Trafficking of Kainate Receptors

Abstract
:1. Introduction
2. Mechanisms of Assembly and Surface Expression of Kainate Receptors
2.1. Homomeric KARs and Trafficking/Retention Motifs
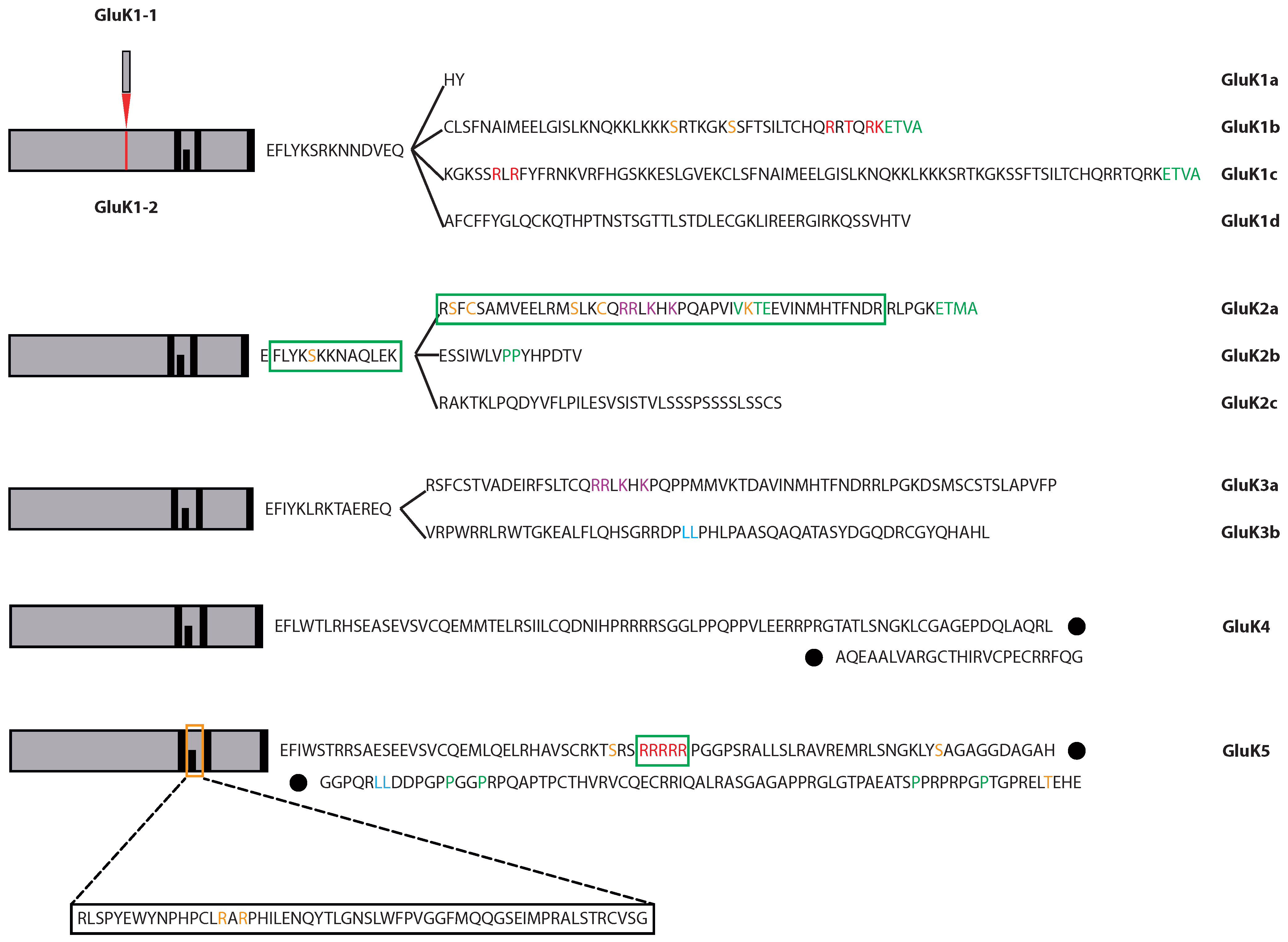
2.1.1. GluK1
2.1.2. GluK2
2.1.3. GluK3
2.1.4. GluK4 and GluK5
2.2. Assembly and Trafficking of Heteromeric KARs
2.2.1. Structural Determinants of Subunit Assembly and ER Exit
2.2.2. Heteromeric Assembly, Trafficking and Endocytosis

3. Activity-Dependent Regulation of Synaptic KAR Abundance
3.1. PKC-Dependent Phosphorylation of KARs and KAR-Mediated LTD
3.2. PKA- and Calcineurin-Mediated Post-Translational Modifications of KARs
3.3. CaMKII-Dependent Regulation of KAR Plasma Membrane Expression
3.4. SUMOylation of KARs

{kind=link}
| Protein | KAR subunit | Target | Reference |
|---|---|---|---|
| Phosphorylation | |||
| CaMKII | GluK5 | S859; S892; T976 | [94] |
| PKA | GluK2(R) | S856; S868 | [88,89,90] |
| PKC | GluK1-2b | S880; S886 | [79] |
| PKC | GluK2a | S846; S868 | [29] |
| PKC | GluK2b | S846 | [29] |
| Palmitoylation | |||
| GluK2 | C858; C871 | [29] | |
| SUMOylation | |||
| Ubc9/PIAS3 | GluK2a | K886 | [96] |
| ND | GluK3a/b | ND | [97] |
4. Proteins Interacting with KARs
4.1. Proteins of the SAP Family
4.2. Other Protein-Protein Interactions Established via PDZ Domains
| Interacting protein | Interacting domain | KAR subunit | Target domain/motif | Regulation of | Reference |
|---|---|---|---|---|---|
| PDZ proteins | |||||
| PSD-95 | ND | GluK1b/c | -ETVA | ND | [79] |
| PDZ1 | GluK2 | -ETMA | Desensitization | [104] | |
| and clustering | [104] | ||||
| GK | GluK5 | ND | Clustering | [104] | |
| SH3 | GluK5 | 914 PPGGPRP 920 | Clustering | [104] | |
| SH3 | GluK5 | 962 PPRPRPGP 969 | Clustering | [104] | |
| SAP-97 | PDZ1 | GluK2 | -ETMA | Clustering | [104,115] |
| SAP-102 | ND | GluK2 | ND | Clustering | [104] |
| ND | GluK5 | ND | Clustering | [104] | |
| PICK1 | PDZ | GluK1b/c | -ETVA | Membrane | [79] |
| anchoring | |||||
| ND | GluK2 | CTD | ND | [79] | |
| GRIP | PDZ4-5 | GluK1b/c | -ETVA | Membrane | [79] |
| anchoring | |||||
| PDZ4-5 | GluK2 | CTD | ND | [79] | |
| Syntenin | PDZ2 | GluK1b/c | -ETVA | ND | [79] |
| ND | GluK2 | CTD | ND | [79] | |
| Cytoskeleton | |||||
| Cadherins | ND | GluK2 | ND | Localization | [136] |
| and trafficking | |||||
| β-catenin | ND | GluK2 | CTD (indirect) | Membrane | [136] |
| dynamics | |||||
| ND | GluK5 | ND | ND | [136] | |
| p120 catenin | ND | GluK2 | ND | ND | [136] |
| Velis | ND | GluK2 | ND | ND | [136] |
| 4.1N | ND | GluK1/2 | MPD | Trafficking | [137] |
| Profilin II | ND | GluK2b | 862 PP 863 | Trafficking | [32,33] |
| Spectrin | ND | GluK2a | CTD | ND | [32] |
| BTB/Kelch proteins | |||||
| KRIP6 | BTB/POZ | GluK2a | residues 842-899 | Gating | [138] |
| Actinfilin | ND | GluK1 | ND | ND | [139] |
| Kelch repeats | GluK2 | CTD | Degradation | [139] | |
| Kinases | |||||
| CASK | ND | GluK2 | -ETMA | ND | [136] |
| Phosphatases | |||||
| Calcineurin | ND | GluK2b | CTD | Function | [32,91] |
| CUB proteins | |||||
| NETO1 | ND | GluK2/3/5 | ND | Trafficking | [128,133] |
| CUB2 | GluK2/3/5 | ND | Interaction | [124,128] | |
| ND | GluK1-5 | ND | Function | [124,130] | |
| 348 RKK 350 | GluK2 | ND | Rectification | [129] | |
| NETO2 | ND | GluK1/2 | ND | Trafficking | [118,126] |
| CUB2 | GluK2/3/5 | ND | Interaction | [124,128] | |
| LDLa | GluK1/2/5 | ND | Function | [125,127,131] | |
| 347 RKK 349 | GluK2 | ND | Rectification | [129] | |
| Other proteins | |||||
| COPI | ND | GluK5 | 862 RRRRR 866 | Trafficking | [35] |
| 14-3-3 | ND | GluK2a/5 | ND | Trafficking | [32,35] |
| SNAP-25 | ND | GluK5 | CTD | Trafficking | [81] |
| Calmodulin | ND | GluK2 | CTD | ND | [32] |
| Contactin | ND | GluK2a | CTD | ND | [32] |
| Dynamin-1 | ND | GluK2a | CTD | ND | [32] |
| Dynamitin | ND | GluK2a | CTD | ND | [32] |
| NSF | ND | GluK2b | CTD | ND | [32] |
| VILIP1 | ND | GluK2b | CTD | ND | [32] |
| VILIP3 | ND | GluK2b | CTD | ND | [32] |
| Ubc9 | ND | GluK2a | 885VKTE888 | Endocytosis | [96] |
| PIAS3 | ND | GluK2a | 885VKTE888 | Endocytosis | [96] |
4.3. CUB Domain-Mediated Interactions with KARs
4.4. Interactions of KARs with Proteins of the BTB/Kelch Family
4.5. Interactions with Cell Adhesion Proteins and the Cytoskeleton
5. Conclusions
Acknowledgments
Author Contributions
Definitions
| AMPA | α-amino-3-hydroxy-5-methylisoxazole- 4-propionic acid |
| APP | amyloid precursor protein |
| BTB | broad-complex, tram-track, and bric-a-brac |
| BTB/POZ | broad-complex, tram-track, and bric-a-brac/poxvirus and zinc finger |
| CaMK | calmodulin-dependent kinase |
| CASK | (calcium/calmodulin-dependent serine protein kinase |
