

Interfacial Enzymes Enable Gram-Positive Microbes to Eat Fatty Acids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Protein–Membrane Association
1.2. Interfacial Enzymes
1.3. Membrane Binding Experimental Methods
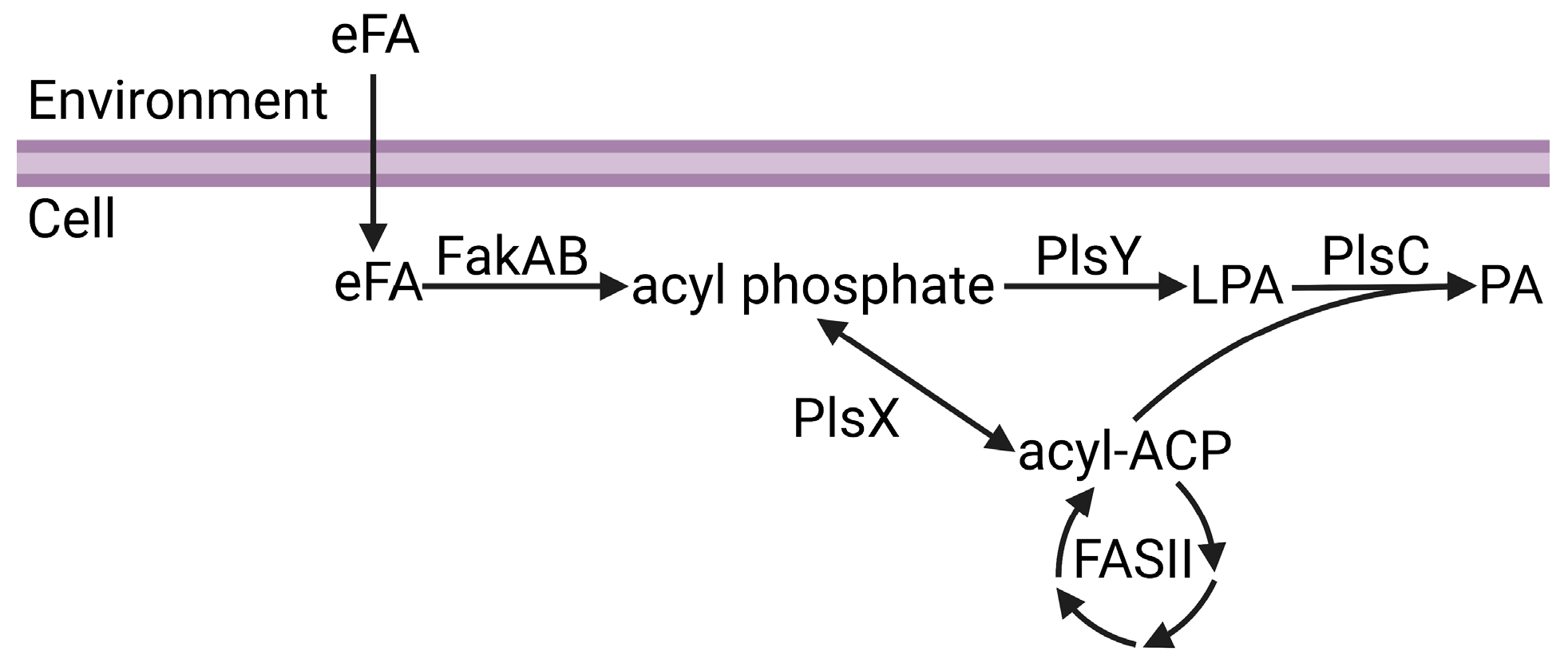
2. Bacterial Fatty Acid Metabolism
2.1. Bacterial Phospholipid Synthesis
2.2. Acyl-ACP:Phosphate Transacylase (PlsX)
2.3. Fatty Acid Kinase (FakAB)
3. Conclusions
4. Discussion
Funding
Data Availability Statement
Conflicts of Interest
References
- Strahl, H.; Errington, J. Bacterial membranes: Structure, domains, and function. Annu. Rev. Microbiol. 2017, 71, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Xu, Y.; Zhang, K.; Xiong, Y.; Li, H.; Cui, L.; Wang, X.; Lou, J.; Zhai, Y.; Sun, F.; et al. Crystal structure of E. coli apolipoprotein N-acyl transferase. Nat. Commun. 2017, 8, 15948. [Google Scholar] [CrossRef]
- Gardiner, J.H.t.; Komazin, G.; Matsuo, M.; Cole, K.; Gotz, F.; Meredith, T.C. Lipoprotein N-Acylation in Staphylococcus aureus Is Catalyzed by a Two-Component Acyl Transferase System. mBio 2020, 11, 01619–01620. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Alonzo, F., 3rd. Bacterial lipolysis of immune-activating ligands promotes evasion of innate defenses. Proc. Natl. Acad. Sci. USA 2019, 116, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelig, J. Thermodynamics of lipid-peptide interactions. Biochim. Biophys. Acta 2004, 1666, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whited, A.M.; Johs, A. The interactions of peripheral membrane proteins with biological membranes. Chem. Phys. Lipids 2015, 192, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmberg, N.J.; Van Buskirk, D.R.; Falke, J.J. Membrane-docking loops of the cPLA2 C2 domain: Detailed structural analysis of the protein-membrane interface via site-directed spin-labeling. Biochemistry 2003, 42, 13227–13240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamsjaeger, R.; Johs, A.; Gries, A.; Gruber, H.J.; Romanin, C.; Prassl, R.; Hinterdorfer, P. Membrane binding of β2-glycoprotein I can be described by a two-state reaction model: An atomic force microscopy and surface plasmon resonance study. Biochem. J. 2005, 389, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Verma, M.L.; Azmi, W.; Kanwar, S.S. Microbial lipases: At the interface of aqueous and non-aqueous media. A review. Acta Microbiol. Immunol. Hung. 2008, 55, 265–294. [Google Scholar] [CrossRef] [Green Version]
- Del Vecchio, K.; Stahelin, R.V. Using Surface Plasmon Resonance to Quantitatively Assess Lipid-Protein Interactions. Methods Mol. Biol. 2016, 1376, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Sahu, I.D.; Lorigan, G.A. Electron paramagnetic resonance as a tool for studying membrane proteins. Biomolecules 2020, 10, 763. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, T.I.; Smirnov, A.I. Peptide-membrane interactions by spin-labeling EPR. Methods Enzymol. 