
Transcriptome Survey of a Marine Food Fish: Asian Seabass (Lates calcarifer)


Abstract
:1. Introduction
2. Experimental Section
2.1. Sample and Library Preparation, Sequencing and Quality Control
2.2. Filtering of Contaminating Reads
2.3. Sequence Assembly, Mapping and Redundancy Removal
2.4. GC Content and Microsatellites

2.5. Generating a Refined Nile Tilapia Sequence Dataset as a Reference for Asian Seabass Transcriptome Annotation
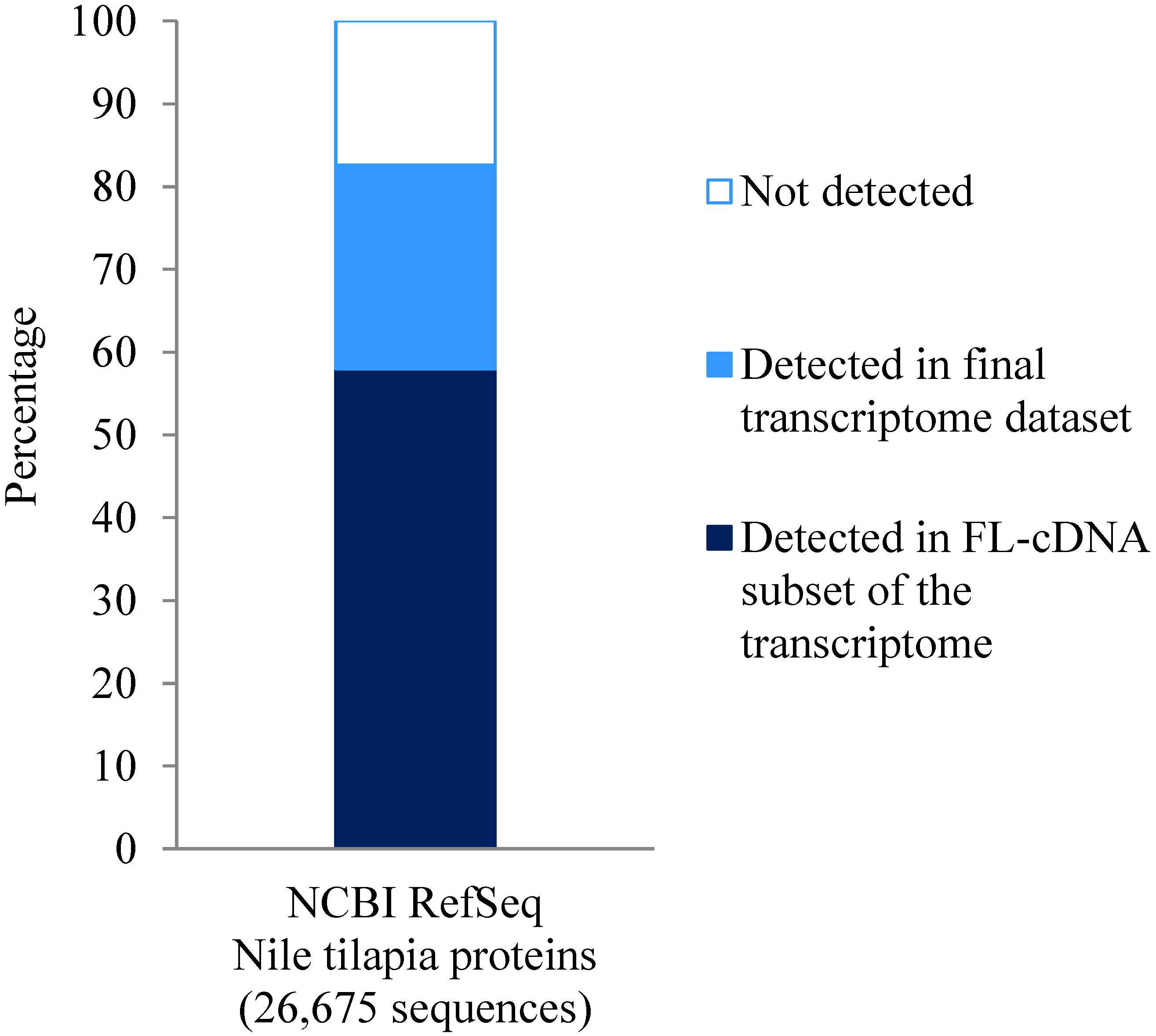
2.6. Sequence Annotation, Estimation of Completeness and Full-Length Sequence Prediction
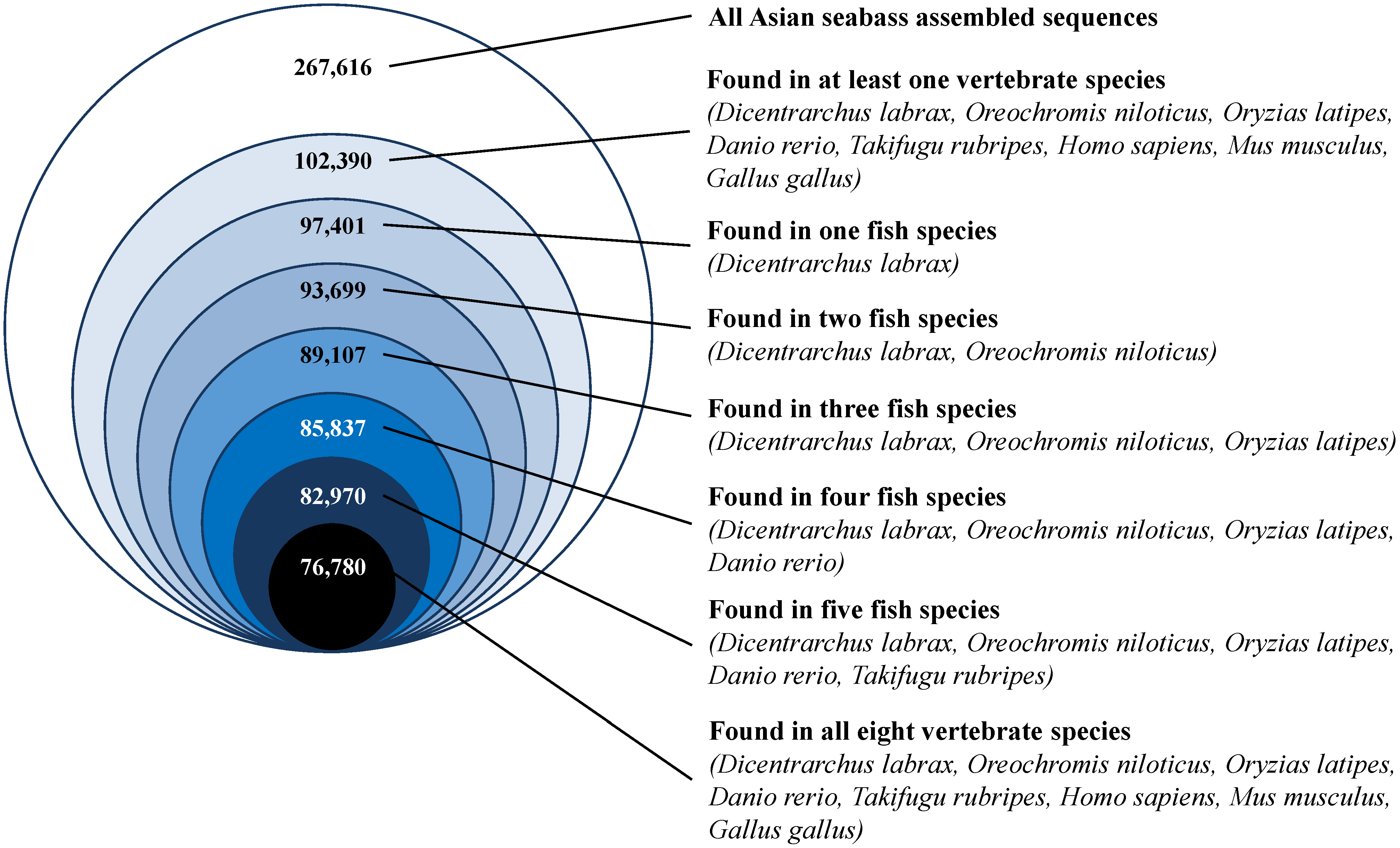
2.7. Sequence Conservation with Other Vertebrates
2.8. Pathway Distribution and Analysis of Organ-Specific Sequences
2.9. Evaluation of Asian Seabass Transcriptome as a Reference for RNA-seq Experiments
3. Results
3.1. Sequencing, Quality Control and Filtering of Reads
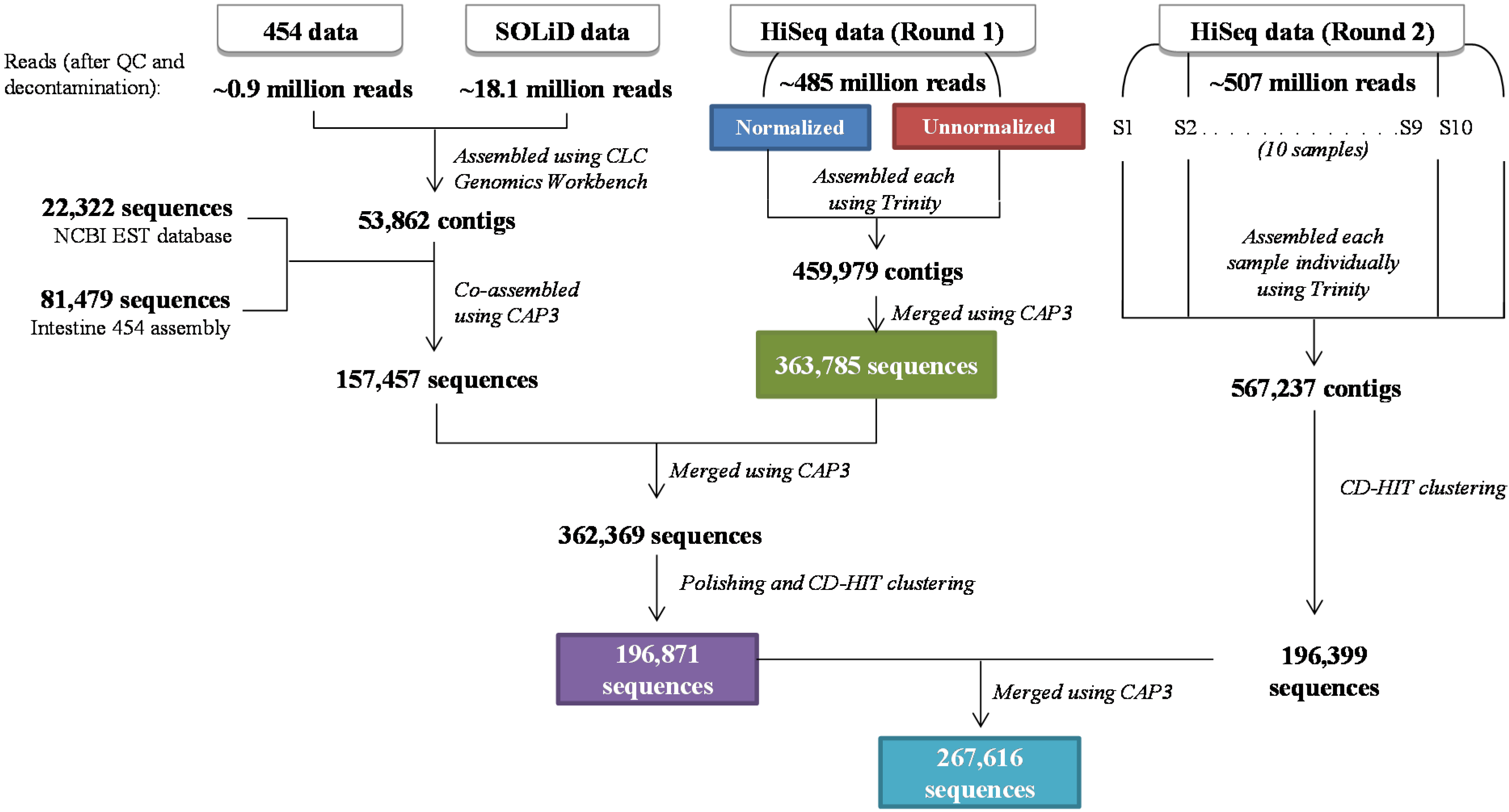
3.2. Assembly of the Asian Seabass Transcriptome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 454 | SOLiD | HiSeq Normalized (HN) | HiSeq Unnormalized (HU) | HiSeq Round 2 (HR2) | |
|---|---|---|---|---|---|
| Raw reads | 1,068,743 | 38,336,497 | 236,141,512 | 284,842,758 | 665,889,628 |
| After adaptor and quality trimming | 1,043,802 | 18,792,575 | 225,382,446 | 262,102,222 | 520,397,768 |
| 98% * | 49% | 95% | 92% | 78% | |
| After rRNA and microbial removal | 908,019 | 18,189,484 | 224,731,908 | 260,937,802 | 507,301,076 |
