
An Electrochemistry and Computational Study at an Electrified Liquid–Liquid Interface for Studying Beta-Amyloid Aggregation




Abstract
:1. Introduction
2. Experimental Section
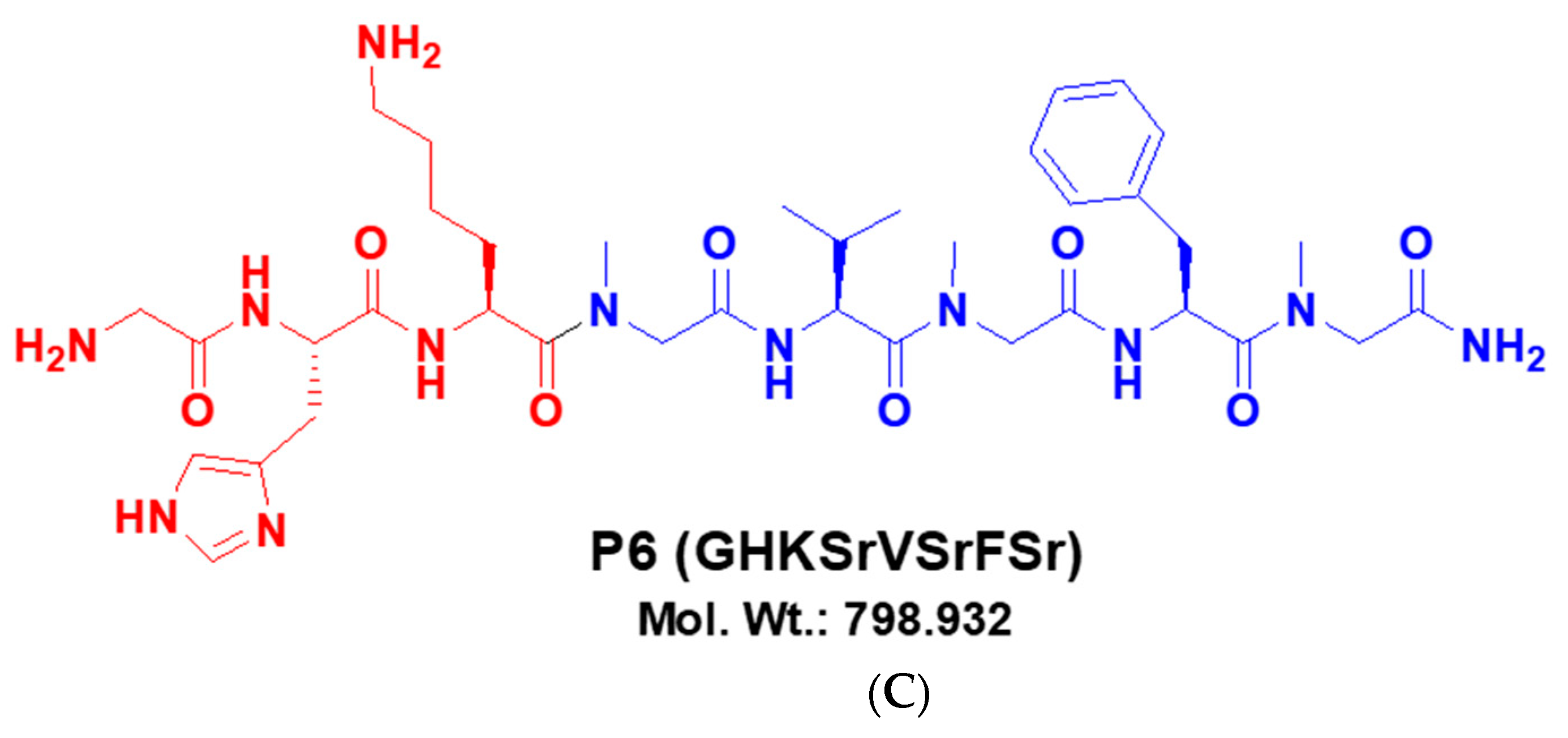
2.1. Reagents and Chemicals
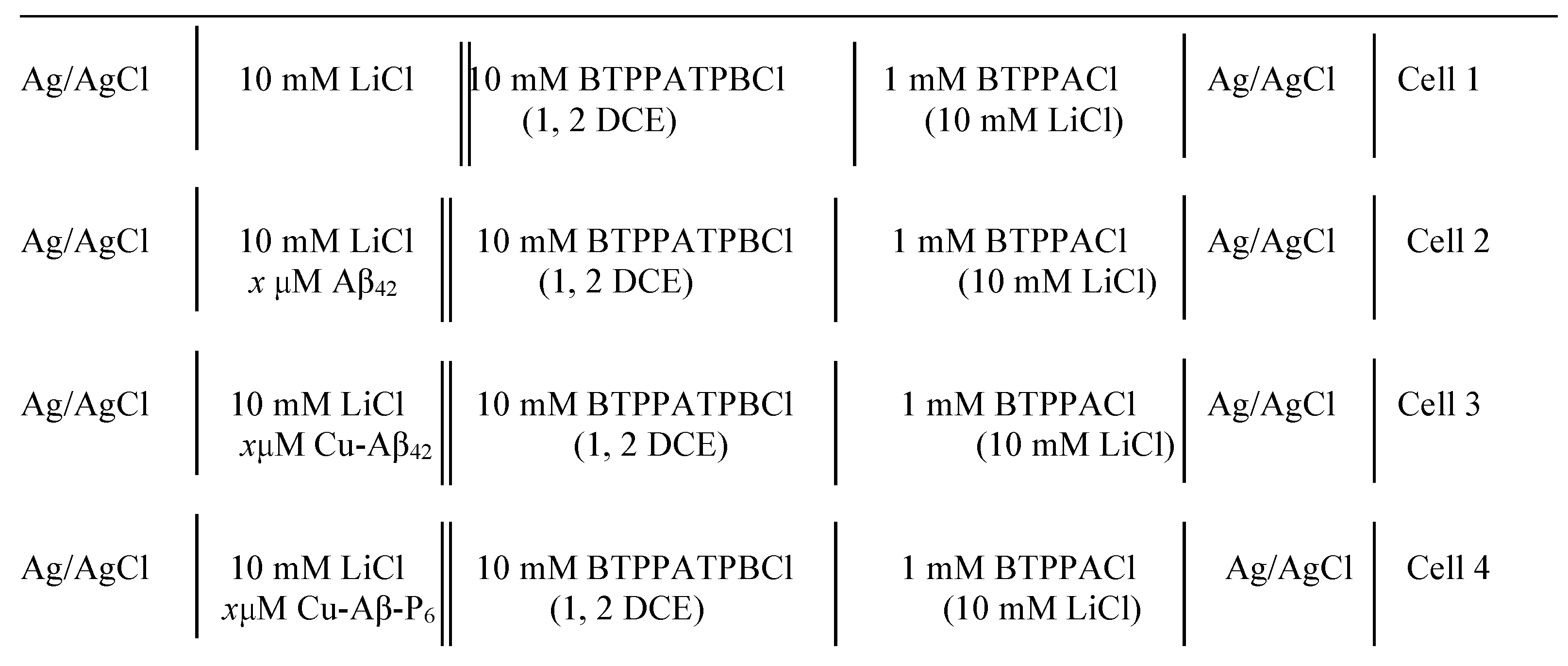
2.2. Electrochemical
2.3. Computational
3. Results and Discussion
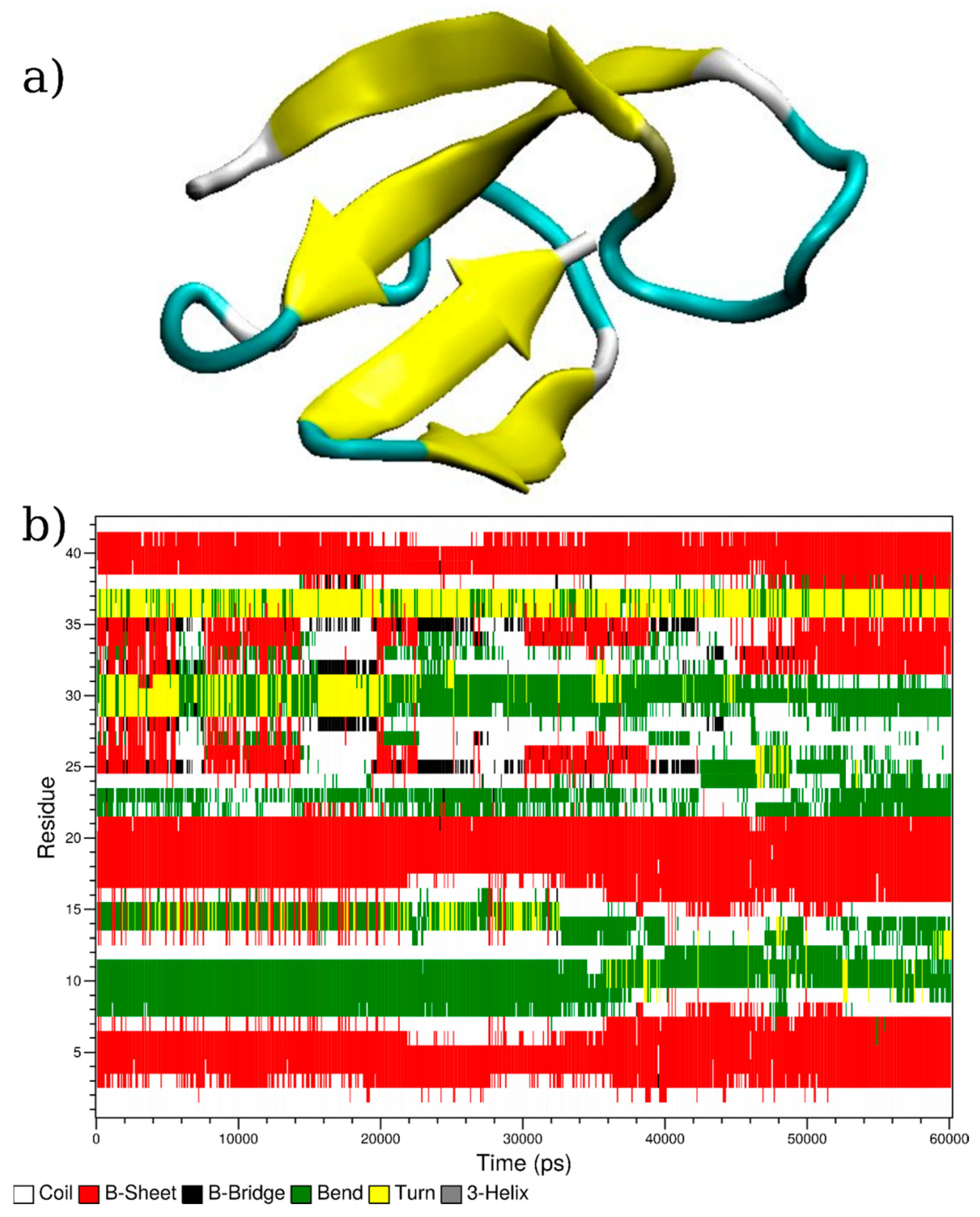
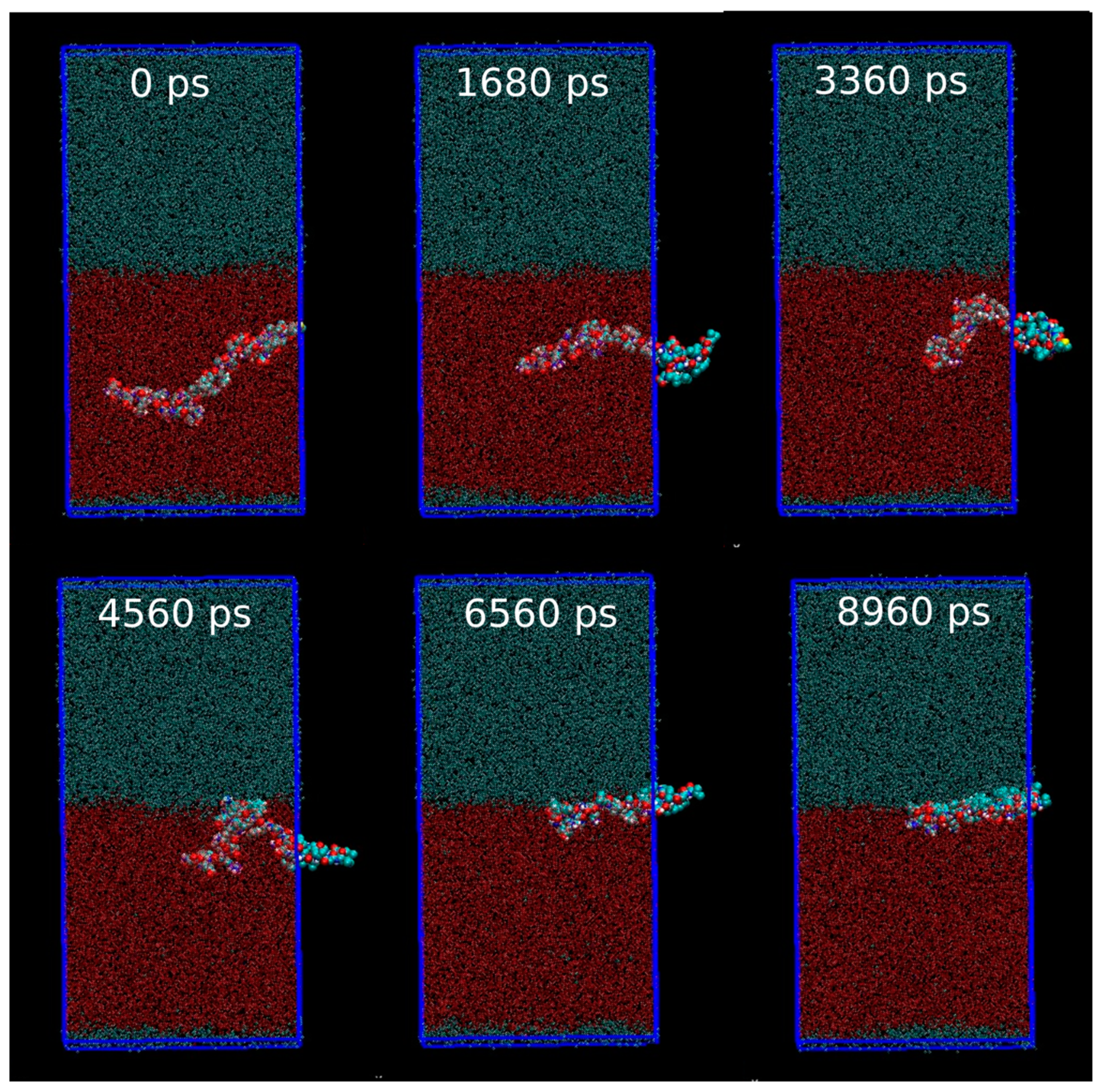
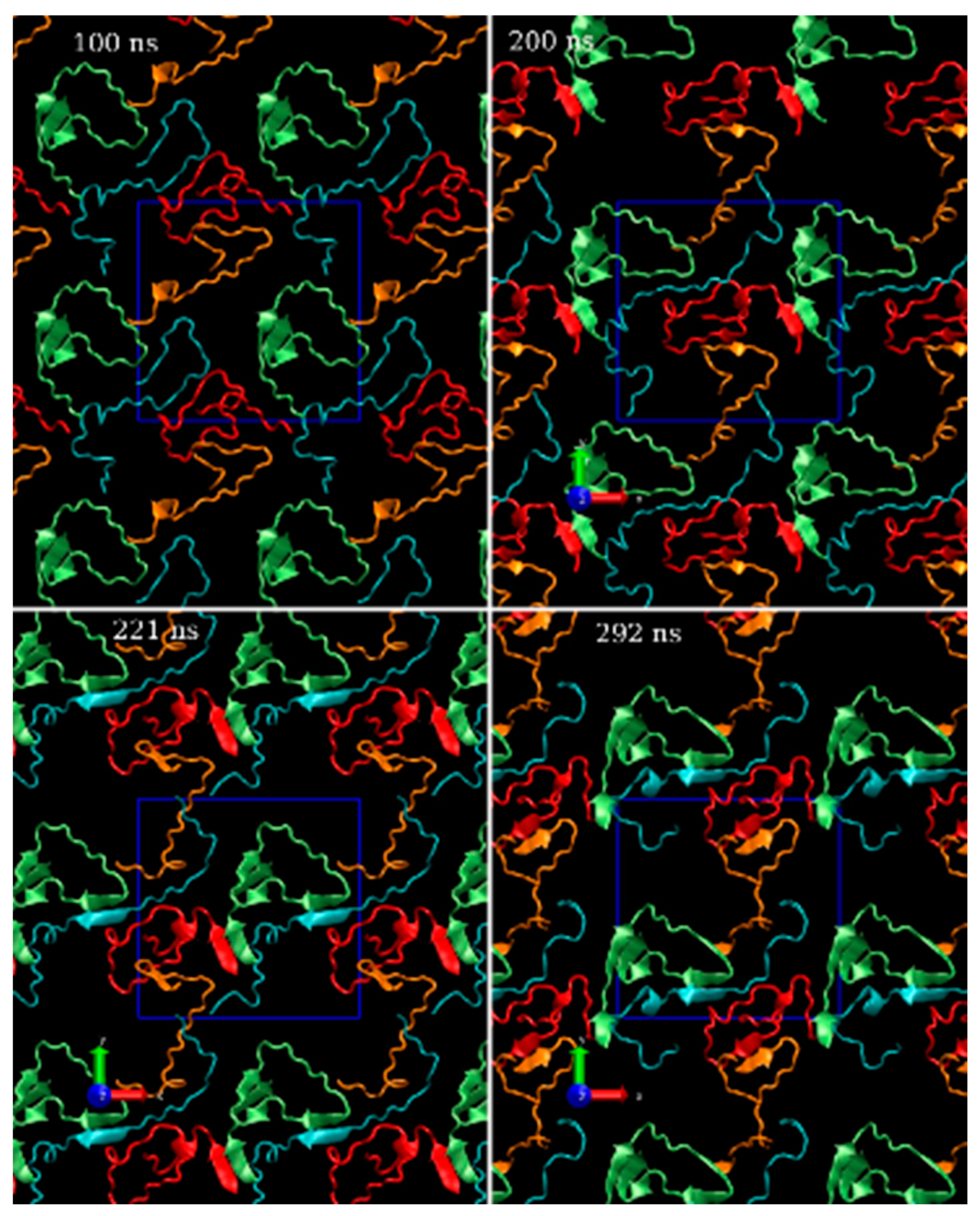
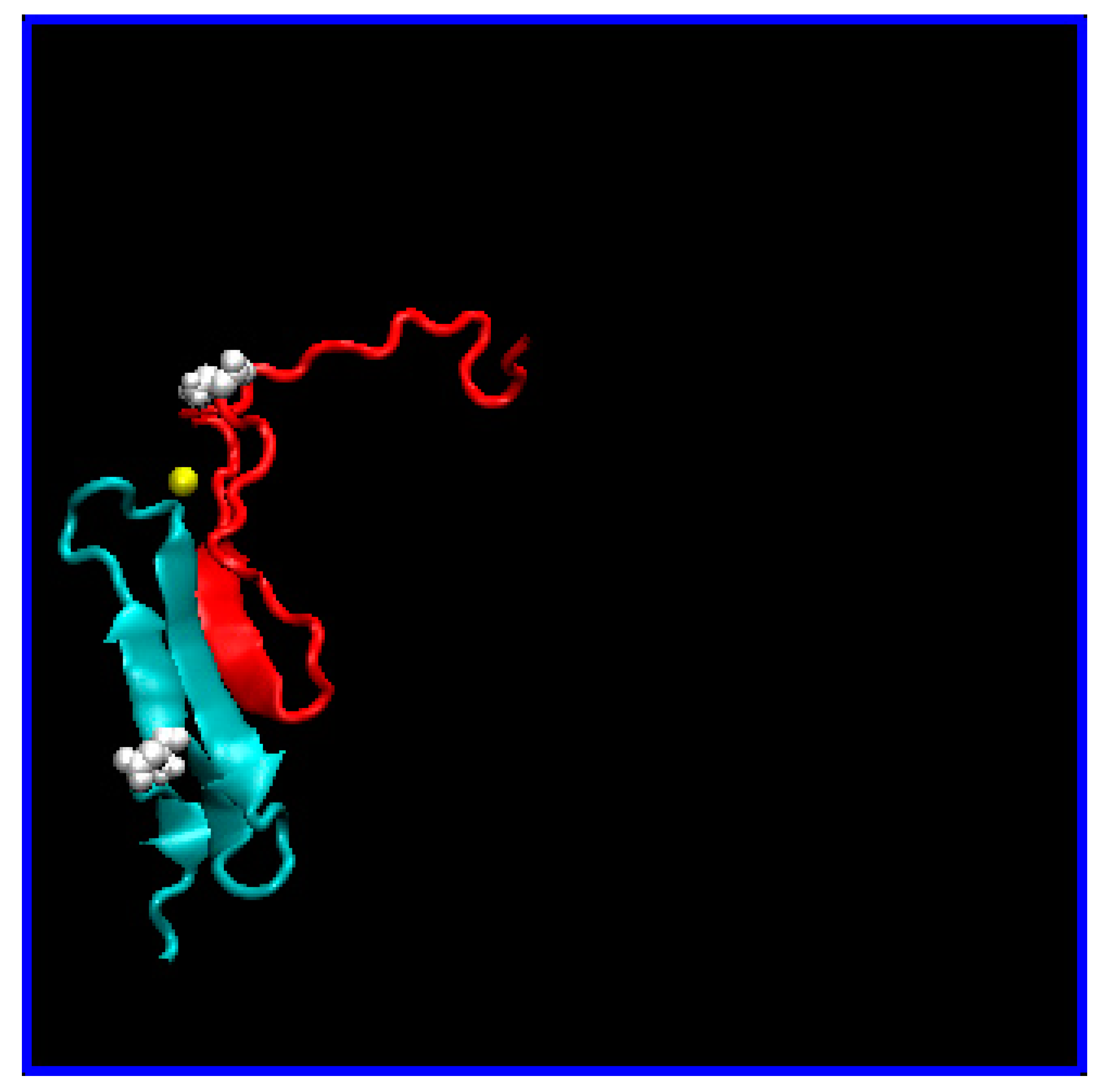
3.1. Computational

3.2. Electrochemical Measurements
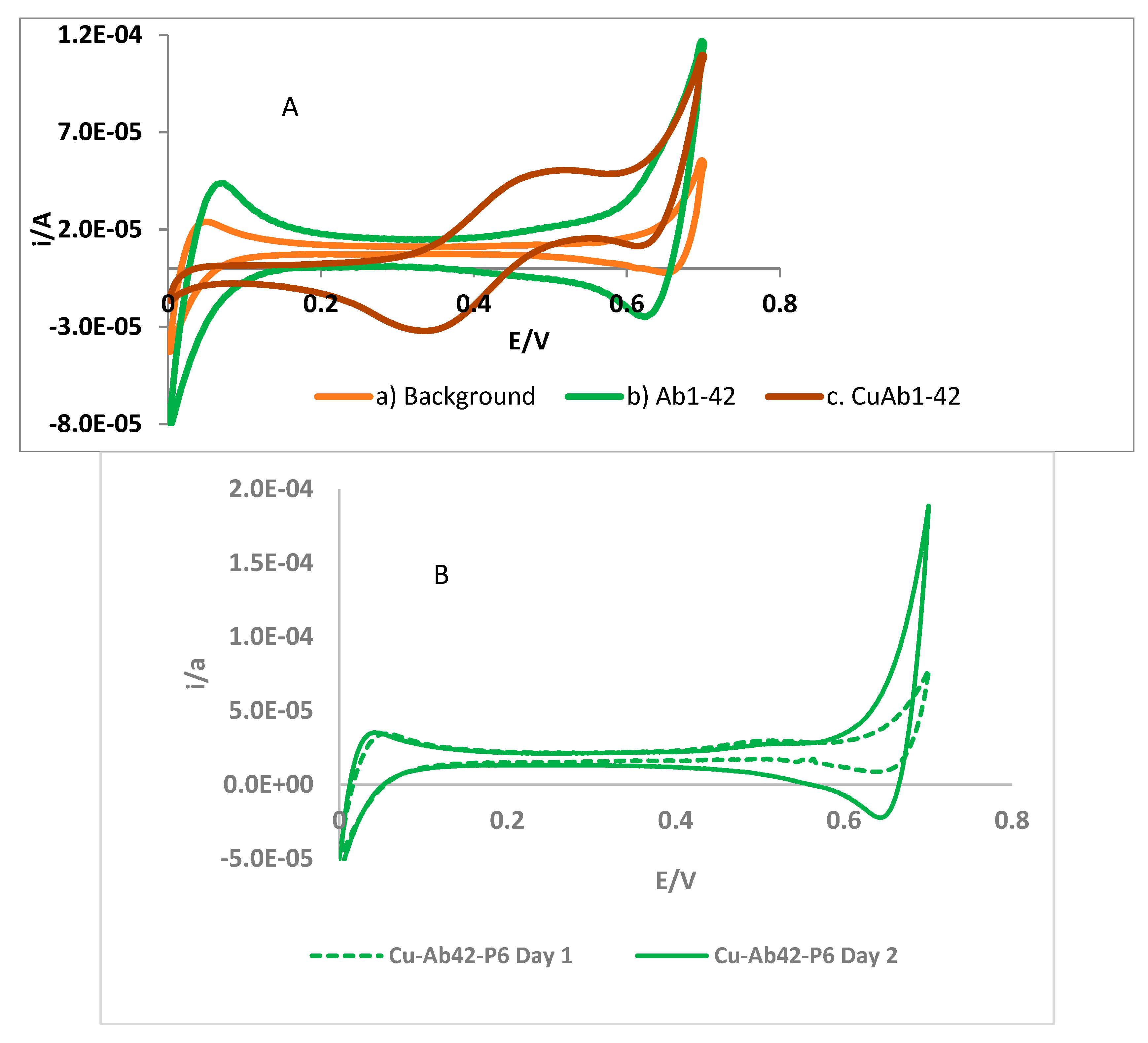
3.2.1. Cyclic Voltammetry at the Liquid–Liquid Interface
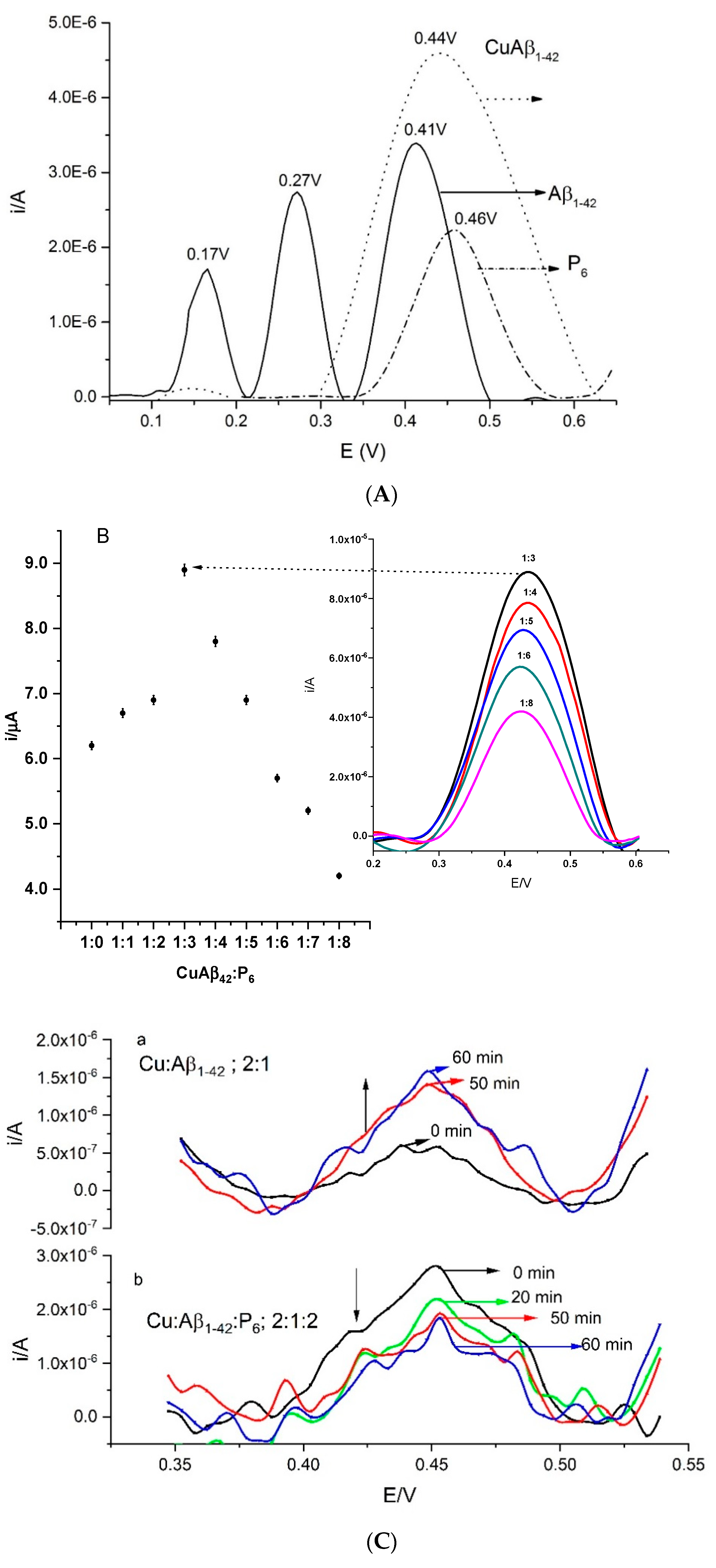
3.2.2. Differential Pulse Voltammetry (DPV) at the Liquid–Liquid Interface
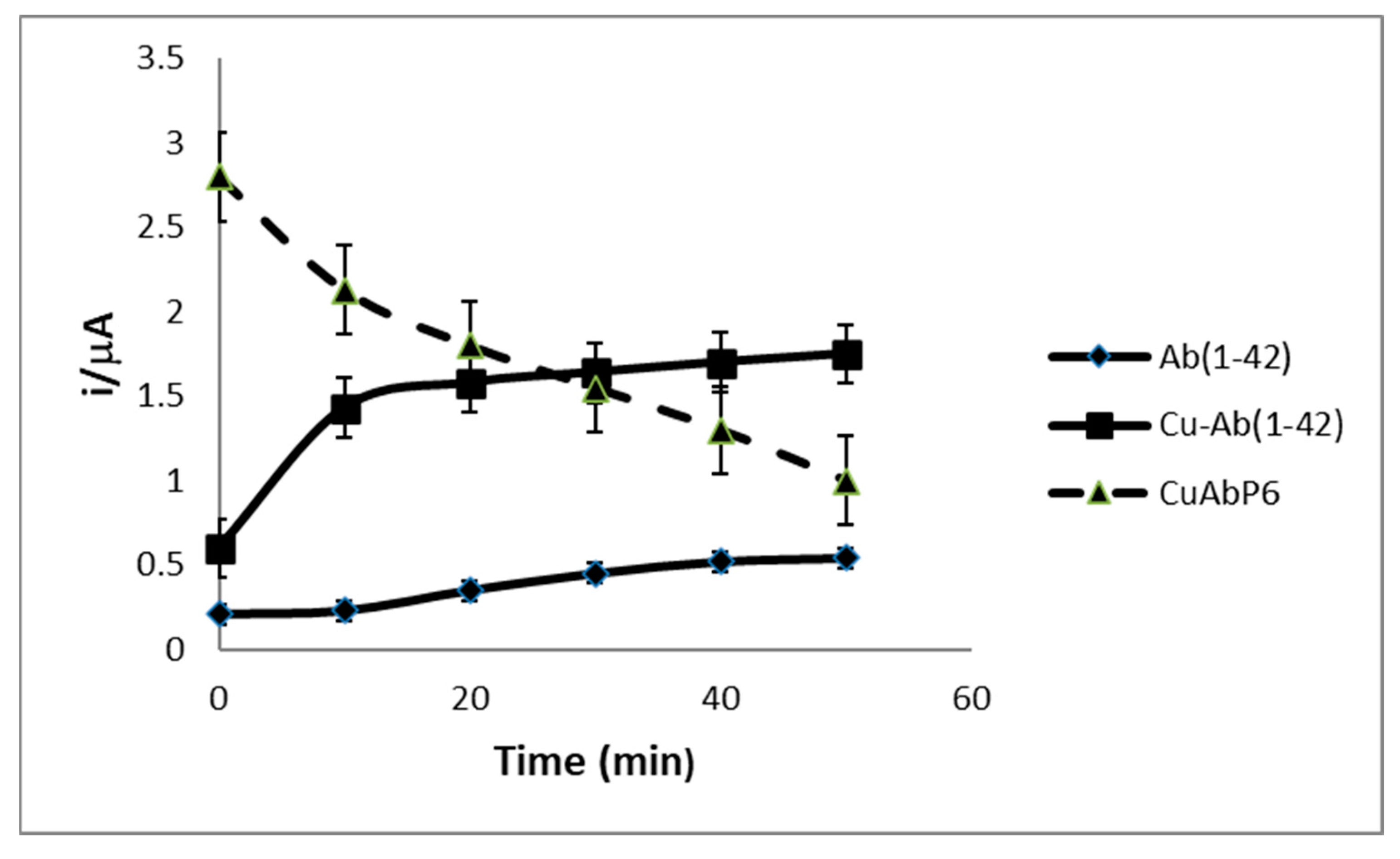
3.3. Binding Constant and Lipophilicity Estimates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hane, F.; Tran, G.; Attwood, S.J.; Leonenko, Z. Cu2+ Affects Amyloid-β (1-42) Aggregation by Increasing Peptide-Peptide Binding Forces. PLoS ONE 2013, 8, e59005. [Google Scholar] [CrossRef] [Green Version]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A Specific Amyloid-β Protein Assembly in the Brain Impairs Memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Finkelstein, D.; Hawco, N.J.; Rath, N.P.; Kim, J.; Mirica, L.M. Bifunctional Compounds for Controlling Metal-Mediated Aggregation of the Aβ42 Peptide. J. Am. Chem. Soc. 2012, 134, 6625–6636. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Kim, J.; Mirica, L.M. The Effect of Cu2+ and Zn2+ on the Aβ42 Peptide Aggregation and Cellular Toxicity. Metallomics 2013, 5, 1529–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Su, B.; Wang, X.; Smith, M.A.; Perry, G. Causes of Oxidative Stress in Alzheimer Disease. Cell. Mol. Life Sci. 2007, 64, 2202–2210. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s Diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef]
- Viles, J.H. Metal Ions and Amyloid Fiber Formation in Neurodegenerative Diseases. Copper, Zinc and Iron in Alzheimer’s, Parkinson’s and Prion Diseases. Coord. Chem. Rev. 2012, 256, 2271–2284. [Google Scholar] [CrossRef]
- Sarell, C.J.; Syme, C.D.; Rigby, S.E.J.; Viles, J.H. Copper(II) Binding to Amyloid-β Fibrils of Alzheimer’s Disease Reveals a Picomolar Affinity: Stoichiometry and Coordination Geometry Are Independent of Aβ Oligomeric Form. Biochemistry 2009, 48, 4388–4402. [Google Scholar] [CrossRef]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of Copper Interactions with Alzheimer Amyloid β Peptides. J. Neurochem. 2000, 75, 1219–1233. [Google Scholar] [CrossRef]
- Rana, M.; Sharma, A.K. Cu and Zn Interactions with Aβ Peptides: Consequence of Coordination on Aggregation and Formation of Neurotoxic Soluble Aβ Oligomers. Metallomics 2019, 11, 64–84. [Google Scholar] [CrossRef] [PubMed]