2015, 564, 219–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opella, S.J.; Marassi, F.M. Applications of NMR to membrane proteins. Arch. Biochem. Biophys. 2017, 628, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Oxenoid, K.; Chou, J.J. A functional NMR for membrane proteins: Dynamics, ligand binding, and allosteric modulation. Protein Sci. 2016, 25, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Pant, S.; Tajkhorshid, E. Microscopic characterization of GRP1 PH domain interaction with anionic membranes. J. Comput. Chem. 2020, 41, 489–499. [Google Scholar] [CrossRef]
- Gullett, J.M.; Cuypers, M.G.; Grace, C.R.; Pant, S.; Subramanian, C.; Tajkhorshid, E.; Rock, C.O.; White, S.W. Identification of structural transitions in bacterial fatty acid binding proteins that permit ligand entry and exit at membranes. J. Biol. Chem. 2022, 298, 101676. [Google Scholar] [CrossRef]
- Parsons, J.B.; Rock, C.O. Bacterial lipids: Metabolism and membrane homeostasis. Prog. Lipid Res. 2013, 52, 249–276. [Google Scholar] [CrossRef] [Green Version]
- White, S.W.; Zheng, J.; Zhang, Y.-M.; Rock, C.O. The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 2005, 74, 791–831. [Google Scholar] [CrossRef]
- Radka, C.D.; Rock, C.O. Mining fatty acid biosynthesis for new antimicrobials. Annu. Rev. Microbiol. 2022, 76, 281–304. [Google Scholar] [CrossRef]
- Wittke, F.; Vincent, C.; Chen, J.; Heller, B.; Kabler, H.; Overcash, J.S.; Leylavergne, F.; Dieppois, G. Afabicin, a first-in-class antistaphylococcal antibiotic, in the treatment of acute bacterial skin and skin structure infections: Clinical noninferiority to vancomycin/linezolid. Antimicrob. Agents Chemother. 2020, 64, e00250-20. [Google Scholar] [CrossRef]
- Vuong, C.; Yeh, A.J.; Cheung, G.Y.; Otto, M. Investigational drugs to treat methicillin-resistant Staphylococcus aureus. Expert Opin. Investig. Drugs 2016, 25, 73–93. [Google Scholar] [CrossRef] [Green Version]
- Quehenberger, O.; Dennis, E.A. The human plasma lipidome. N. Engl. J. Med. 2011, 365, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Radka, C.D.; Batte, J.L.; Frank, M.W.; Rosch, J.W.; Rock, C.O. Oleate hydratase (OhyA) is a virulence determinant in Staphylococcus aureus. Microbiol. Spectr. 2021, 9, e0154621. [Google Scholar] [CrossRef]
- Brinster, S.; Lamberet, G.; Staels, B.; Trieu-Cuot, P.; Gruss, A.; Poyart, C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 2009, 458, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Morvan, C.; Halpern, D.; Kenanian, G.; Pathania, A.; Anba-Mondoloni, J.; Lamberet, G.; Gruss, A.; Gloux, K. The Staphylococcus aureus FASII bypass escape route from FASII inhibitors. Biochimie 2017, 141, 40–46. [Google Scholar] [CrossRef]