| CNS | central nervous system |
| COPI | coatomer protein complex I |
| COS | CV-1 (simian) in origin, carrying SV40 genetic material |
| CTD | C-terminal domain |
| CUB | complement C1r/C1s, Uegf, Bmp1 |
| Cul3 | Cullin 3 |
| DG | dorsal root ganglia |
| EndoH | endoglycosidase H |
| EPSC | excitatory postsynaptic current |
| ER | endoplasmic reticulum |
| GABA | γ-aminobutyric acid |
| GK | guanylate kinase |
| GRIP | glutamate receptor interacting protein 1 |
| iGluR | ionotropic glutamate receptor |
| JNK | c-Jun N-terminal kinase |
| KA | kainate |
| KAR | kainate receptor |
| KCC2 | potassium chloride cotransporter 2 |
| KRIP6 | kainate receptor-interacting protein for GluR6 |
| LBD | ligand binding domain |
| LDLa | low-density lipoprotein class a |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MAGUK | membrane-associated guanylate kinase |
| mGluR | metabotropic glutamate receptor |
| MLK | mixed-lineage kinase |
| Nbea | neurobeachin |
| NETO | neuropilin and tolloid-like |
| NMDA | N-methyl-D-aspartate |
| NTD | N-terminal domain |
| PDZ | post synaptic density protein (PSD-95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1) |
| PICK1 | protein interacting with PRKCA 1 |
| PKA | cAMP-dependent protein kinase |
| PKC | protein kinase C |
| PSD | postsynaptic density |
| RNA | ribonucleic acid |
| RNAi | RNA interference |
| SAP | synapse-associated protein |
| SENP | sentrin-specific protease |
| SH3 | Src homology 3 |
| SNAP | synaptosomal-associated protein |
| SUMO | small ubiquitin-related modifier |
| TMD | transmembrane domain |
Conflicts of Interest
References
- Lodge, D. The history of the pharmacology and cloning of ionotropic glutamate receptors and the development of idiosyncratic nomenclature. Neuropharmacology 2009, 56, 6–21. [Google Scholar]
- Bennett, J.A.; Dingledine, R. Topology profile for a glutamate receptor: Three transmembrane domains and a channel-lining reentrant membrane loop. Neuron 1995, 14, 373–384. [Google Scholar]
- Hollmann, M.; Maron, C.; Heinemann, S. N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron 1994, 13, 1331–1343. [Google Scholar]
- Wo, Z.G.; Oswald, R.E. Transmembrane topology of two kainate receptor subunits revealed by N-glycosylation. Proc. Natl. Acad. Sci. USA 1994, 91, 7154–7158. [Google Scholar]
- Wood, M.W.; VanDongen, H.M.; VanDongen, A.M. Structural conservation of ion conduction pathways in K channels and glutamate receptors. Proc. Natl. Acad. Sci. USA 1995, 92, 4882–4886. [Google Scholar]
- Stern-Bach, Y.; Bettler, B.; Hartley, M.; Sheppard, P.O.; O’Hara, P.J.; Heinemann, S.F. Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron 1994, 13, 1345–1357. [Google Scholar]
- Paas, Y. The macro- and microarchitectures of the ligand-binding domain of glutamate receptors. Trends Neurosci. 1998, 21, 117–125. [Google Scholar]
- Rodríguez-Moreno, A.; Sihra, T.S. Kainate receptors with a metabotropic modus operandi. Trends Neurosci. 2007, 30, 630–637. [Google Scholar]
- Contractor, A.; Mulle, C.; Swanson, G.T. Kainate receptors coming of age: Milestones of two decades of research. Trends Neurosci. 2011, 34, 154–163. [Google Scholar]
- Carta, M.; Fièvre, S.; Gorlewicz, A.; Mulle, C. Kainate receptors in the hippocampus. Eur. J. Neurosci. 2014, 39, 1835–1844. [Google Scholar]
- Wisden, W.; Seeburg, P.H. A complex mosaic of high-affinity kainate receptors in rat brain. J. Neurosci. 1993, 13, 3582–3598. [Google Scholar]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar]
- Meador-Woodruff, J.H.; Davis, K.L.; Haroutunian, V. Abnormal kainate receptor expression in prefrontal cortex in schizophrenia. Neuropsychopharmacology 2001, 24, 545–552. [Google Scholar]
- Schiffer, H.H.; Heinemann, S.F. Association of the human kainate receptor GluR7 gene (GRIK3) with recurrent major depressive disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 20–26. [Google Scholar]
- Jamain, S.; Betancur, C.; Quach, H.; Philippe, A.; Fellous, M.; Giros, B.; Gillberg, C.; Leboyer, M.; Bourgeron, T. Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry 2002, 7, 302–310. [Google Scholar]
- Pinheiro, P.; Mulle, C. Kainate receptors. Cell Tissue Res. 2006, 326, 457–482. [Google Scholar]
- Zerangue, N.; Schwappach, B.; Jan, Y.N.; Jan, L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 1999, 22, 537–548. [Google Scholar]
- Standley, S.; Roche, K.W.; McCallum, J.; Sans, N.; Wenthold, R.J. PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 2000, 28, 887–898. [Google Scholar]
- Scott, D.B.; Blanpied, T.A.; Swanson, G.T.; Zhang, C.; Ehlers, M.D. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J. Neurosci. 2001, 21, 3063–3072. [Google Scholar]