- Rauk, A. The Chemistry of Alzheimer’s Disease. Chem. Soc. Rev. 2009, 38, 2698–2715. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.R.; Moss, M.A.; Reed, D.K.; Hoh, J.H.; Rosenberry, T.L. Rapid Assembly of Amyloid-β Peptide at a Liquid/Liquid Interface Produces Unstable β-Sheet Fibers. Biochemistry 2005, 44, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Hureau, C.; Balland, V.; Coppel, Y.; Solari, P.L.; Fonda, E.; Faller, P. Importance of Dynamical Processes in the Coordination Chemistry and Redox Conversion of Copper Amyloid-β Complexes. J. Biol. Inorg. Chem. 2009, 14, 995–1000. [Google Scholar] [CrossRef]
- Ghosh, C.; Dey, S.G. Ligand-Field and Ligand-Binding Analysis of the Active Site of Copper-Bound Aβ Associated with Alzheimer’s Disease. Inorg. Chem. 2013, 52, 1318–1327. [Google Scholar] [CrossRef]
- Faller, P. Copper and Zinc Binding to Amyloid-β: Coordination, Dynamics, Aggregation, Reactivity and Metal-Ion Transfer. ChemBioChem 2009, 10, 2837–2845. [Google Scholar] [CrossRef]
- Zou, J.; Kajita, K.; Sugimoto, N. Cu2+ Inhibits the Aggregation of Amyloid β-Peptide(1-42) in Vitro. Angew Chem. Int. Ed. Engl. 2001, 40, 2274–2277. [Google Scholar] [CrossRef]
- Yoshiike, Y.; Tanemura, K.; Murayama, O.; Akagi, T.; Murayama, M.; Sato, S.; Sun, X.; Tanaka, N.; Takashima, A. New Insights on How Metals Disrupt Amyloid β-Aggregation and Their Effects on Amyloid-β Cytotoxicity *. J. Biol. Chem. 2001, 276, 32293–32299. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, M.; Salvietti, E.; Guidotti, M.; Casini, A.; Bellandi, S.; Foresti, M.L.; Gabbiani, C.; Pozzi, A.; Zatta, P.; Messori, L. Trace Copper(II) or Zinc(II) Ions Drastically Modify the Aggregation Behavior of Amyloid-β 1-42: An AFM Study. J. Alzheimer’s Dis. 2010, 19, 1323–1329. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Østergaard, J.; Rozlosnik, N.; Gammelgaard, B.; Heegaard, N.H.H. Cu(II) Mediates Kinetically Distinct, Non-Amyloidogenic Aggregation of Amyloid-β Peptides *. J. Biol. Chem. 2011, 286, 26952–26963. [Google Scholar] [CrossRef] [Green Version]
- Atwood, C.S.; Moir, R.D.; Huang, X.; Scarpa, R.C.; Bacarra, N.M.E.; Romano, D.M.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Dramatic Aggregation of Alzheimer Aβ by Cu(II) Is Induced by Conditions Representing Physiological Acidosis *. J. Biol. Chem. 1998, 273, 12817–12826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarell, C.J.; Wilkinson, S.R.; Viles, J.H. Substoichiometric Levels of Cu2+ Ions Accelerate the Kinetics of Fiber Formation and Promote Cell Toxicity of Amyloid-β from Alzheimer Disease *. J. Biol. Chem. 2010, 285, 41533–41540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekhar, K.; Madhu, C.; Govindaraju, T. Natural Tripeptide-Based Inhibitor of Multifaceted Amyloid β Toxicity. ACS Chem. Neurosci. 2016, 7, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Reymond, F.; Fermín, D.; Lee, H.J.; Girault, H.H. Electrochemistry at Liquid/Liquid Interfaces: Methodology and Potential Applications. Electrochim. Acta 2000, 45, 2647–2662. [Google Scholar] [CrossRef]