- Kenanian, G.; Morvan, C.; Weckel, A.; Pathania, A.; Anba-Mondoloni, J.; Halpern, D.; Gaillard, M.; Solgadi, A.; Dupont, L.; Henry, C.; et al. Permissive fatty acid incorporation promotes staphylococcal adaptation to FASII antibiotics in host environments. Cell Rep. 2019, 29, 3974–3982.e3974. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Lounis, N.; Gilissen, R.; Guillemont, J.; Simmen, K.; Andries, K.; Koul, A. Essentiality of FASII pathway for Staphylococcus aureus. Nature 2010, 463, E3, discussion E4. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Lu, Y.-J.; Zhang, Y.-M.; Grimes, K.D.; Qi, J.; Lee, R.E.; Rock, C.O. Acyl-phosphates initiate membrane phospholipid synthesis in Gram-positive pathogens. Mol. Cell 2006, 23, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Rock, C.O. Phosphatidic acid synthesis in bacteria. Biochim. Biophys. Acta 2013, 1831, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoletti, L.; Lu, Y.-J.; Schujman, G.E.; de Mendoza, D.; Rock, C.O. Coupling of fatty acid and phospholipid synthesis in Bacillus subtilis. J. Bacteriol. 2007, 189, 5816–5824. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-M.; Rock, C.O. Acyltransferases in bacterial glycerophospholipid synthesis. J. Lipid Res 2008, 49, 1867–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.B.; Broussard, T.C.; Bose, J.L.; Rosch, J.W.; Jackson, P.; Subramanian, C.; Rock, C.O. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2014, 111, 10532–10537. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Tang, Y.; Wu, Y.; Zhao, S.; Bao, J.; Luo, Y.; Li, D. Structural insights into the committed step of bacterial phospholipid biosynthesis. Nat. Commun. 2017, 8, 1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.M.; Yao, J.; Gajewski, S.; Kumar, G.; Martin, E.W.; Rock, C.O.; White, S.W. A two-helix motif positions the lysophosphatidic acid acyltransferase active site for catalysis within the membrane bilayer. Nat. Struct. Mol. Biol. 2017, 24, 666–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Dai, X.; Qin, M.; Guo, Z. Identification of an amphipathic peptide sensor of the Bacillus subtilis fluid membrane microdomains. Commun. Biol. 2019, 2, 316. [Google Scholar] [CrossRef] [Green Version]
- Sastre, D.E.; Bisson-Filho, A.; de Mendoza, D.; Gueiros-Filho, F.J. Revisiting the cell biology of the acyl-ACP: Phosphate transacylase PlsX suggests that the phospholipid synthesis and cell division machineries are not coupled in Bacillus subtilis. Mol. Microbiol. 2016, 100, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Badger, J.; Sauder, J.M.; Adams, J.M.; Antonysamy, S.; Bain, K.; Bergseid, M.G.; Buchanan, S.G.; Buchanan, M.D.; Batiyenko, Y.; Christopher, J.A.; et al. Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins 2005, 60, 787–796. [Google Scholar] [CrossRef]
- Kim, Y.; Li, H.; Binkowski, T.A.; Holzle, D.; Joachimiak, A. Crystal structure of fatty acid/phospholipid synthesis protein PlsX from Enterococcus faecalis. J. Struct. Funct. Genomics 2009, 10, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre, D.E.; Pulschen, A.A.; Basso, L.G.M.; Benites Pariente, J.S.; Marques Netto, C.G.C.; Machinandiarena, F.; Albanesi, D.; Navarro, M.; de Mendoza, D.; Gueiros-Filho, F.J. The phosphatidic acid pathway enzyme PlsX plays both catalytic and channeling roles in bacterial phospholipid synthesis. J. Biol. Chem. 2020, 295, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Sastre, D.E.; Basso, L.G.M.; Trastoy, B.; Cifuente, J.O.; Contreras, X.; Gueiros-Filho, F.; de Mendoza, D.; Navarro, M.; Guerin, M.E. Membrane fluidity adjusts the insertion of the transacylase PlsX to regulate phospholipid biosynthesis in Gram-positive bacteria. J. Biol. Chem. 2020, 295, 2136–2147. [Google Scholar] [CrossRef]