| 85% | 47% | 95% | 92% | 76% |
| Assembly | Number of Contigs | Number of Contigs after cd-hit-est | Number of Contigs ≥1 kb * | Average Contig Length | Maximum Contig Length |
|---|---|---|---|---|---|
| HiSeq Normalized (HN) | 194,957 | 106,768 | 28,548 (27%) | 885 | 17,036 |
| HiSeq Unnormalized (HU) | 265,022 | 126,377 | 40,752 (32%) | 1082 | 30,061 |
| HiSeq Round 1 (HR1) | 363,785 | 182,911 | 47,458 (26%) | 927 | 31,251 |
| Multiplatform (MP) | 362,369 | 196,871 | 51,947 (26%) | 965 | 31,251 |
3.3. GC-Content and Microsatellite Distribution
| Total length (bp) | 262,023,963 |
| Number of contigs | 267,616 |
| Number of contigs ≥1 kb | 70,588 |
| Min length (bp) | 200 |
| Max length (bp) | 31,251 |
| Average length (bp) | 979 |
| Total GC count (bp) | 121,345,916 |
| GC-content (%) | 46.31 |
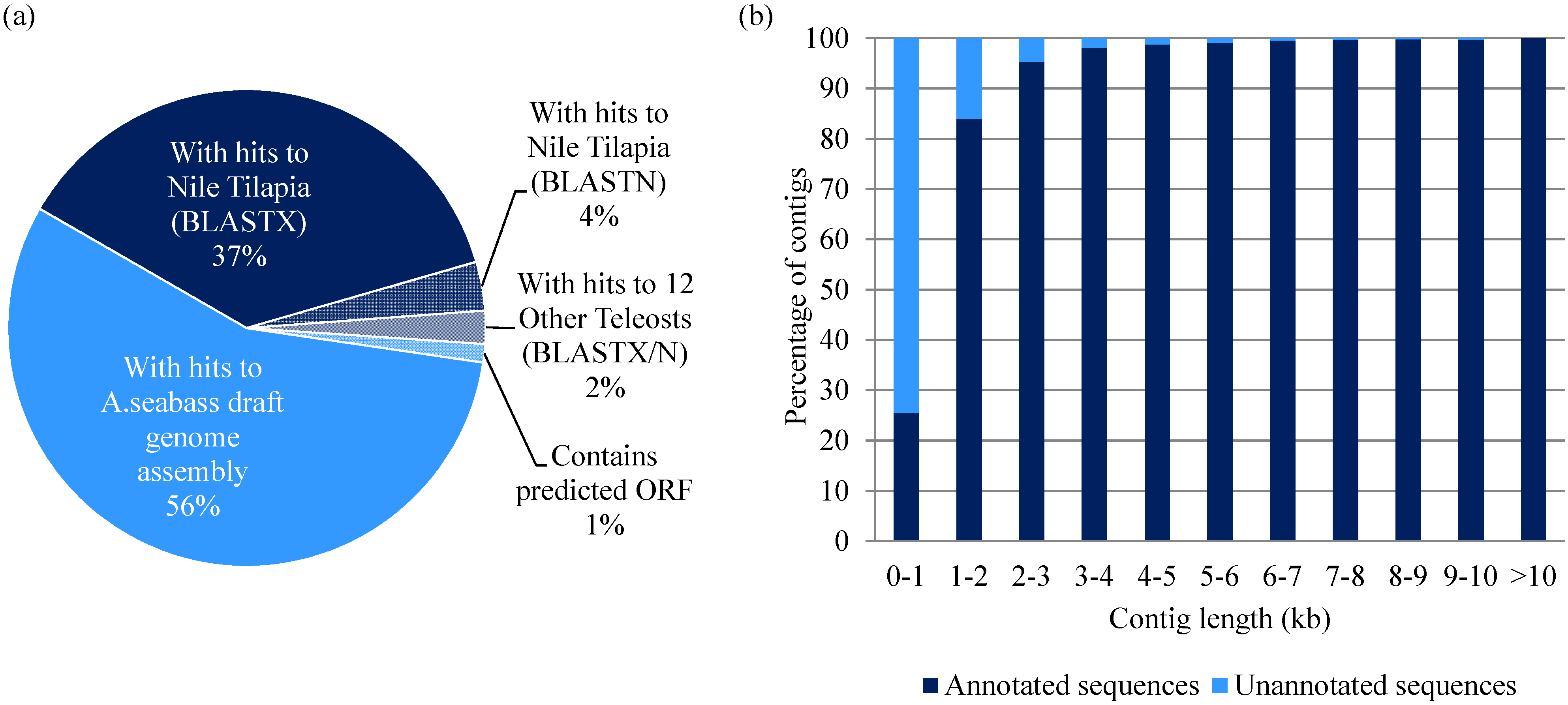
3.4. Sequence Annotation of Transcriptome Contigs and Prediction of Full-Length cDNAs


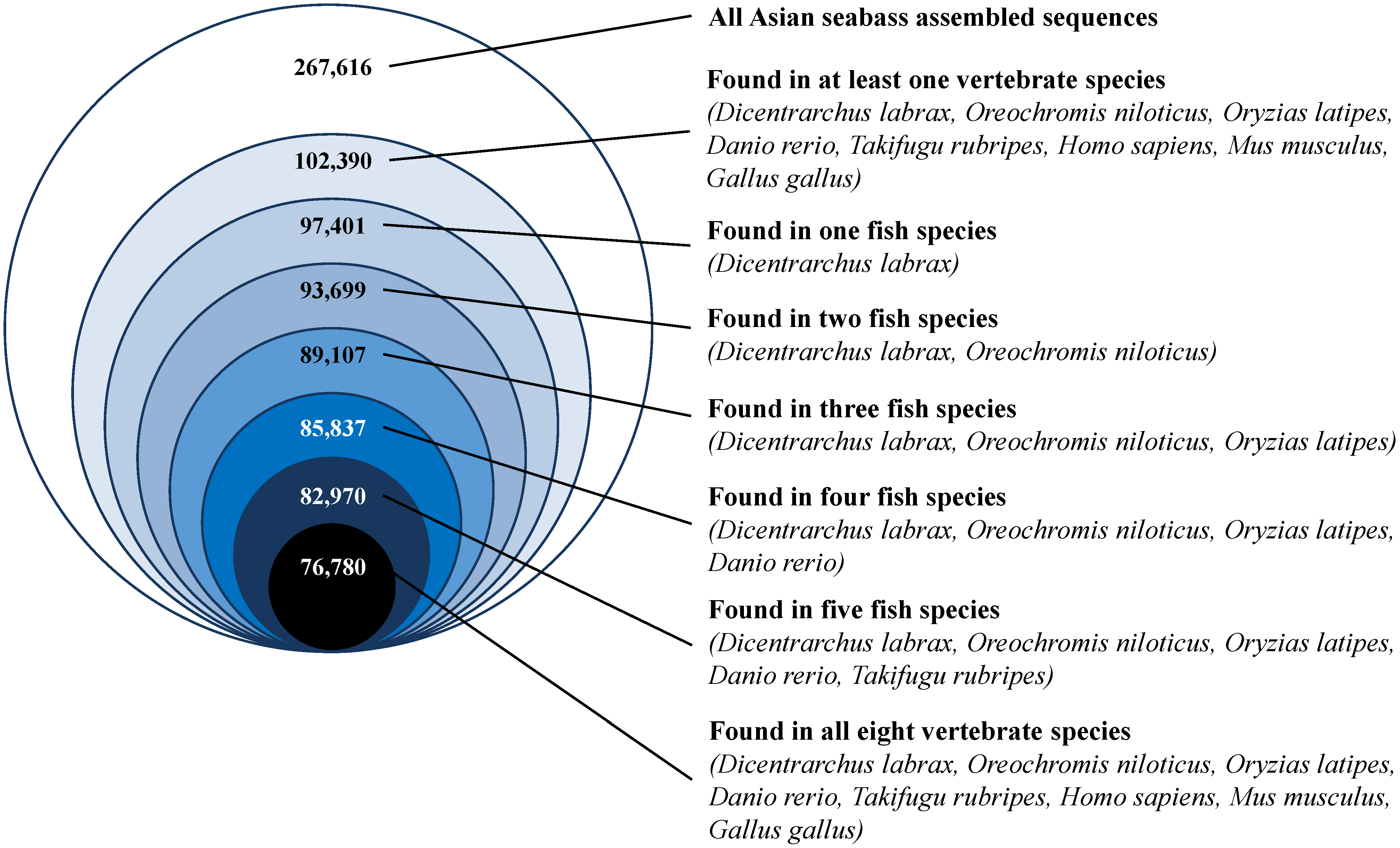
3.5. Sequence Conservation with Other Vertebrate Species
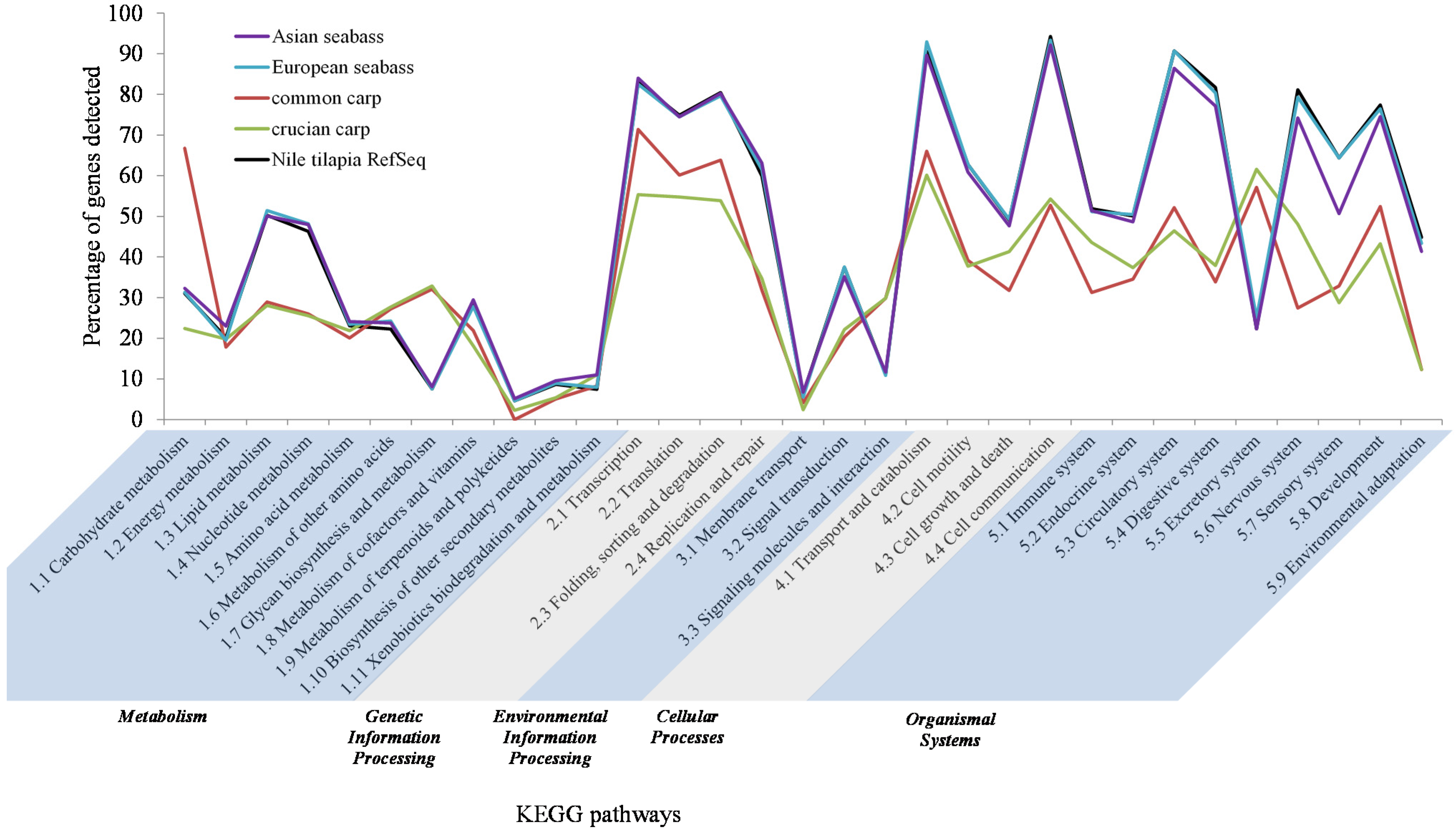
3.6. Pathway Distribution of Transcriptome Contigs
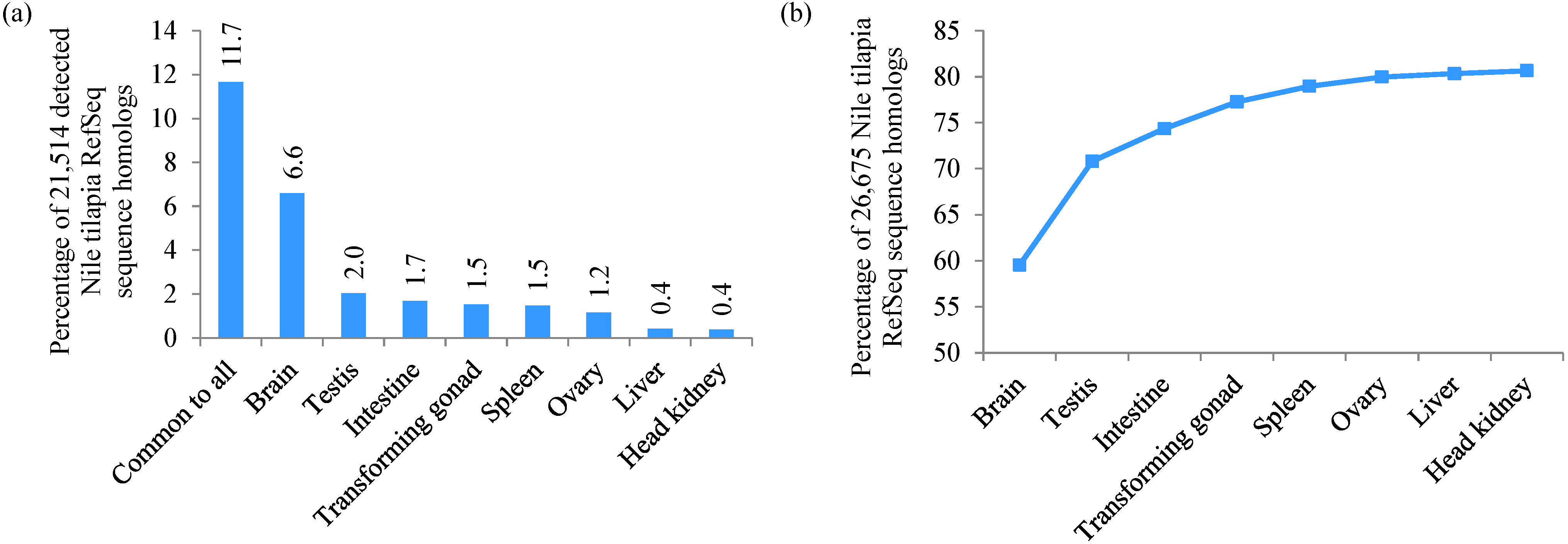
3.7. Analysis of Organ-Specific Transcripts

3.8. Application of the Asian Seabass Transcriptome for RNA-seq Experiments

| Transcripts | Differential Expression (vs. Ovary) | Gene Symbol | Sex-Related Role or Expression * | Reference |