- Ren, Z.; Riley, N.J.; Garcia, E.P.; Sanders, J.M.; Swanson, G.T.; Marshall, J. Multiple trafficking signals regulate kainate receptor KA2 subunit surface expression. J. Neurosci. 2003, 23, 6608–6616. [Google Scholar]
- Jaskolski, F.; Coussen, F.; Nagarajan, N.; Normand, E.; Rosenmund, C.; Mulle, C. Subunit composition and alternative splicing regulate membrane delivery of kainate receptors. J. Neurosci. 2004, 24, 2506–2515. [Google Scholar]
- Huyghe, D.; Veran, J.; Labrousse, V.F.; Perrais, D.; Mulle, C.; Coussen, F. Endocytosis of the glutamate receptor subunit GluK3 controls polarized trafficking. J. Neurosci. 2011, 31, 11645–11654. [Google Scholar]
- Ren, Z.; Riley, N.J.; Needleman, L.A.; Sanders, J.M.; Swanson, G.T.; Marshall, J. Cell surface expression of GluR5 kainate receptors is regulated by an endoplasmic reticulum retention signal. J. Biol. Chem. 2003, 278, 52700–52709. [Google Scholar]
- Nasu-Nishimura, Y.; Hurtado, D.; Braud, S.; Tang, T.T.T.; Isaac, J.T.R.; Roche, K.W. Identification of an endoplasmic reticulum-retention motif in an intracellular loop of the kainate receptor subunit KA2. J. Neurosci. 2006, 26, 7014–7021. [Google Scholar]
- Sommer, B.; Burnashev, N.; Verdoorn, T.A.; Keinänen, K.; Sakmann, B.; Seeburg, P.H. A glutamate receptor channel with high affinity for domoate and kainate. EMBO J. 1992, 11, 1651–1656. [Google Scholar]
- Bettler, B.; Boulter, J.; Hermans-Borgmeyer, I.; O’Shea-Greenfield, A.; Deneris, E.S.; Moll, C.; Borgmeyer, U.; Hollmann, M.; Heinemann, S. Cloning of a novel glutamate receptor subunit, GluR5: Expression in the nervous system during development. Neuron 1990, 5, 583–595. [Google Scholar]
- Gregor, P.; O’Hara, B.F.; Yang, X.; Uhl, G.R. Expression and novel subunit isoforms of glutamate receptor genes GluR5 and GluR6. Neuroreport 1993, 4, 1343–1346. [Google Scholar]
- Yan, S.; Sanders, J.M.; Xu, J.; Zhu, Y.; Contractor, A.; Swanson, G.T. A C-terminal determinant of GluR6 kainate receptor trafficking. J. Neurosci. 2004, 24, 679–691. [Google Scholar]
- Pickering, D.S.; Taverna, F.A.; Salter, M.W.; Hampson, D.R. Palmitoylation of the GluR6 kainate receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 12090–12094. [Google Scholar]
- Nasu-Nishimura, Y.; Jaffe, H.; Isaac, J.T.R.; Roche, K.W. Differential regulation of kainate receptor trafficking by phosphorylation of distinct sites on GluR6. J. Biol. Chem. 2010, 285, 2847–2856. [Google Scholar]
- Birbach, A. Profilin, a multi-modal regulator of neuronal plasticity. Bioessays 2008, 30, 994–1002. [Google Scholar]
- Coussen, F.; Perrais, D.; Jaskolski, F.; Sachidhanandam, S.; Normand, E.; Bockaert, J.; Marin, P.; Mulle, C. Co-assembly of two GluR6 kainate receptor splice variants within a functional protein complex. Neuron 2005, 47, 555–566. [Google Scholar]
- Mondin, M.; Carta, M.; Normand, E.; Mulle, C.; Coussen, F. Profilin II regulates the exocytosis of kainate glutamate receptors. J. Biol. Chem. 2010, 285, 40060–40071. [Google Scholar]
- Jaskolski, F.; Normand, E.; Mulle, C.; Coussen, F. Differential trafficking of GluR7 kainate receptor subunit splice variants. J. Biol. Chem. 2005, 280, 22968–22976. [Google Scholar]
- Vivithanaporn, P.; Yan, S.; Swanson, G.T. Intracellular trafficking of KA2 kainate receptors mediated by interactions with coatomer protein complex I (COPI) and 14-3-3 chaperone systems. J. Biol. Chem. 2006, 281, 15475–15484. [Google Scholar]
- Michelsen, K.; Yuan, H.; Schwappach, B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 2005, 6, 717–722. [Google Scholar]
- Ayalon, G.; Stern-Bach, Y. Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron 2001, 31, 103–113. [Google Scholar]
- Kumar, J.; Schuck, P.; Mayer, M.L. Structure and assembly mechanism for heteromeric kainate receptors. Neuron 2011, 71, 319–331. [Google Scholar]
- Plested, A.J.R.; Mayer, M.L. Structure and mechanism of kainate receptor modulation by anions. Neuron 2007, 53, 829–841. [Google Scholar]
- Mah, S.J.; Cornell, E.; Mitchell, N.A.; Fleck, M.W. Glutamate receptor trafficking: Endoplasmic reticulum quality control involves ligand binding and receptor function. J. Neurosci. 2005, 25, 2215–2225. [Google Scholar]
- Valluru, L.; Xu, J.; Zhu, Y.; Yan, S.; Contractor, A.; Swanson, G.T. Ligand binding is a critical requirement for plasma membrane expression of heteromeric kainate receptors. J. Biol. Chem. 2005, 280, 6085–6093. [Google Scholar]
- Fisher, J.L.; Housley, P.R. Agonist binding to the GluK5 subunit is sufficient for functional surface expression of heteromeric GluK2/GluK5 kainate receptors. Cell. Mol. Neurobiol. 2013, 33, 1099–1108. [Google Scholar]
- Perrais, D.; Veran, J.; Mulle, C. Gating and permeation of kainate receptors: Differences unveiled. Trends Pharmacol. Sci. 2010, 31, 516–522. [Google Scholar]