- Subramanian, C.; Cuypers, M.G.; Radka, C.D.; White, S.W.; Rock, C.O. Domain architecture and catalysis of the Staphylococcus aureus fatty acid kinase. J. Biol. Chem. 2022, 298, 101993. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zang, N.; Lou, N.; Xu, Y.; Sun, J.; Huang, M.; Zhang, H.; Lu, H.; Zhou, C.; Feng, Y. Structure and mechanism for streptococcal fatty acid kinase (Fak) system dedicated to host fatty acid scavenging. Sci. Adv. 2022, 8, eabq3944. [Google Scholar] [CrossRef] [PubMed]
- Gullett, J.M.; Cuypers, M.G.; Frank, M.W.; White, S.W.; Rock, C.O. A fatty acid-binding protein of Streptococcus pneumoniae facilitates the acquisition of host polyunsaturated fatty acids. J. Biol. Chem. 2019, 294, 16416–16428. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, M.G.; Subramanian, C.; Gullett, J.M.; Frank, M.W.; White, S.W.; Rock, C.O. Acyl chain selectivity and physiological roles of Staphylococcus aureus fatty acid binding proteins. J. Biol. Chem. 2019, 294, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Broussard, T.C.; Miller, D.J.; Jackson, P.; Nourse, A.; White, S.W.; Rock, C.O. Biochemical roles for conserved residues in the bacterial fatty acid binding protein family. J. Biol. Chem. 2016, 291, 6292–6303. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, N.; Stahelin, R.V.; Langlois, R.E.; Cho, W.; Lu, H. Structural bioinformatics prediction of membrane-binding proteins. J. Mol. Biol. 2006, 359, 486–495. [Google Scholar] [CrossRef] [Green Version]
- Chatzigoulas, A.; Cournia, Z. Predicting protein-membrane interfaces of peripheral membrane proteins using ensemble machine learning. Brief. Bioinform. 2022, 23, bbab518. [Google Scholar] [CrossRef]
- Chatzigoulas, A.; Cournia, Z. DREAMM: A web-based server for drugging protein-membrane interfaces as a novel workflow for targeted drug design. Bioinformatics 2022, 38, 5449–5451. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radka, C.D. Interfacial Enzymes Enable Gram-Positive Microbes to Eat Fatty Acids. Membranes 2023, 13, 423. https://doi.org/10.3390/membranes13040423
Radka CD. Interfacial Enzymes Enable Gram-Positive Microbes to Eat Fatty Acids. Membranes. 2023; 13(4):423. https://doi.org/10.3390/membranes13040423
Chicago/Turabian StyleRadka, Christopher D. 2023. "Interfacial Enzymes Enable Gram-Positive Microbes to Eat Fatty Acids" Membranes 13, no. 4: 423. https://doi.org/10.3390/membranes13040423
APA StyleRadka, C. D. (2023). Interfacial Enzymes Enable Gram-Positive Microbes to Eat Fatty Acids. Membranes, 13(4), 423. https://doi.org/10.3390/membranes13040423