|---|---|---|---|---|
| Contig9986 | Testis down | dnd | DJOT | [30] |
| Head_kidney_comp8086_c0_seq1 | Testis down | tp53 | UO | [8] |
| Ovary_comp51781_c4_seq1 | Testis down | zp2 | UO | [8] |
| Contig9942 | Testis down | nanos3 | DJOT | [30] |
| MP_Contig36467 | Testis down | stra6 | Differentially expressed during gonad transformation | [30] |
| Contig29413 | Testis down | zp2 | UO | [8] |
| Contig31358 | Testis down | dvl2 | UO | [8] |
| Contig26426 | Testis down | cyp26a1 | UO | [8] |
| Testis_comp111815_c0_seq1 | Testis up | piwil1 | UT | [8] |
| Brain_comp205525_c3_seq2 | Testis up | sept6 | UT | [8] |
| Transiting_gonad_comp205341_c0_seq2 | Testis up | ar | UT | [8] |
| Contig32418 | Testis up | tdrd7 | UT | [8] |
| Contig15683 | Testis up | esr1 | UT | [8] |
| Contig31060 | Testis up | sycp3 | UJOT | [31] |
| Contig19652 | Testis up | wt1b | Influence on PGC number during gonad development | [32] |
| Testis_comp102806_c0_seq1 | Testis up | cyp17a1 | UT | [8] |
| Liver_comp139786_c2_seq1 | Testis up | nr5a2 | UT | [8] |
| Spleen_comp222831_c1_seq3 | Testis up | peli1 | UT | [30] |
| Testis_comp113396_c0_seq1 | Testis up | star | DJOT | [30] |
| Contig5958 | Testis up | cyp17a2 | Facilitate enzymatic reactions in the gonads | [33] |
| Testis_comp195172_c0_seq1 | Testis up | odf3b | Differentially expressed between wild type and PGC-depleted morphants at 22 dpf | [34] |
4. Discussion
5. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vij, S.; Kathiresan, P.; Gopikrishna, G.; Lau, D.; Saju, J.; Shamsudheen, K.V.; Vinaya Kumar, K.; Basheer, V.S.; Gopalakrishnan, A.; Hossain, M.S.; et al. Barcoding of Asian seabass across its geographic range provides evidence for its bifurcation into two distinct species. Front. Mar. Sci. 2014, 1, 30. [Google Scholar] [CrossRef]
- Moore, R. Spawning and early life history of burramundi, Lates calcarifer (Bloch), in Papua New Guinea. Mar. Freshw. Res. 1982, 33, 647–661. [Google Scholar] [CrossRef]
- Guiguen, Y.; Cauty, C.; Fostier, A.; Fuchs, J.; Jalabert, B. Reproductive cycle and sex inversion of the seabass, Lates calcarifer, reared in sea cages in French Polynesia: Histological and morphometric description. Environ. Biol. Fish. 1994, 39, 231–247. [Google Scholar] [CrossRef]
- Moore, R. Natural sex inversion in the giant perch (Lates calcarifer). Mar. Freshw. Res. 1979, 30, 803–813. [Google Scholar] [CrossRef]
- Asian seabass Sequencing Consortium. The Asian seabass (Lates calcarifer) sequencing project. Manuscript in preparation. 2015. [Google Scholar]
- Lin, G.; Lo, L.C.; Zhu, Z.Y.; Feng, F.; Chou, R.; Yue, G.H. The complete mitochondrial genome sequence and characterization of single-nucleotide polymorphisms in the control region of the Asian seabass (Lates calcarifer). Mar. Biotechnol. (N.Y.) 2006, 8, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.S.; Thevasagayam, N.M.; Sridatta, P.S.; Komissarov, A.S.; Saju, J.M.; Ngoh, S.Y.; Jiang, J.; Shen, X.; Orban, L. Primary analysis of repeat elements of the Asian seabass (Lates calcarifer) transcriptome and genome. Front. Genet. 