

Mixed-Matrix Organo-Silica–Hydrotalcite Membrane for CO2 Separation Part 1: Synthesis and Analytical Description



Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation
2.1.1. Hydrotalcite
2.1.2. Organo-Silica Matrix
2.1.3. Mixed-Matrix
2.2. Analytical Methods
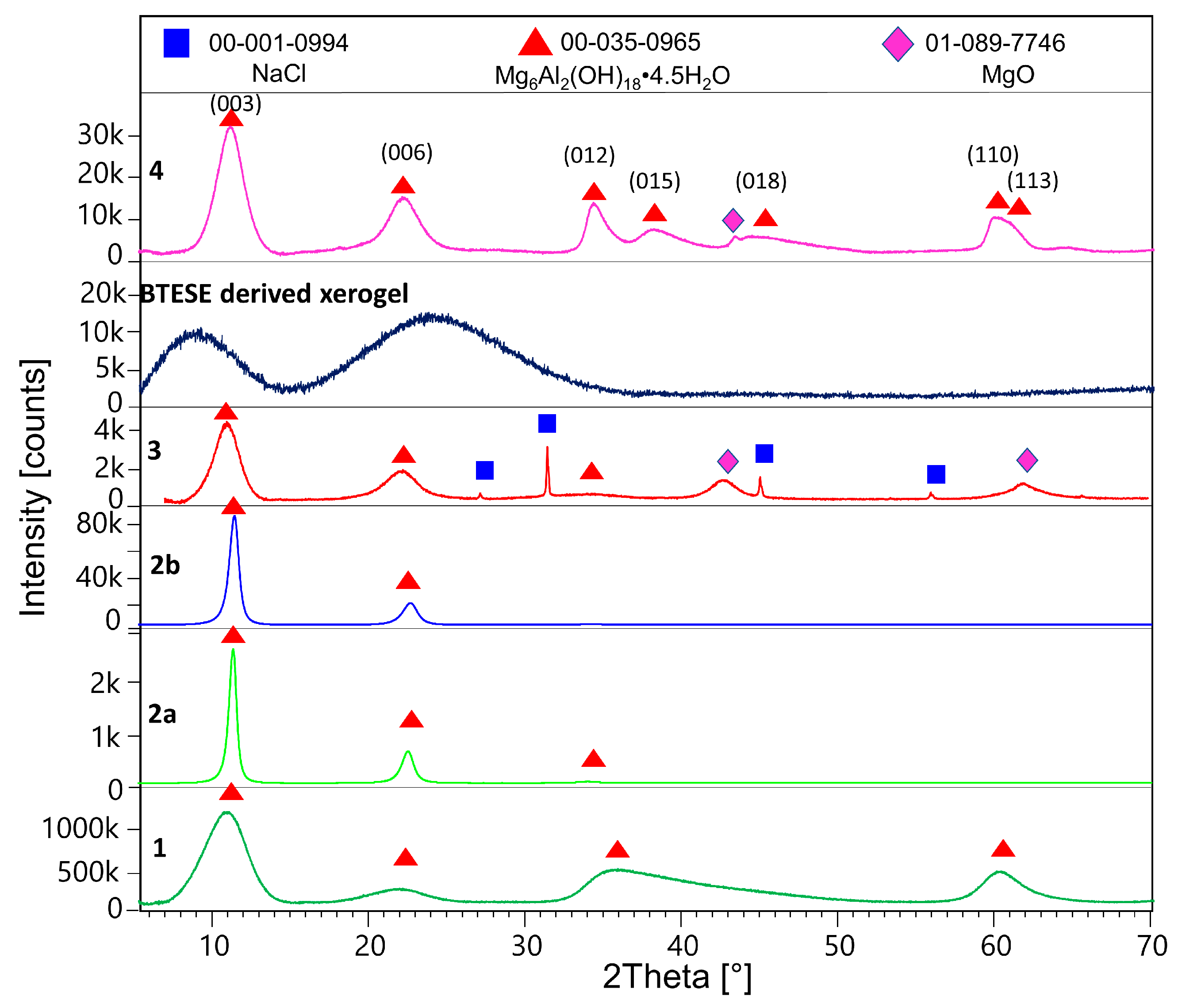
2.2.1. X-ray Diffraction (XRD)
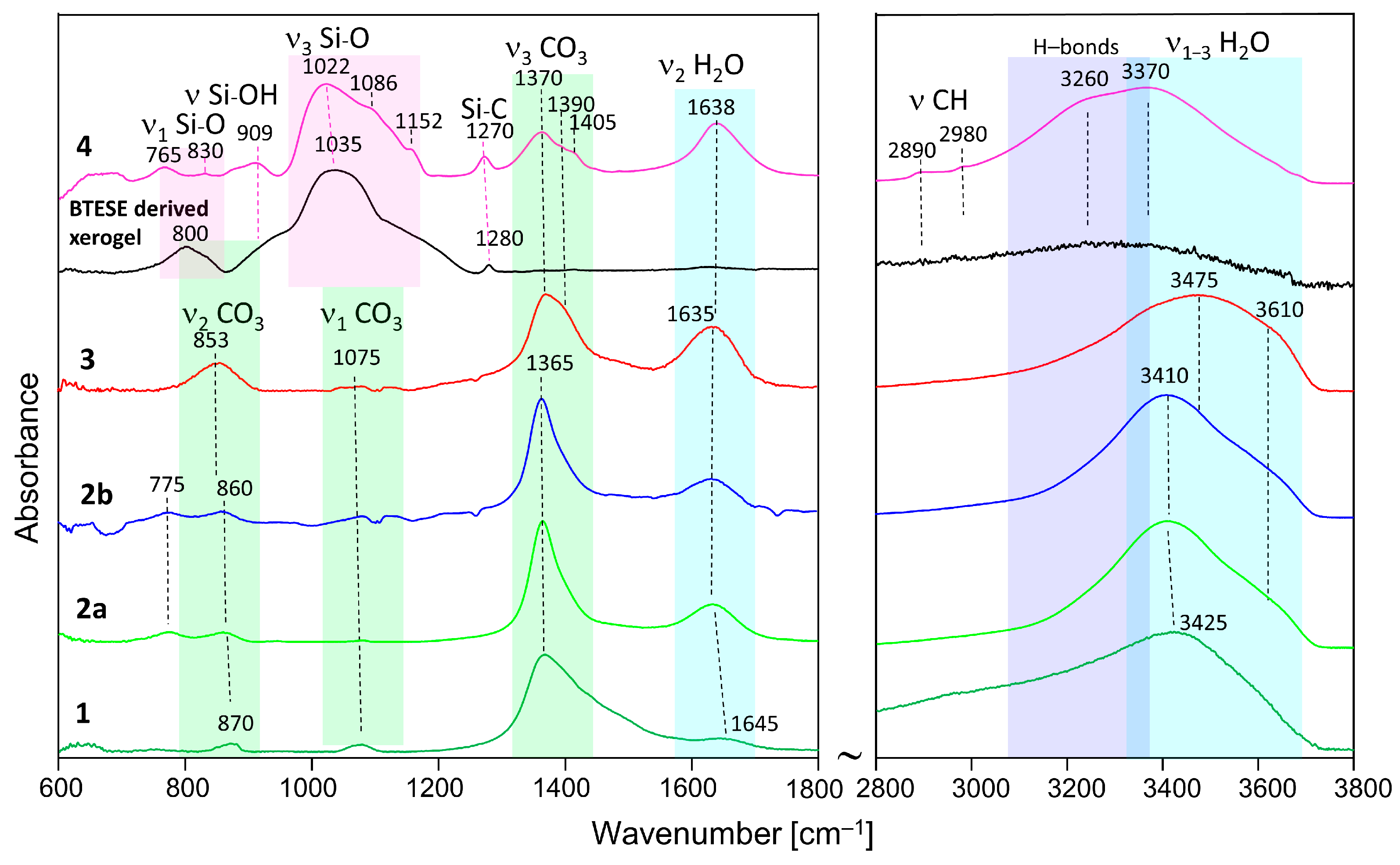
2.2.2. IR Analysis
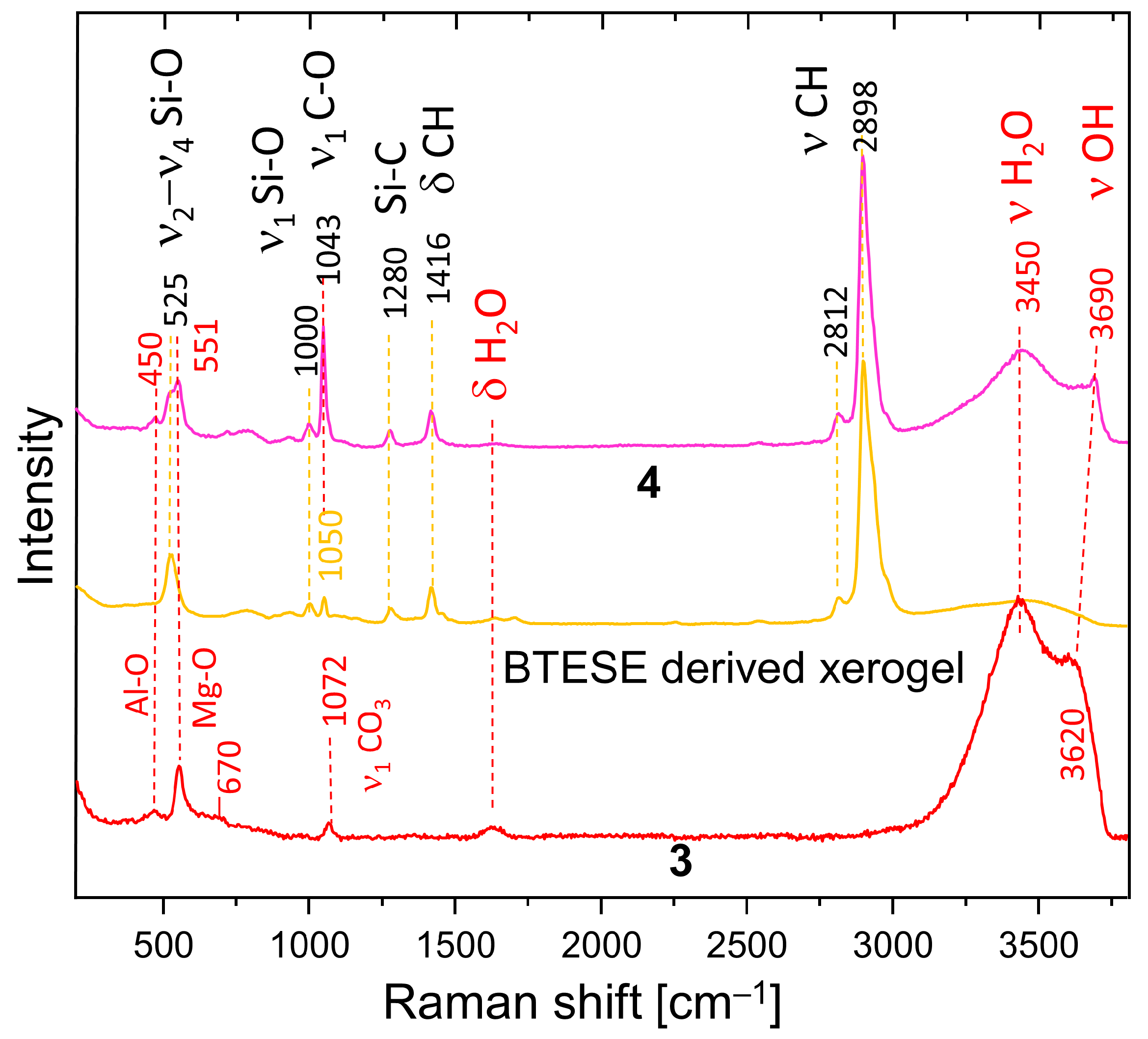
2.2.3. Raman Spectroscopy (RS)
2.2.4. Thermal Analysis (TA)
3. Results and Discussion
3.1. XRD Results
3.2. IR Spectroscopy
3.3. Raman Spectroscopy
3.4. Thermal Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bünger, L.; Kurtz, T.; Garbev, K.; Stemmermann, P.; Stapf, D. Mixed-Matrix Organo–Silica–Hydrotalcite Membrane for CO2 Separation Part 2: Permeation and Selectivity Study. Membranes 2024, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Hochstetter, C. Untersuchung über die Zusammensetzung einiger Mineralien. J. Prakt. Chem. 1842, 27, 375–378. [Google Scholar] [CrossRef]
- Mills, S.J.; Christy, A.G.; Schmitt, R.T. The creation of neotypes for hydrotalcite. Mineral. Mag. 2016, 80, 1023–1029. [Google Scholar] [CrossRef]
- Allmann, R.; Jepsen, H.P. Die Struktur des Hydrotalkits. Neues Jb. Miner. Monat. 1969, 1969, 544–551. [Google Scholar]
- Bellotto, M.; Rebours, B.; Clause, O.; Lynch, J.; Bazin, D.; Elkaim, E. A reexamination of hydrotalcite crystal chemistry. J. Phys. Chem. 1996, 100, 8527–8534. [Google Scholar] [CrossRef]
- Zhitova, E.; Krivovichev, S.; Pekov, I.; Greenwell, H. Crystal chemistry of natural layered double hydroxides. 5. Single-crystal structure refinement of hydrotalcite, [Mg6Al2(OH)16](CO3)(H2O)4. Mineral. Mag. 2019, 83, 269–280. [Google Scholar] [CrossRef]
- Evans, D.G.; Slade, R.C.T. Structural Aspects of Layered Double Hydroxides. In Layered Double Hydroxides. Structure and Bonding; Duan, X., Evans, D.G., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 119. [Google Scholar] [CrossRef]