- Gill, M.B.; Vivithanaporn, P.; Swanson, G.T. Glutamate binding and conformational flexibility of ligand-binding domains are critical early determinants of efficient kainate receptor biogenesis. J. Biol. Chem. 2009, 284, 14503–14512. [Google Scholar]
- Priel, A.; Selak, S.; Lerma, J.; Stern-Bach, Y. Block of kainate receptor desensitization uncovers a key trafficking checkpoint. Neuron 2006, 52, 1037–1046. [Google Scholar]
- Vivithanaporn, P.; Lash, L.L.; Marszalec, W.; Swanson, G.T. Critical roles for the M3-S2 transduction linker domain in kainate receptor assembly and post-assembly trafficking. J. Neurosci. 2007, 27, 10423–10433. [Google Scholar]
- Ball, S.M.; Atlason, P.T.; Shittu-Balogun, O.O.; Molnár, E. Assembly and intracellular distribution of kainate receptors is determined by RNA editing and subunit composition. J. Neurochem. 2010, 114, 1805–1818. [Google Scholar]
- Reiner, A.; Arant, R.J.; Isacoff, E.Y. Assembly stoichiometry of the GluK2/GluK5 kainate receptor complex. Cell Rep. 2012, 1, 234–240. [Google Scholar]
- Ma-Högemeier, Z.L.; Körber, C.; Werner, M.; Racine, D.; Muth-Köhne, E.; Tapken, D.; Hollmann, M. Oligomerization in the endoplasmic reticulum and intracellular trafficking of kainate receptors are subunit-dependent but not editing-dependent. J. Neurochem. 2010, 113, 1403–1415. [Google Scholar]
- Barberis, A.; Sachidhanandam, S.; Mulle, C. GluR6/KA2 kainate receptors mediate slow-deactivating currents. J. Neurosci. 2008, 28, 6402–6406. [Google Scholar]
- Gallyas, F., Jr.; Ball, S.M.; Molnar, E. Assembly and cell surface expression of KA-2 subunit-containing kainate receptors. J. Neurochem. 2003, 86, 1414–1427. [Google Scholar]
- Hayes, D.M.; Braud, S.; Hurtado, D.E.; McCallum, J.; Standley, S.; Isaac, J.T.R.; Roche, K.W. Trafficking and surface expression of the glutamate receptor subunit, KA2. Biochem. Biophys. Res. Commun. 2003, 310, 8–13. [Google Scholar]
- O’Kelly, I.; Butler, M.H.; Zilberberg, N.; Goldstein, S.A.N. Forward transport. 14-3-3 binding overcomes retention in endoplasmic reticulum by dibasic signals. Cell 2002, 111, 577–588. [Google Scholar]
- Yuan, H.; Michelsen, K.; Schwappach, B. 14-3-3 dimers probe the assembly status of multimeric membrane proteins. Curr. Biol. 2003, 13, 638–646. [Google Scholar]
- Jeanclos, E.M.; Lin, L.; Treuil, M.W.; Rao, J.; DeCoster, M.A.; Anand, R. The chaperone protein 14-3-3eta interacts with the nicotinic acetylcholine receptor alpha 4 subunit. Evidence for a dynamic role in subunit stabilization. J. Biol. Chem. 2001, 276, 28281–28290. [Google Scholar]
- Rajan, S.; Preisig-Müller, R.; Wischmeyer, E.; Nehring, R.; Hanley, P.J.; Renigunta, V.; Musset, B.; Schlichthörl, G.; Derst, C.; Karschin, A.; et al. Interaction with 14-3-3 proteins promotes functional expression of the potassium channels TASK-1 and TASK-3. J. Physiol. 2002, 545, 13–26. [Google Scholar]
- Sun, C.; Qiao, H.; Zhou, Q.; Wang, Y.; Wu, Y.; Zhou, Y.; Li, Y. Modulation of GluK2a subunit-containing kainate receptors by 14-3-3 proteins. J. Biol. Chem. 2013, 288, 24676–24690. [Google Scholar]
- Christensen, J.K.; Paternain, A.V.; Selak, S.; Ahring, P.K.; Lerma, J. A mosaic of functional kainate receptors in hippocampal interneurons. J. Neurosci. 2004, 24, 8986–8993. [Google Scholar]
- Ruiz, A.; Sachidhanandam, S.; Utvik, J.K.; Coussen, F.; Mulle, C. Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J. Neurosci. 2005, 25, 11710–11718. [Google Scholar]
- Darstein, M.; Petralia, R.S.; Swanson, G.T.; Wenthold, R.J.; Heinemann, S.F. Distribution of kainate receptor subunits at hippocampal mossy fiber synapses. J. Neurosci. 2003, 23, 8013–8019. [Google Scholar]
- Kayadjanian, N.; Lee, H.S.; Piña-Crespo, J.; Heinemann, S.F. Localization of glutamate receptors to distal dendrites depends on subunit composition and the kinesin motor protein KIF17. Mol. Cell. Neurosci. 2007, 34, 219–230. [Google Scholar]
- Pinheiro, P.S.; Perrais, D.; Coussen, F.; Barhanin, J.; Bettler, B.; Mann, J.R.; Malva, J.O.; Heinemann, S.F.; Mulle, C. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc. Natl. Acad. Sci. USA 2007, 104, 12181–12186. [Google Scholar]
- Nair, R.; Lauks, J.; Jung, S.; Cooke, N.E.; de Wit, H.; Brose, N.; Kilimann, M.W.; Verhage, M.; Rhee, J. Neurobeachin regulates neurotransmitter receptor trafficking to synapses. J. Cell Biol. 2013, 200, 61–80. [Google Scholar]
- Wang, X.; Herberg, F.W.; Laue, M.M.; Wullner, C.; Hu, B.; Petrasch-Parwez, E.; Kilimann, M.W. Neurobeachin: A protein kinase A-anchoring, beige/Chediak-higashi protein homolog implicated in neuronal membrane traffic. J. Neurosci. 2000, 20, 8551–8565. [Google Scholar]