2014, 5, 223. [Google Scholar] [CrossRef] [PubMed]
- Ravi, P.; Jiang, J.; Liew, W.C.; Orban, L. Small-scale transcriptomics reveals differences among gonadal stages in Asian seabass (Lates calcarifer). Reprod. Biol. Endocrinol. 2014, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.H.; Liu, P.; Liu, F.; Lin, G.; Sun, F.; Tu, R.; Yue, G.H. Analysis of stress-responsive transcriptome in the intestine of Asian seabass (Lates calcarifer) using RNA-seq. DNA Res. 2013, 20, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Furmanek, T.; Kryvi, H.; Krossoy, C.; Totland, G.; Grotmol, S.; Wargelius, A. Transcriptome sequencing of Atlantic salmon (Salmo salar L.) notochord prior to development of the vertebrae provides clues to regulation of positional fate, chordoblast lineage and mineralisation. BMC Genomics 2014, 15, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, P.; Liu, G.; Xu, J.; Wang, X.; Li, J.; Zhao, Z.; Zhang, X.; Zhang, Y.; Xu, P.; Sun, X. Characterization of common carp transcriptome: Sequencing, de novo assembly, annotation and comparative genomics. PLoS ONE 2012, 7, e35152. [Google Scholar] [CrossRef] [PubMed]