- Ram Reddy, M.K.; Xu, Z.P.; Lu, G.Q.; Diniz da Costa, J.C. Layered Double Hydroxides for CO2 Capture: Structure Evolution and Regeneration. Ind. Eng. Chem. Res. 2006, 45, 7504–7509. [Google Scholar] [CrossRef]
- León, M.; Díaz, E.; Bennici, S.; Vega, A.; Ordóñez, S.; Auroux, A. Adsorption of CO2 on Hydrotalcite-Derived Mixed Oxides: Sorption Mechanisms and Consequences for Adsorption Irreversibility. Ind. Eng. Chem. Res. 2010, 49, 3663–3671. [Google Scholar] [CrossRef]
- Wang, Q.; Tay, H.H.; Ng, D.J.W.; Chen, L.; Liu, Y.; Chang, J.; Zhong, Z.; Luo, J.; Borgna, A. The effect of trivalent cations on the performance of Mg-M-CO3 layered double hydroxides for high-temperature CO2 capture. Chem. Sus. Chem. 2010, 3, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Bublinski, M. CO2-Abtrennung aus Synthesegasen mit Hydrotalciten unter Hochtemperatur-Hochdruckbedingungen. Ph.D. Thesis, University of Stuttgart, Stuttgart, Germany, 2017. [Google Scholar] [CrossRef]
- Adachi-Pagano, M.; Forano, C.; Besse, J.-P. Delamination of layered double hydroxides by use of surfactants. Chem. Commun. 2000, 1, 91–92. [Google Scholar] [CrossRef]
- Xie, J.; Khalid, Z.; Oh, J.-M. Recent advances in the synthesis of layered double hydroxides nanosheets. B. Kor. Chem. Soc. 2022, 44, 100–111. [Google Scholar] [CrossRef]
- Shirin, V.A.; Sankar, R.; Johnson, A.P.; Gangadharappa, H.; Pramod, K. Advanced drug delivery applications of layered double hydroxide. J. Control. Release 2021, 330, 398–426. [Google Scholar] [CrossRef]
- Mao, N.; Zhou, C.H.; Tong, D.S.; Yu, W.H.; Lin, C.C. Cynthia Lin, Exfoliation of layered double hydroxide solids into functional nanosheets. Appl. Clay Sci. 2017, 144, 60–78. [Google Scholar] [CrossRef]
- Kim, T.W.; Sahimi, M.; Tsotsis, T.T. The Preparation and Characterization of Hydrotalcite Thin Films. Ind. Eng. Chem. Res. 2009, 48, 5794–5801. [Google Scholar] [CrossRef]