- Su, Y.; Balice-Gordon, R.J.; Hess, D.M.; Landsman, D.S.; Minarcik, J.; Golden, J.; Hurwitz, I.; Liebhaber, S.A.; Cooke, N.E. Neurobeachin is essential for neuromuscular synaptic transmission. J. Neurosci. 2004, 24, 3627–3636. [Google Scholar]
- Medrihan, L.; Rohlmann, A.; Fairless, R.; Andrae, J.; Döring, M.; Missler, M.; Zhang, W.; Kilimann, M.W. Neurobeachin, a protein implicated in membrane protein traffic and autism, is required for the formation and functioning of central synapses. J. Physiol. 2009, 587, 5095–5106. [Google Scholar]
- Niesmann, K.; Breuer, D.; Brockhaus, J.; Born, G.; Wolff, I.; Reissner, C.; Kilimann, M.W.; Rohlmann, A.; Missler, M. Dendritic spine formation and synaptic function require neurobeachin. Nat. Commun. 2011, 2, 557. [Google Scholar]
- Murata, Y.; Constantine-Paton, M. Postsynaptic density scaffold SAP102 regulates cortical synapse development through EphB and PAK signaling pathway. J. Neurosci. 2013, 33, 5040–5052. [Google Scholar]
- Lauks, J.; Klemmer, P.; Farzana, F.; Karupothula, R.; Zalm, R.; Cooke, N.E.; Li, K.W.; Smit, A.B.; Toonen, R.; Verhage, M. Synapse associated protein 102 (SAP102) binds the C-terminal part of the scaffolding protein neurobeachin. PLoS One 2012, 7, e39420. [Google Scholar]
- Schiffer, H.H.; Swanson, G.T.; Heinemann, S.F. Rat GluR7 and a carboxy-terminal splice variant, GluR7b, are functional kainate receptor subunits with a low sensitivity to glutamate. Neuron 1997, 19, 1141–1146. [Google Scholar]
- Egebjerg, J.; Bettler, B.; Hermans-Borgmeyer, I.; Heinemann, S. Cloning of a cDNA for a glutamate receptor subunit activated by kainate but not AMPA. Nature 1991, 351, 745–748. [Google Scholar]
- Bettler, B.; Egebjerg, J.; Sharma, G.; Pecht, G.; Hermans-Borgmeyer, I.; Moll, C.; Stevens, C.F.; Heinemann, S. Cloning of a putative glutamate receptor: A low affinity kainate-binding subunit. Neuron 1992, 8, 257–265. [Google Scholar]
- Werner, P.; Voigt, M.; Keinänen, K.; Wisden, W.; Seeburg, P.H. Cloning of a putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells. Nature 1991, 351, 742–744. [Google Scholar]
- Herb, A.; Burnashev, N.; Werner, P.; Sakmann, B.; Wisden, W.; Seeburg, P.H. The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron 1992, 8, 775–785. [Google Scholar]
- Barbon, A.; Vallini, I.; Barlati, S. Genomic organization of the human GRIK2 gene and evidence for multiple splicing variants. Gene 2001, 274, 187–197. [Google Scholar]
- Dildy-Mayfield, J.E.; Harris, R.A. Activation of protein kinase C inhibits kainate-induced currents in oocytes expressing glutamate receptor subunits. J. Neurochem. 1994, 62, 1639–1642. [Google Scholar]
- Park, Y.; Jo, J.; Isaac, J.T.R.; Cho, K. Long-term depression of kainate receptor-mediated synaptic transmission. Neuron 2006, 49, 95–106. [Google Scholar]
- Staudinger, J.; Lu, J.; Olson, E.N. Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-alpha. J. Biol. Chem. 1997, 272, 32019–32024. [Google Scholar]
- Hirbec, H.; Francis, J.C.; Lauri, S.E.; Braithwaite, S.P.; Coussen, F.; Mulle, C.; Dev, K.K.; Coutinho, V.; Meyer, G.; Isaac, J.T.R.; et al. Rapid and differential regulation of AMPA and kainate receptors at hippocampal mossy fibre synapses by PICK1 and GRIP. Neuron 2003, 37, 625–638. [Google Scholar]
- Cho, K.; Francis, J.C.; Hirbec, H.; Dev, K.; Brown, M.W.; Henley, J.M.; Bashir, Z.I. Regulation of kainate receptors by protein kinase C and metabotropic glutamate receptors. J. Physiol. 2003, 548, 723–730. [Google Scholar]
- Selak, S.; Paternain, A.V.; Aller, M.I.; Aller, I.M.; Picó, E.; Rivera, R.; Lerma, J. A role for SNAP25 in internalization of kainate receptors and synaptic plasticity. Neuron 2009, 63, 357–371. [Google Scholar]
- Zhang, Z.; Wang, D.; Sun, T.; Xu, J.; Chiang, H.C.; Shin, W.; Wu, L.G. The SNARE proteins SNAP25 and synaptobrevin are involved in endocytosis at hippocampal synapses. J. Neurosci. 2013, 33, 9169–9175. [Google Scholar]
- Rivera, R.; Rozas, J.L.; Lerma, J. PKC-dependent autoregulation of membrane kainate receptors. EMBO J. 2007, 26, 4359–4367. [Google Scholar]
- Klee, C.B.; Crouch, T.H.; Krinks, M.H. Calcineurin: A calcium- and calmodulin-binding protein of the nervous system. Proc. Natl. Acad. Sci. USA 1979, 76, 6270–6273. [Google Scholar]
- Kohout, S.C.; Corbalán-García, S.; Torrecillas, A.; Goméz-Fernandéz, J.C.; Falke, J.J. C2 domains of protein kinase C isoforms alpha, beta, and gamma: Activation parameters and calcium stoichiometries of the membrane-bound state. Biochemistry 2002, 41, 11411–11424. [Google Scholar]
- Stewart, A.A.; Ingebritsen, T.S.; Manalan, A.; Klee, C.B.; Cohen, P. Discovery of a Ca2+- and calmodulin-dependent protein phosphatase: Probable identity with calcineurin (CaM-BP80). FEBS Lett. 1982, 137, 80–84. [Google Scholar]