- Magnanou, E.; Klopp, C.; Noirot, C.; Besseau, L.; Falcón, J. Generation and characterization of the sea bass Dicentrarchus labrax brain and liver transcriptomes. Gene 2014, 544, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Kaitetzidou, E.; Xiang, J.; Antonopoulou, E.; Tsigenopoulos, C.S.; Sarropoulou, E. Dynamics of gene expression patterns during early development of the European Sea Bass (Dicentrarchus labrax). Physiol. Genomics 2015, 47, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Rexroad, C.; Wang, J.; Thorgaard, G.; Yao, J. Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics 2010, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Pereiro, P.; Balseiro, P.; Romero, A.; Dios, S.; Forn-Cuni, G.; Fuste, B.; Planas, J.V.; Beltran, S.; Novoa, B.; Figueras, A. High-throughput sequence analysis of turbot (Scophthalmus maximus) transcriptome using 454-pyrosequencing for the discovery of antiviral immune genes. PLoS ONE 2012, 7, e35369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reading, B.; Chapman, R.; Schaff, J.; Scholl, E.; Opperman, C.; Sullivan, C. An ovary transcriptome for all maturational stages of the striped bass (Morone saxatilis), a highly advanced perciform fish. BMC Res. Notes 2012, 5, 111. [Google Scholar] [CrossRef] [PubMed]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef] [PubMed]