- Fajrina, N.; Yusof, N.; Ismail, A.; Jaafar, J.; Aziz, F.; Salleh, W.; Nordin, N. MgAl-CO3 layered double hydroxide as potential filler in substrate layer of composite membrane for enhanced carbon dioxide separation. J. Environ. Chem. Eng. 2021, 9, 106164. [Google Scholar] [CrossRef]
- Huang, N.; Wang, C.; Chen, C. Ethylene vinyl acetate copolymer/Mg-Al-layered double hydroxide nanocomposite membranes applied in CO2/N2 gas separation. Polym. Compos. 2021, 42, 4065–4072. [Google Scholar] [CrossRef]
- Wiheeb, A.D.; Shakir, S.W.; Othman, M.R. Synthesis and Characterization of Mesoporous Hydrotalcite-Alumina Membrane for Carbon Dioxide Enrichment. IOP Conf. Ser. Mater. Sci. Eng. 2018, 454, 12107. [Google Scholar] [CrossRef]
- de Vos, R.M.; Verweij, H. High-selectivity, high-flux silica membranes for gas separation. Science 1998, 279, 1710–1711. [Google Scholar] [CrossRef]
- Elshof, J.E.T. Hybrid Materials for Molecular Sieves. In Handbook of Sol-Gel Science and Technology, Living Reference Work; Klein, L.C., Aparicio, M., Jitianu, A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–27. [Google Scholar] [CrossRef]
- Kanezashi, M.; Matsugasako, R.; Tawarayama, H.; Nagasawa, H.; Tsuru, T. Pore size tuning of sol-gel-derived triethoxysilane (TRIES) membranes for gas separation. J. Membr. Sci. 2017, 524, 64–72. [Google Scholar] [CrossRef]
- Van Gestel, T.; Velterop, F.; Meulenberg, W.A. Meulenberg, Zirconia-supported hybrid organosilica microporous membranes for CO2 separation and pervaporation. Sep. Purif. Technol. 2021, 259, 118114. [Google Scholar] [CrossRef]
- Castricum, H.L.; Kreiter, R.; van Veen, H.M.; Blank, D.H.; Vente, J.F.; Elshof, J.E.T. High-performance hybrid pervaporation membranes with superior hydrothermal and acid stability. J. Membr. Sci. 2008, 324, 111–118. [Google Scholar] [CrossRef]