- Herzog, G.; Kam, V.; Arrigan, D.W.M. Electrochemical Behaviour of Haemoglobin at the Liquid/Liquid Interface. Electrochim. Acta 2008, 53, 7204–7209. [Google Scholar] [CrossRef]
- Amemiya, S.; Yang, X.; Wazenegger, T.L. Voltammetry of the Phase Transfer of Polypeptide Protamines across Polarized Liquid/Liquid Interfaces. J. Am. Chem. Soc. 2003, 125, 11832–11833. [Google Scholar] [CrossRef]
- Trojánek, A.; Langmaier, J.; Samcová, E.; Samec, Z. Counterion Binding to Protamine Polyion at a Polarised Liquid–Liquid Interface. J. Electroanal. Chem. 2007, 603, 235–242. [Google Scholar] [CrossRef]
- Vagin, M.Y.; Malyh, E.V.; Larionova, N.I.; Karyakin, A.A. Spontaneous and Facilitated Micelles Formation at Liquid|liquid Interface: Towards Amperometric Detection of Redox Inactive Proteins. Electrochem. Commun. 2003, 5, 329–333. [Google Scholar] [CrossRef]
- Kivlehan, F.; Lanyon, Y.H.; Arrigan, D.W.M. Electrochemical Study of Insulin at the Polarized Liquid−Liquid Interface. Langmuir 2008, 24, 9876–9882. [Google Scholar] [CrossRef]
- Scanlon, M.D.; Jennings, E.; Arrigan, D.W.M. Electrochemical Behaviour of Hen-Egg-White Lysozyme at the Polarised Water/1, 2-Dichloroethane Interface. Phys. Chem. Chem. Phys. 2009, 11, 2272–2280. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution Structure of the Alzheimer Amyloid β-Peptide (1-42) in an Apolar Microenvironment. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef]