- PRINSEQ. PReprocessing and INformation of SEQuence data. Available online: http://prinseq.sourceforge.net/ (accessed on 12 May 2013).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Bowtie: An ultrafast memory-efficient short read aligner. Available online: http://bowtie-bio.sourceforge.net/index.shtml (accessed on 12 May 2013).
- Asian seabass (Lates calcarifer) Transcriptome Project. Available online: http://laszlo.tll.org.sg/asb_transcriptome/ (accessed on 8 May 2015).
- Scripts: Average_GC_Content_Analysis. Available online: https://github.com/ramadatta/Scripts/tree/master/Average_GC_Content_Analysis (accessed on 29 January 2015).
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Full-LengtherNEXT (0.0.8). Available online: https://www.omniref.com/ruby/gems/full_lengther_next/0.0.8 (accessed on 20 March 2014).
- Tine, M.; Kuhl, H.; Gagnaire, P.A.; Louro, B.; Desmarais, E.; Martins, R.S.; Hecht, J.; Knaust, F.; Belkhir, K.; Klages, S.; et al. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat. Commun. 2014, 5, 5770. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Scripts—KEGG_KAAS_Table. Available online: https://github.com/ramadatta/Scripts/tree/master/KEGG_KAAS_Table (accessed on 29 January 2015).
- Liao, X.; Cheng, L.; Xu, P.; Lu, G.; Wachholtz, M.; Sun, X.; Chen, S. Transcriptome analysis of crucian carp (Carassius auratus), an important aquaculture and hypoxia-tolerant species. PLoS ONE 2013, 8, e62308. [Google Scholar] [CrossRef] [PubMed]
- Ensembl Biomart. Available online: http://www.ensembl.org/biomart/martview (accessed on 30 April 2015).