- Yu, L.; Kanezashi, M.; Nagasawa, H.; Guo, M.; Moriyama, N.; Ito, K.; Tsuru, T. Tailoring Ultramicroporosity To Maximize CO2 Transport within Pyrimidine-Bridged Organosilica Membranes. ACS Appl. Mater. Interfaces 2019, 11, 7164–7173. [Google Scholar] [CrossRef]
- Castricum, H.L.; Paradis, G.G.; Mittelmeijer-Hazeleger, M.C.; Kreiter, R.; Vente, J.F.; Elshof, J.E.T. Tailoring the separation behavior of hybrid organosilica membranes by adjusting the structure of the organic bridging group. Adv. Funct. Mater. 2011, 21, 2319–2329. [Google Scholar] [CrossRef]
- Kanezashi, M.; Yada, K.; Yoshioka, T.; Tsuru, T. Design of silica networks for development of highly permeable hydrogen separation membranes with hydrothermal stability. J. Am. Chem. Soc. 2009, 131, 414–415. [Google Scholar] [CrossRef]
- Moriyama, N.; Nagasawa, H.; Kanezashi, M.; Ito, K.; Tsuru, T. Bis(triethoxysilyl)ethane (BTESE)-derived silica membranes: Pore formation mechanism and gas permeation properties. J. Sol-Gel Sci. Technol. 2018, 86, 63–72. [Google Scholar] [CrossRef]
- Agirre, I.; Arias, P.L.; Castricum, H.L.; Creatore, M.; Elshof, J.E.T.; Paradis, G.G.; Ngamou, P.H.; van Veen, H.M.; Vente, J.F. Hybrid organosilica membranes and processes: Status and outlook. Sep. Purif. Technol. 2014, 121, 2–12. [Google Scholar] [CrossRef]
- Leroux, F.; Adachi-Pagano, M.; Intissar, M.; Chauvière, S.; Forano, C.; Besse, J.-P. Delamination and restacking of layered double hydroxides. J. Mater. Chem. 2001, 11, 105–112. [Google Scholar] [CrossRef]
- Piccinni, M.; Bellani, S.; Bianca, G.; Bonaccorso, F. Nickel-Iron Layered Double Hydroxide Dispersions in Ethanol Stabilized by Acetate Anions. Inorg. Chem. 2022, 61, 4598–4608. [Google Scholar] [CrossRef]
- Othman, M.R.; Helwani, Z.; Martunus; Fernando, W.J.N. Synthetic hydrotalcites from different routes and their application as catalysts and gas adsorbents: A review. Appl. Organometal. Chem. 2009, 23, 335–346. [Google Scholar] [CrossRef]