- Perrino, B.A.; Ng, L.Y.; Soderling, T.R. Calcium regulation of calcineurin phosphatase activity by its B subunit and calmodulin. Role of the autoinhibitory domain. J. Biol. Chem. 1995, 270, 340–346. [Google Scholar]
- Wang, L.Y.; Taverna, F.A.; Huang, X.P.; MacDonald, J.F.; Hampson, D.R. Phosphorylation and modulation of a kainate receptor (GluR6) by cAMP-dependent protein kinase. Science 1993, 259, 1173–1175. [Google Scholar]
- Raymond, L.A.; Blackstone, C.D.; Huganir, R.L. Phosphorylation and modulation of recombinant GluR6 glutamate receptors by cAMP-dependent protein kinase. Nature 1993, 361, 637–641. [Google Scholar]
- Kornreich, B.G.; Niu, L.; Roberson, M.S.; Oswald, R.E. Identification of C-terminal domain residues involved in protein kinase A-mediated potentiation of kainate receptor subtype 6. Neuroscience 2007, 146, 1158–1168. [Google Scholar]
- Traynelis, S.F.; Wahl, P. Control of rat GluR6 glutamate receptor open probability by protein kinase A and calcineurin. J. Physiol. 1997, 503, 513–531. [Google Scholar]
- Ghetti, A.; Heinemann, S.F. NMDA-Dependent modulation of hippocampal kainate receptors by calcineurin and Ca(2+)/calmodulin-dependent protein kinase. J. Neurosci. 2000, 20, 2766–2773. [Google Scholar]
- Rebola, N.; Sachidhanandam, S.; Perrais, D.; Cunha, R.A.; Mulle, C. Short-term plasticity of kainate receptor-mediated EPSCs induced by NMDA receptors at hippocampal mossy fiber synapses. J. Neurosci. 2007, 27, 3987–3993. [Google Scholar]
- Carta, M.; Opazo, P.; Veran, J.; Athané, A.; Choquet, D.; Coussen, F.; Mulle, C. CaMKII-dependent phosphorylation of GluK5 mediates plasticity of kainate receptors. EMBO J. 2013, 32, 496–510. [Google Scholar]
- Caporale, N.; Dan, Y. Spike timing-dependent plasticity: A Hebbian learning rule. Annu. Rev. Neurosci. 2008, 31, 25–46. [Google Scholar]
- Martin, S.; Nishimune, A.; Mellor, J.R.; Henley, J.M. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature 2007, 447, 321–325. [Google Scholar]
- Wilkinson, K.A.; Nishimune, A.; Henley, J.M. Analysis of SUMO-1 modification of neuronal proteins containing consensus SUMOylation motifs. Neurosci. Lett. 2008, 436, 239–244. [Google Scholar]
- Lüscher, C.; Xia, H.; Beattie, E.C.; Carroll, R.C.; von Zastrow, M.; Malenka, R.C.; Nicoll, R.A. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron 1999, 24, 649–658. [Google Scholar]
- Borgdorff, A.J.; Choquet, D. Regulation of AMPA receptor lateral movements. Nature 2002, 417, 649–653. [Google Scholar]
- Martin, S.; Henley, J.M. Activity-dependent endocytic sorting of kainate receptors to recycling or degradation pathways. EMBO J. 2004, 23, 4749–4759. [Google Scholar]
- González-González, I.M.; Henley, J.M. Postsynaptic kainate receptor recycling and surface expression are regulated by metabotropic autoreceptor signaling. Traffic 2013, 14, 810–822. [Google Scholar]
- Konopacki, F.A.; Jaafari, N.; Rocca, D.L.; Wilkinson, K.A.; Chamberlain, S.; Rubin, P.; Kantamneni, S.; Mellor, J.R.; Henley, J.M. Agonist-induced PKC phosphorylation regulates GluK2 SUMOylation and kainate receptor endocytosis. Proc. Natl. Acad. Sci. USA 2011, 108, 19772–19777. [Google Scholar]
- Chamberlain, S.E.L.; González-González, I.M.; Wilkinson, K.A.; Konopacki, F.A.; Kantamneni, S.; Henley, J.M.; Mellor, J.R. SUMOylation and phosphorylation of GluK2 regulate kainate receptor trafficking and synaptic plasticity. Nat. Neurosci. 2012, 15, 845–852. [Google Scholar]
- Garcia, E.P.; Mehta, S.; Blair, L.A.; Wells, D.G.; Shang, J.; Fukushima, T.; Fallon, J.R.; Garner, C.C.; Marshall, J. SAP90 binds and clusters kainate receptors causing incomplete desensitization. Neuron 1998, 21, 727–739. [Google Scholar]
- Bowie, D.; Garcia, E.P.; Marshall, J.; Traynelis, S.F.; Lange, G.D. Allosteric regulation and spatial distribution of kainate receptors bound to ancillary proteins. J. Physiol. 2003, 547, 373–385. [Google Scholar]
- Savinainen, A.; Garcia, E.P.; Dorow, D.; Marshall, J.; Liu, Y.F. Kainate receptor activation induces mixed lineage kinase-mediated cellular signaling cascades via post-synaptic density protein 95. J. Biol. Chem. 2001, 276, 11382–11386. [Google Scholar]
- Hirai, S.; Katoh, M.; Terada, M.; Kyriakis, J.M.; Zon, L.I.; Rana, A.; Avruch, J.; Ohno, S. MST/MLK2, a member of the mixed lineage kinase family, directly phosphorylates and activates SEK1, an activator of c-Jun N-terminal kinase/stress-activated protein kinase. J. Biol. Chem. 1997, 272, 15167–15173. [Google Scholar]
- Nagata, K.; Puls, A.; Futter, C.; Aspenstrom, P.; Schaefer, E.; Nakata, T.; Hirokawa, N.; Hall, A. The MAP kinase kinase kinase MLK2 co-localizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO J. 1998, 17, 149–158. [Google Scholar]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar]
