.; Xia, Y. Targeting HER2 Alterations in Non-Small-Cell Lung Cancer: A Comprehensive Review. JCO Precis. Oncol. 2020, 4, 411–425. [Google Scholar] [CrossRef]
- La Salvia, A.; Lopez-Gomez, V.; Garcia-Carbonero, R. HER2-Targeted Therapy: An Emerging Strategy in Advanced Colorectal Cancer. Expert. Opin. Investig. Drugs 2019, 28, 29–38. [Google Scholar] [CrossRef]
- Greally, M.; Kelly, C.M.; Cercek, A. HER2: An Emerging Target in Colorectal Cancer. Curr. Probl. Cancer 2018, 42, 560–571. [Google Scholar] [CrossRef]
- Brückl, W.M.; Reck, M.; Griesinger, F.; Schäfer, H.; Kortsik, C.; Gaska, T.; Rawluk, J.; Krüger, S.; Kokowski, K.; Budweiser, S.; et al. Afatinib as First-Line Treatment in Patients with EGFR-Mutated Non-Small Cell Lung Cancer in Routine Clinical Practice. Ther. Adv. Med. Oncol. 2021, 13, 17588359211012361. [Google Scholar] [CrossRef]
- Harvey, R.D.; Adams, V.R.; Beardslee, T.; Medina, P. Afatinib for the Treatment of EGFR Mutation-Positive NSCLC: A Review of Clinical Findings. J. Oncol. Pharm. Pract. 2020, 26, 1461–1474. [Google Scholar] [CrossRef]
- Riedesser, J.E.; Ebert, M.P.; Betge, J. Precision Medicine for Metastatic Colorectal Cancer in Clinical Practice. Ther. Adv. Med. Oncol. 2022, 14, 175883592110727. [Google Scholar] [CrossRef]
- Gómez-España, M.A.; Gallego, J.; González-Flores, E.; Maurel, J.; Páez, D.; Sastre, J.; Aparicio, J.; Benavides, M.; Feliu, J.; Vera, R. SEOM Clinical Guidelines for Diagnosis and Treatment of Metastatic Colorectal Cancer (2018). Clin. Transl. Oncol. 2019, 21, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Dervenis, C.; Xynos, E.; Sotiropoulos, G.; Gouvas, N.; Boukovinas, I.; Agalianos, C.; Androulakis, N.; Athanasiadis, A.; Christodoulou, C.; Chrysou, E.; et al. Clinical Practice Guidelines for the Management of Metastatic Colorectal Cancer: A Consensus Statement of the Hellenic Society of Medical Oncologists (HeSMO). Ann. Gastroenterol. 2016, 29, 390–416. [Google Scholar] [CrossRef]
- Labianca, R.; Nordlinger, B.; Beretta, G.D.; Mosconi, S.; Mandalà, M.; Cervantes, A.; Arnold, D. Early Colon Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2013, 24 (Suppl. 6), vi64-72. [Google Scholar] [CrossRef]
- Jiao, Z.; Cao, S.; Li, J.; Hu, N.; Gong, Y.; Wang, L.; Jin, S. Clinical Associations of Preoperative and Postoperative Serum CEA and Lung Cancer Outcome. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- Arrieta, O.; Villarreal-Garza, C.; Martínez-Barrera, L.; Morales, M.; Dorantes-Gallareta, Y.; Peña-Curiel, O.; Contreras-Reyes, S.; Macedo-Pérez, E.O.; Alatorre-Alexander, J. Usefulness of Serum Carcinoembryonic Antigen (CEA) in Evaluating Response to Chemotherapy in Patients with Advanced Non Small-Cell Lung Cancer: A Prospective Cohort Study. BMC Cancer 2013, 13. [Google Scholar] [CrossRef] [Green Version]
- Xing, H.; Wang, J.; Wang, Y.; Tong, M.; Hu, H.; Huang, C.; Li, D. Diagnostic Value of CA 19-9 and Carcinoembryonic Antigen for Pancreatic Cancer: A Meta-Analysis. Gastroenterol. Res. Pract. 2018, 2018, 8704751. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Xu, W.; Ji, S.; Zhang, B.; Ni, Q.; Xu, J.; Yu, X. Diagnostic and Prognostic Value of Carcinoembryonic Antigen in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Onco. Targets Ther. 2017, 10, 4591–4598. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, A.; Reuben, J.M.; Lanza, F.; Anfossi, S.; Cappelletti, M.R.; Zanotti, L.; Gobbi, A.; Senti, C.; Brambilla, P.; Milani, M.; et al. Breast Cancer Circulating Biomarkers: Advantages, Drawbacks, and New Insights. Tumour Biol. 2015, 36, 6653–6665. [Google Scholar] [CrossRef] [PubMed]
- Chourin, S.; Georgescu, D.; Gray, C.; Guillemet, C.; Loeb, A.; Veyret, C.; Basuyau, J.P. Value of CA 15-3 Determination in the Initial Management of Breast Cancer Patients. Ann. Oncol. 2009, 20, 962–964. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Koh, H.K.; Chie, E.K.; Oh, D.Y.; Bang, Y.J.; Nam, E.M.; Kim, K. Change in Carbohydrate Antigen 19-9 Level as a Prognostic Marker of Overall Survival in Locally Advanced Pancreatic Cancer Treated with Concurrent Chemoradiotherapy. Int. J. Clin. Oncol. 2017, 22, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Stiksma, J.; Grootendorst, D.C.; Van Der Linden, P.W.G. CA 19-9 as a Marker in Addition to CEA to Monitor Colorectal Cancer. Clin. Colorectal Cancer 2014, 13, 239–244. [Google Scholar] [CrossRef]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. CA125 and Ovarian Cancer: A Comprehensive Review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef]
- Funston, G.; Van Melle, M.; Baun, M.L.L.; Jensen, H.; Helsper, C.; Emery, J.; Crosbie, E.J.; Thompson, M.; Hamilton, W.; Walter, F.M. Variation in the Initial Assessment and Investigation for Ovarian Cancer in Symptomatic Women: A Systematic Review of International Guidelines. BMC Cancer 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Mckertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging New Therapeutic Antibody Derivatives for Cancer Treatment. Signal Transduct. Target. Ther. 2022, 7, 1–28. [Google Scholar] [CrossRef]
- Karcini, A.; Lazar, I.M. The SKBR3 Cell-Membrane Proteome Reveals Telltales of Aberrant Cancer Cell Proliferation and Targets for Precision Medicine Applications. Sci. Rep. 2022, 12, 10847. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Jong, E.; Kocer, A. Current Methods for Identifying Plasma Membrane Proteins as Cancer Biomarkers. Membranes 2023, 13, 409. https://doi.org/10.3390/membranes13040409
de Jong E, Kocer A. Current Methods for Identifying Plasma Membrane Proteins as Cancer Biomarkers. Membranes. 2023; 13(4):409. https://doi.org/10.3390/membranes13040409
Chicago/Turabian Stylede Jong, Edwin, and Armagan Kocer. 2023. "Current Methods for Identifying Plasma Membrane Proteins as Cancer Biomarkers" Membranes 13, no. 4: 409. https://doi.org/10.3390/membranes13040409