- Gursky, J.A.; Blough, S.D.; Luna, C.; Gomez, C.; Luevano, A.N.; Gardner, E.A. Gardner, Particle-particle interactions between layered double hydroxide nanoparticles. J. Am. Chem. Soc. 2006, 128, 8376–8377. [Google Scholar] [CrossRef]
- Gardner, E.; Huntoon, K.M.; Pinnavaia, T.J. Direct Synthesis of Alkoxide-Intercalated Derivatives of Hydrocalcite-like Layered Double Hydroxides: Precursors for the Formation of Colloidal Layered Double Hydroxide Suspensions and Transparent Thin Films. Adv. Mater. 2001, 13, 1263. [Google Scholar] [CrossRef]
- Merz, D.; Dregert, O.; Garbev, K.; Stemmermann, P. A reliable quantitative TA-FTIR method for cementitious material characterization. In Proceedings of the GEFTA-STK-Joint Meeting on Thermal Analysis and Calorimetry, Saarbrücken, Germany, 10–12 October 2012; Volume 53. [Google Scholar]
- Kloprogge, J.T.; Wharton, D.; Hickey, L.; Frost, R.L. Infrared and Raman study of interlayer anions CO32−, NO3−, SO42− and ClO4− in Mg/Al-hydrotalcite. Am. Mineral. 2002, 87, 623–629. [Google Scholar] [CrossRef]
- Frost, R.L.; Spratt, H.J.; Palmer, S.J. Infrared and near-infrared spectroscopic study of synthetic hydrotalcites with variable divalent/trivalent cationic ratios. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2009, 72, 984–988. [Google Scholar] [CrossRef]
- Hernandez-Moreno, M.J.; Ulibarri, M.A.; Rendon, J.L.; Serna, C.J. IR characteristics of hydrotalcite-like compounds. Phys. Chem. Miner. 1985, 12, 34–38. [Google Scholar] [CrossRef]
- Mališová, M.; Horňáček, M.; Mikulec, J.; Hudec, P.; Jorík, V. FTIR study of hydrotalcite. Acta Chim. Slov. 2018, 11, 147–166. [Google Scholar] [CrossRef]
- Coenen, K.; Gallucci, F.; Mezari, B.; Hensen, E.; van Sint Annaland, M. An in-situ IR study on the adsorption of CO2 and H2O on hydrotalcites. J. CO2 Util. 2018, 24, 228–239. [Google Scholar] [CrossRef]