- Dérijard, B.; Raingeaud, J.; Barrett, T.; Wu, I.H.; Han, J.; Ulevitch, R.J.; Davis, R.J. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 1995, 267, 682–685. [Google Scholar]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar]
- Tian, H.; Zhang, Q.G.; Zhu, G.X.; Pei, D.S.; Guan, Q.H.; Zhang, G.Y. Activation of c-Jun NH2-terminal kinase 3 is mediated by the GluR6.PSD-95.MLK3 signaling module following cerebral ischemia in rat hippocampus. Brain Res. 2005, 1061, 57–66. [Google Scholar]
- Pei, D.S.; Wang, X.T.; Liu, Y.; Sun, Y.F.; Guan, Q.H.; Wang, W.; Yan, J.Z.; Zong, Y.Y.; Xu, T.L.; Zhang, G.Y. Neuroprotection against ischaemic brain injury by a GluR6-9c peptide containing the TAT protein transduction sequence. Brain 2006, 129, 465–479. [Google Scholar]
- Liu, X.M.; Pei, D.S.; Guan, Q.H.; Sun, Y.F.; Wang, X.T.; Zhang, Q.X.; Zhang, G.Y. Neuroprotection of Tat-GluR6-9c against neuronal death induced by kainate in rat hippocampus via nuclear and non-nuclear pathways. J. Biol. Chem. 2006, 281, 17432–17445. [Google Scholar]
- Mehta, S.; Wu, H.; Garner, C.C.; Marshall, J. Molecular mechanisms regulating the differential association of kainate receptor subunits with SAP90/PSD-95 and SAP97. J. Biol. Chem. 2001, 276, 16092–16099. [Google Scholar]
- Müller, B.M.; Kistner, U.; Veh, R.W.; Cases-Langhoff, C.; Becker, B.; Gundelfinger, E.D.; Garner, C.C. Molecular characterization and spatial distribution of SAP97, a novel presynaptic protein homologous to SAP90 and the Drosophila discs-large tumor suppressor protein. J. Neurosci. 1995, 15, 2354–2366. [Google Scholar]
- Rao, A.; Kim, E.; Sheng, M.; Craig, A.M. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J. Neurosci. 1998, 18, 1217–1229. [Google Scholar]
- Tang, M.; Ivakine, E.; Mahadevan, V.; Salter, M.W.; McInnes, R.R. Neto2 interacts with the scaffolding protein GRIP and regulates synaptic abundance of kainate receptors. PLoS One 2012, 7, e51433. [Google Scholar]
- Setou, M.; Seog, D.H.; Tanaka, Y.; Kanai, Y.; Takei, Y.; Kawagishi, M.; Hirokawa, N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature 2002, 417, 83–87. [Google Scholar]
- Ng, D.; Pitcher, G.M.; Szilard, R.K.; Sertié, A.; Kanisek, M.; Clapcote, S.J.; Lipina, T.; Kalia, L.V.; Joo, D.; McKerlie, C.; et al. Neto1 is a novel CUB-domain NMDA receptor-interacting protein required for synaptic plasticity and learning. PLoS Biol. 2009, 7, e41. [Google Scholar]
- Stöhr, H.; Berger, C.; Fröhlich, S.; Weber, B.H.F. A novel gene encoding a putative transmembrane protein with two extracellular CUB domains and a low-density lipoprotein class A module: Isolation of alternatively spliced isoforms in retina and brain. Gene 2002, 286, 223–231. [Google Scholar]
- Michishita, M.; Ikeda, T.; Nakashiba, T.; Ogawa, M.; Tashiro, K.; Honjo, T.; Doi, K.; Itohara, S.; Endo, S. A novel gene, Btcl1, encoding CUB and LDLa domains is expressed in restricted areas of mouse brain. Biochem. Biophys. Res. Commun. 2003, 306, 680–686. [Google Scholar]
- Michishita, M.; Ikeda, T.; Nakashiba, T.; Ogawa, M.; Tashiro, K.; Honjo, T.; Doi, K.; Itohara, S.; Endo, S. Expression of Btcl2, a novel member of Btcl gene family, during development of the central nervous system. Dev. Brain Res. 2004, 153, 135–142. [Google Scholar]
- Straub, C.; Hunt, D.L.; Yamasaki, M.; Kim, K.S.; Watanabe, M.; Castillo, P.E.; Tomita, S. Distinct functions of kainate receptors in the brain are determined by the auxiliary subunit Neto1. Nat. Neurosci. 2011, 14, 866–873. [Google Scholar]
- Straub, C.; Zhang, W.; Howe, J.R. Neto2 modulation of kainate receptors with different subunit compositions. J. Neurosci. 2011, 31, 8078–8082. [Google Scholar]
- Copits, B.A.; Robbins, J.S.; Frausto, S.; Swanson, G.T. Synaptic targeting and functional modulation of GluK1 kainate receptors by the auxiliary neuropilin and tolloid-like (NETO) proteins. J. Neurosci. 2011, 31, 7334–7340. [Google Scholar]
- Zhang, W.; St-Gelais, F.; Grabner, C.P.; Trinidad, J.C.; Sumioka, A.; Morimoto-Tomita, M.; Kim, K.S.; Straub, C.; Burlingame, A.L.; Howe, J.R.; et al. A transmembrane accessory subunit that modulates kainate-type glutamate receptors. Neuron 2009, 61, 385–396. [Google Scholar]
- Tang, M.; Pelkey, K.A.; Ng, D.; Ivakine, E.; McBain, C.J.; Salter, M.W.; McInnes, R.R. Neto1 is an auxiliary subunit of native synaptic kainate receptors. J. Neurosci. 2011, 31, 10009–10018. [Google Scholar]
- Fisher, J.L.; Mott, D.D. Neto1 is an auxiliary subunit of native synaptic kainate receptors. J. Neurosci. 2012, 32, 12928–12933. [Google Scholar]