- Strodel, B.; Lee, J.W.L.; Whittleston, C.S.; Wales, D.J. Transmembrane Structures for Alzheimer’s Aβ1−42 Oligomers. J. Am. Chem. Soc. 2010, 132, 13300–13312. [Google Scholar] [CrossRef]
- Mutter, S.T.; Turner, M.; Deeth, R.J.; Platts, J.A. Molecular Dynamics Simulations of Copper Binding to Amyloid-β Glu22 Mutants. Heliyon 2020, 6, e03071. [Google Scholar] [CrossRef] [Green Version]
- Raffa, D.F.; Rauk, A. Molecular Dynamics Study of the Beta Amyloid Peptide of Alzheimer’s Disease and Its Divalent Copper Complexes. J. Phys. Chem. B 2007, 111, 3789–3799. [Google Scholar] [CrossRef]
- Streltsov, V.A.; Titmuss, S.J.; Epa, V.C.; Barnham, K.J.; Masters, C.L.; Varghese, J.N. The Structure of the Amyloid-β Peptide High-Affinity Copper II Binding Site in Alzheimer Disease. Biophys. J. 2008, 95, 3447–3456. [Google Scholar] [CrossRef] [Green Version]
- Legleiter, J.; Thakkar, R.; Velásquez-Silva, A.; Miranda-Carvajal, I.; Whitaker, S.; Tomich, J.; Comer, J. Design of Peptides That Fold and Self-Assemble on Graphite. J. Chem. Inf. Model. 2022, 62, 4066–4082. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A Series of PDB-Related Databanks for Everyday Needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Balland, V.; Hureau, C.; Savéant, J.