- Dawes, A.; Mason, N.J.; Fraser, H. Using the C-O stretch to unravel the nature of hydrogen bonding in low-temperature solid methanol-water condensates. Phys. Chem. Chem. Phys. PCCP 2016, 18, 1245–1257. [Google Scholar] [CrossRef] [PubMed]
- Plyler, E.K. Infrared spectra of methanol, ethanol, and n-propanol. J. Res. Nat. Bur.Stand. 1952, 48, 281–286. [Google Scholar] [CrossRef]
- Rosales-Reina, B.; Cruz-Quesada, G.; Padilla-Postigo, N.; Irigoyen-Razquin, M.; Alonso-Martínez, E.; López-Ramón, M.V.; Espinal-Viguri, M.; Garrido, J.J. Tunability of Hybrid Silica Xerogels: Surface Chemistry and Porous Texture Based on the Aromatic Precursor. Gels 2023, 9, 382. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Hwang, M.S.; Kim, H.J.; Kim, J.Y.; Lee, Y. Infrared spectroscopy study of low-dielectric-constant fluorine-incorporated and carbon-incorporated silicon oxide films. J. Appl. Phys. 2001, 90, 3367–3370. [Google Scholar] [CrossRef]
- Meng, L.; Kanezashi, M.; Wang, J.; Tsuru, T. Permeation properties of BTESE–TEOS organosilica membranes and application to O2/SO2 gas separation. J. Membr. Sci. 2015, 496, 211–218. [Google Scholar] [CrossRef]
- Wahab, M.A.; Kim, I.; Ha, C.-S. Hybrid periodic mesoporous organosilica materials prepared from 1,2-bis (triethoxysilyl) ethane and (3-cyanopropyl) triethoxysilane. Micropor. Mesopor. Mat. 2004, 69, 19–27. [Google Scholar] [CrossRef]
- Keefe, C.D.; Gillis, E.A.L.; MacDonald, L. Improper Hydrogen-Bonding CH Center Dot Y Interactions in Binary Methanol Systems As Studied by FTIR and Raman Spectroscopy. J. Phys. Chem. A 2009, 113, 2544–2550. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, Y.; Lin, K.; Hu, N.; Zhou, X.; Liu, S. Complete Raman spectral assignment of methanol in the C-H stretching region. J. Phys. Chem. A 2013, 117, 4377–4384. [Google Scholar] [CrossRef]
- Ishikawa, K.; Fujima, N.; Komura, H. First—order Raman scattering in MgO microcrystals. J. Appl. Phys. 1985, 57, 973–975. [Google Scholar] [CrossRef]