- Fisher, J.L.; Mott, D.D. Modulation of homomeric and heteromeric kainate receptors by the auxiliary subunit Neto1. J. Physiol. 2013, 591, 4711–4724. [Google Scholar]
- Zhang, W.; Devi, S.P.S.; Tomita, S.; Howe, J.R. Auxiliary proteins promote modal gating of AMPA- and kainate-type glutamate receptors. Eur. J. Neurosci. 2014, 39, 1138–1147. [Google Scholar]
- Chen, L.; Chetkovich, D.M.; Petralia, R.S.; Sweeney, N.T.; Kawasaki, Y.; Wenthold, R.J.; Bredt, D.S.; Nicoll, R.A. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 2000, 408, 936–943. [Google Scholar]
- Wyeth, M.S.; Pelkey, K.A.; Petralia, R.S.; Salter, M.W.; McInnes, R.R.; McBain, C.J. Neto auxiliary protein interactions regulate kainate and NMDA receptor subunit localization at mossy fiber-CA3 pyramidal cell synapses. J. Neurosci. 2014, 34, 622–628. [Google Scholar]
- Cousins, S.L.; Innocent, N.; Stephenson, F.A. Neto1 associates with the NMDA receptor/amyloid precursor protein complex. J. Neurochem. 2013, 126, 554–564. [Google Scholar]
- Ivakine, E.A.; Acton, B.A.; Mahadevan, V.; Ormond, J.; Tang, M.; Pressey, J.C.; Huang, M.Y.; Ng, D.; Delpire, E.; Salter, M.W.; Woodin, M.A.; McInnes, R.R. Neto2 is a KCC2 interacting protein required for neuronal Cl- regulation in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 3561–3566. [Google Scholar]
- Coussen, F.; Normand, E.; Marchal, C.; Costet, P.; Choquet, D.; Lambert, M.; Mège, R.M.; Mulle, C. Recruitment of the kainate receptor subunit glutamate receptor 6 by cadherin/catenin complexes. J. Neurosci. 2002, 22, 6426–6436. [Google Scholar]
- Copits, B.A.; Swanson, G.T. Kainate receptor post-translational modifications differentially regulate association with 4.1N to control activity-dependent receptor endocytosis. J. Biol. Chem. 2013, 288, 8952–8965. [Google Scholar]
- Laezza, F.; Wilding, T.J.; Sequeira, S.; Coussen, F.; Zhang, X.Z.; Hill-Robinson, R.; Mulle, C.; Huettner, J.E.; Craig, A.M. KRIP6: A novel BTB/kelch protein regulating function of kainate receptors. Mol. Cell. Neurosci. 2007, 34, 539–550. [Google Scholar]
- Salinas, G.D.; Blair, L.A.C.; Needleman, L.A.; Gonzales, J.D.; Chen, Y.; Li, M.; Singer, J.D.; Marshall, J. Actinfilin is a Cul3 substrate adaptor, linking GluR6 kainate receptor subunits to the ubiquitin-proteasome pathway. J. Biol. Chem. 2006, 281, 40164–40173. [Google Scholar]
- Laezza, F.; Wilding, T.J.; Sequeira, S.; Craig, A.M.; Huettner, J.E. The BTB/kelch protein, KRIP6, modulates the interaction of PICK1 with GluR6 kainate receptors. Neuropharmacology 2008, 55, 1131–1139. [Google Scholar]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar]
- Jo, K.; Derin, R.; Li, M.; Bredt, D.S. Characterization of MALS/Velis-1, -2, and -3: A family of mammalian LIN-7 homologs enriched at brain synapses in association with the postsynaptic density-95/NMDA receptor postsynaptic complex. J. Neurosci. 1999, 19, 4189–4199. [Google Scholar]
- Perego, C.; Vanoni, C.; Villa, A.; Longhi, R.; Kaech, S.M.; Fröhli, E.; Hajnal, A.; Kim, S.K.; Pietrini, G. PDZ-mediated interactions retain the epithelial GABA transporter on the basolateral surface of polarized epithelial cells. EMBO J. 1999, 18, 2384–2393. [Google Scholar]
- Perego, C.; Vanoni, C.; Massari, S.; Longhi, R.; Pietrini, G. Mammalian LIN-7 PDZ proteins associate with beta-catenin at the cell-cell junctions of epithelia and neurons. EMBO J. 2000, 19, 3978–3989. [Google Scholar]
- Meyer, G.; Varoqueaux, F.; Neeb, A.; Oschlies, M.; Brose, N. The complexity of PDZ domain-mediated interactions at glutamatergic synapses: A case study on neuroligin. Neuropharmacology 2004, 47, 724–733. [Google Scholar]
- Budreck, E.C.; Kwon, O.B.; Jung, J.H.; Baudouin, S.; Thommen, A.; Kim, H.S.; Fukazawa, Y.; Harada, H.; Tabuchi, K.; Shigemoto, R.; et al. Neuroligin-1 controls synaptic abundance of NMDA-type glutamate receptors through extracellular coupling. Proc. Natl. Acad. Sci. USA 2013, 110, 725–730. [Google Scholar]
- Bennett, V. The spectrin-actin junction of erythrocyte membrane skeletons. Biochim. Biophys. Acta 1989, 988, 107–121. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pahl, S.; Tapken, D.; Haering, S.C.; Hollmann, M. Trafficking of Kainate Receptors. Membranes 2014, 4, 565-595. https://doi.org/10.3390/membranes4030565
Pahl S, Tapken D, Haering SC, Hollmann M. Trafficking of Kainate Receptors. Membranes. 2014; 4(3):565-595. https://doi.org/10.3390/membranes4030565
Chicago/Turabian StylePahl, Steffen, Daniel Tapken, Simon C. Haering, and Michael Hollmann. 2014. "Trafficking of Kainate Receptors" Membranes 4, no. 3: 565-595. https://doi.org/10.3390/membranes4030565
APA StylePahl, S., Tapken, D., Haering, S. C., & Hollmann, M. (2014). Trafficking of Kainate Receptors. Membranes, 4(3), 565-595. https://doi.org/10.3390/membranes4030565