-M. Electrochemical and Homogeneous Electron Transfers to the Alzheimer Amyloid-β Copper Complex Follow a Preorganization Mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 17113–17118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, J.; Hoffmann, S.V.; Day, L.; Lee, T.-H.; Augustin, M.A.; Aguilar, M.-I.; Wooster, T.J. Conformational Changes of α-Lactalbumin Adsorbed at Oil–Water Interfaces: Interplay between Protein Structure and Emulsion Stability. Langmuir 2012, 28, 2357–2367. [Google Scholar] [CrossRef]
- Jiang, D.; Rauda, I.; Han, S.; Chen, S.; Zhou, F. Aggregation Pathways of the Amyloid β(1-42) Peptide Depend on Its Colloidal Stability and Ordered β-Sheet Stacking. Langmuir 2012, 28, 12711–12721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, J.T.; Heegaard, N.H.H. Analysis of Protein Aggregation in Neurodegenerative Disease. Anal. Chem. 2013, 85, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Wofsy, C.; Goldstein, B. Interpretation of Scatchard Plots for Aggregating Receptor Systems. Math. Biosci. 1992, 112, 115–154. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Shah, K.; Hough, O.; Hynynen, K. Focused Ultrasound-Mediated Drug Delivery through the Blood–Brain Barrier. Expert Rev. Neurother. 2015, 15, 477–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, S.; Gillespie, J.R.; Shin, B.; Saxena, S. The Second Cu(II)-Binding Site in a Proton-Rich Environment Interferes with the Aggregation of Amyloid-β(1−40) into Amyloid Fibrils. Biochemistry 2009, 48, 10724–10732. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