- Slotznick, S.P.; Shim, S.-H. In situ Raman spectroscopy measurements of MgAl2O4 spinel up to 1400 °C. Am. Mineral. 2008, 93, 470–476. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Frost, R.L. Infrared emission spectroscopic study of the thermal transformation of Mg-, Ni- and Co-hydrotalcite catalysts. Appl. Catal. A 1999, 184, 61–71. [Google Scholar] [CrossRef]
- Matsui, K.; Satoh, H.; Kyoto, M. Raman Spectra of Silica Gel Prepared from Triethoxysilane and Tetraethoxysilane by the Sol-Gel Method. J. Ceram. Soc. Jpn 1998, 106, 528–530. [Google Scholar] [CrossRef]
- Kanezaki, E. Effect of atomic ratio Mg/Al in layers of Mg and Al Layered double hydroxide on thermal stability of hydrotalcite-like layered structure by means of in situ high temperature powder X-ray diffraction. Mat. Res. Bull. 1998, 33, 773–778. [Google Scholar] [CrossRef]
- Frost, R.L.; Martens, W.; Ding, Z.; Kloprogge, J.T. DSC and high-resolution TG of synthesized hydrotalcites of Mg and Zn. J. Therm. Anal. Calorim. 2003, 71, 429–438. [Google Scholar] [CrossRef]
- Ke, X.; Bernal, S.A.; Provis, J.L. Uptake of chloride and carbonate by Mg-Al and Ca-Al layered double hydroxides in simulated pore solutions of alkali-activated slag cement. Cem. Concr. Res. 2017, 100, 1–13. [Google Scholar] [CrossRef]
- Kanezashi, M.; Yada, K.; Yoshioka, T.; Tsuru, T. Organic–inorganic hybrid silica membranes with controlled silica network size: Preparation and gas permeation characteristics. J. Membr. Sci. 2010, 348, 310–318. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| XRD Results | Samples | ||||
|---|---|---|---|---|---|
| u.c.p. | 1 | 2a | 2b | 3 | 4 |
| a (Å) | 3.07(1) | 3.066(1) | 3.14(1) | 3.10(5) | 3.064(1) |
| c (Å) 3R | 25.25(1) | 23.683(2) | 23.557(2) | 24.22(1) | 24.25(1) |
| c (Å) 2H | 16.833(7) | 15.788(1) | 15.705(2) | 16.19(1) | 16.181(7) |
| crystal size (nm) | 1.6(1) | 10.6(2) | 8.42(7) | 3.2(1) | 3.4(1) |
| Hydrotalcite | BTESE, HTlc-Modified Organo-Silica Membrane | |||||
|---|---|---|---|---|---|---|
| Sample | 1 | 2a,b | 3 | Sample | BTESE | 4 |
| Band Assignment | Band Positions (cm−1) | Band Assignment | Band Positions (cm−1) | |||
| ν Mg(Al)-O | 630 | ν Mg(Al)-O | 650–700 | |||
| OH libration | 775 | OH libration | 765 | |||
| ν2 (CO3) | 870 | 860 | 853 | ν1 Si-O | 800 | 830 |
| ν1 (CO3) | 1075 | 1075 | 1075 | Si–OH | 950sh | 909 |
| ν3 (CO3) | 1365 | 1365 | 1370 | ν3 Si-O | 1035s | 1022 |
| M,Bν3 (CO3), ν2 CH3 | 1400–1500 | 1390 | ν1 (CO3) | 1086 | ||
| δ H2O | 1645 | 1635 | 1635 | ν3 Si-O | 1150sh | 1152 |
| H-bonding | 3000–3400 | 3100–3400 | 3200–3400 | ν Si-C | 1280 | 1270 |
| ν1 OH(H2O) | 3425 | 3420 | 3475 | ν3 (CO3) 1370 M,Bν3 (CO3) 1405 | ||
| δ H2O | 1638 | |||||
| ν-CH2 | 2890 | |||||
| ν-CH3 | 2980 | |||||
| H-bonding ν1 OH(H2O | 3260 3370 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bünger, L.; Garbev, K.; Ullrich, A.; Stemmermann, P.; Stapf, D. Mixed-Matrix Organo-Silica–Hydrotalcite Membrane for CO2 Separation Part 1: Synthesis and Analytical Description. Membranes 2024, 14, 170. https://doi.org/10.3390/membranes14080170
Bünger L, Garbev K, Ullrich A, Stemmermann P, Stapf D. Mixed-Matrix Organo-Silica–Hydrotalcite Membrane for CO2 Separation Part 1: Synthesis and Analytical Description. Membranes. 2024; 14(8):170. https://doi.org/10.3390/membranes14080170
Chicago/Turabian StyleBünger, Lucas, Krassimir Garbev, Angela Ullrich, Peter Stemmermann, and Dieter Stapf. 2024. "Mixed-Matrix Organo-Silica–Hydrotalcite Membrane for CO2 Separation Part 1: Synthesis and Analytical Description" Membranes 14, no. 8: 170. https://doi.org/10.3390/membranes14080170
APA StyleBünger, L., Garbev, K., Ullrich, A., Stemmermann, P., & Stapf, D. (2024). Mixed-Matrix Organo-Silica–Hydrotalcite Membrane for CO2 Separation Part 1: Synthesis and Analytical Description. Membranes, 14(8), 170. https://doi.org/10.3390/membranes14080170