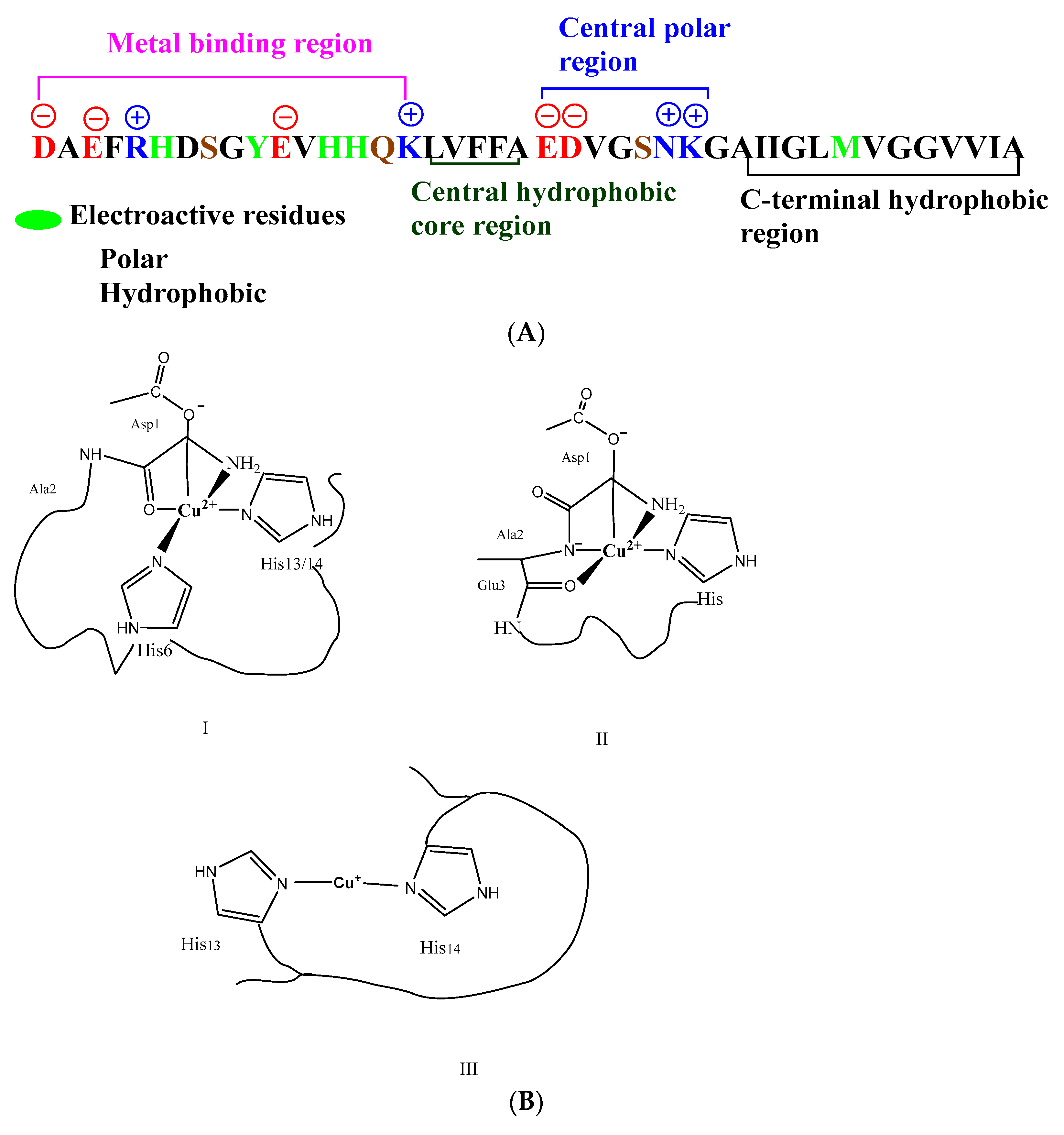{kind=link}
{kind=link}
{kind=link}
| Complexes | |||
|---|---|---|---|
| Cu-Aβ42 | −21.11 ± 0.06 | −0.22 ± 0.07 | 4.04 ± 0.3 |
| Cu-Aβ42 (>10 min) | −58.86 ± 0.07 | −0.61 ± 0.09 | 11.27 ± 0.5 |
| Cu-Aβ-P6 | −16.40 ± 0.07 | −0.17 ± 0.09 | 3.14 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silwane, B.; Wilson, M.; Kataky, R. An Electrochemistry and Computational Study at an Electrified Liquid–Liquid Interface for Studying Beta-Amyloid Aggregation. Membranes 2023, 13, 584. https://doi.org/10.3390/membranes13060584
Silwane B, Wilson M, Kataky R. An Electrochemistry and Computational Study at an Electrified Liquid–Liquid Interface for Studying Beta-Amyloid Aggregation. Membranes. 2023; 13(6):584. https://doi.org/10.3390/membranes13060584
Chicago/Turabian StyleSilwane, Bongiwe, Mark Wilson, and Ritu Kataky. 2023. "An Electrochemistry and Computational Study at an Electrified Liquid–Liquid Interface for Studying Beta-Amyloid Aggregation" Membranes 13, no. 6: 584. https://doi.org/10.3390/membranes13060584
APA StyleSilwane, B., Wilson, M., & Kataky, R. (2023). An Electrochemistry and Computational Study at an Electrified Liquid–Liquid Interface for Studying Beta-Amyloid Aggregation. Membranes, 13(6), 584. https://doi.org/10.3390/membranes13060584