- Jiang, J. Functional Genomic Analysis of Gonad Development in the Protandrous Asian Seabass. Ph.D. Thesis, National University of Singapore, Singapore, 2014. [Google Scholar]
- Sreenivasan, R.; Jiang, J.; Wang, X.; Bártfai, R.; Kwan, H.Y.; Christoffels, A.; Orbán, L. Gonad differentiation in zebrafish is regulated by the canonical wnt signaling pathway. Biol. Reprod. 2014, 90, 45. [Google Scholar] [CrossRef] [PubMed]
- Klüver, N.; Herpin, A.; Braasch, I.; Drieβle, J.; Schartl, M. Regulatory back-up circuit of medaka Wt1 co-orthologs ensures PGC maintenance. Dev. Biol. 2009, 325, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-Y.; Wang, D.-S.; Kobayashi, T.; Yano, A.; Paul-Prasanth, B.; Suzuki, A.; Sakai, F.; Nagahama, Y. A novel type of P450c17 lacking the lyase activity is responsible for C21-steroid biosynthesis in the fish ovary and head kidney. Endocrinology 2007, 148, 4282–4291. [Google Scholar] [CrossRef] [PubMed]
- Tzung, K.-W.; Goto, R.; Saju, J.M.; Sreenivasan, R.; Saito, T.; Arai, K.; Yamaha, E.; Hossain, M.S.; Calvert, M.E.; Orbán, L. Early depletion of primordial germ cells in zebrafish promotes testis formation. Stem Cell Rep. 2015, 4, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.L.; Centner, T.; Fornoff, F.; Geach, A.J.; Gotthardt, M.; McNabb, M.; Witt, C.C.; Labeit, D.; Gregorio, C.C.; Granzier, H.; et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 2001, 89, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, P.; Wang, B.; Zhao, L.; Wang, J.; Zhao, Z.; Zhang, Y.; Li, J.; Xu, P.; Sun, X. Transcriptome sequencing and analysis of wild Amur Ide (Leuciscus waleckii) inhabiting an extreme alkaline-saline lake reveals insights into stress adaptation. PLoS ONE 2013, 8, e59703. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.B. Principles of transcriptome analysis and gene expression quantification: An RNA-seq tutorial. Mol. Ecol. Resour. 2013, 13, 559–572. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, S.T.; Emrich, S.J. Assessing de novo transcriptome assembly metrics for consistency and utility. BMC Genomics 2013, 14, 465. [Google Scholar] [CrossRef] [PubMed]
- DeWoody, J.A.; Abts, K.C.; Fahey, A.L.; Ji, Y.; Kimble, S.J.; Marra, N.J.; Wijayawardena, B.K.; Willoughby, J.R. Of contigs and quagmires: Next-generation sequencing pitfalls associated with transcriptomic studies. Mol. Ecol. Resour. 2013, 13, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Zhou, Z.; Waldbieser, G.; Sun, F.; Lu, J.; Zhang, J.; Jiang, Y.; Zhang, H.; Wang, X.; et al. Efficient assembly and annotation of the transcriptome of catfish by RNA-Seq analysis of a doubled haploid homozygote. BMC Genomics 2012, 13, 595. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, H.K.; Harrison, P.W.; Zachar, G.; Szekely, T.; Mank, J.E. The plover neurotranscriptome assembly: Transcriptomic analysis in an ecological model species without a reference genome. Mol. Ecol. Resour. 2013, 13, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hui, J.H.; Chan, T.F.; Chu, K.H. De novo transcriptome sequencing of the snail Echinolittorina malaccana: Identification of genes responsive to thermal stress and development of genetic markers for population studies. Mar. Biotechnol. (N.Y.) 2014, 16, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Au, K.F.; Sebastiano, V.; Afshar, P.T.; Durruthy, J.D.; Lee, L.; Williams, B.A.; van Bakel, H.; Schadt, E.E.; Reijo-Pera, R.A.; Underwood, J.G.; et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, E4821–E4830. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Wang, Z.; Li, G. The impacts of read length and transcriptome complexity for de novo assembly: A simulation study. PLoS ONE 2014, 9, e94825. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Miyata, M.; Chan, C.; Ngoh, S.Y.; Liew, W.C.; Saju, J.M.; Ng, K.S.; Wong, F.S.; Lee, Y.S.; Chang, S.F.; et al. Differential transcriptomic response in the spleen and head kidney following vaccination and infection of Asian seabass with Streptococcus iniae. PLoS ONE 2014, 9, e99128. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Bernatchez, L.; Beheregaray, L.B. RNA-seq analysis reveals extensive transcriptional plasticity to temperature stress in a freshwater fish species. BMC Genomics 2013, 14, 375. [Google Scholar] [CrossRef] [PubMed]
- Uren Webster, T.M.; Bury, N.; van Aerle, R.; Santos, E.M. Global transcriptome profiling reveals molecular mechanisms of metal tolerance in a chronically exposed wild population of brown trout. Environ. Sci. Technol. 2013, 47, 8869–8877. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thevasagayam, N.M.; Sridatta, P.S.R.; Jiang, J.; Tong, A.; Saju, J.M.; Kathiresan, P.; Kwan, H.Y.; Ngoh, S.Y.; Liew, W.C.; Kuznetsova, I.S.; et al. Transcriptome Survey of a Marine Food Fish: Asian Seabass (Lates calcarifer). J. Mar. Sci. Eng. 2015, 3, 382-400. https://doi.org/10.3390/jmse3020382
Thevasagayam NM, Sridatta PSR, Jiang J, Tong A, Saju JM, Kathiresan P, Kwan HY, Ngoh SY, Liew WC, Kuznetsova IS, et al. Transcriptome Survey of a Marine Food Fish: Asian Seabass (Lates calcarifer). Journal of Marine Science and Engineering. 2015; 3(2):382-400. https://doi.org/10.3390/jmse3020382
Chicago/Turabian StyleThevasagayam, Natascha M., Prakki S.R. Sridatta, Junhui Jiang, Amy Tong, Jolly M. Saju, Purushothaman Kathiresan, Hsiao Yuen Kwan, Si Yan Ngoh, Woei Chang Liew, Inna S. Kuznetsova, and et al. 2015. "Transcriptome Survey of a Marine Food Fish: Asian Seabass (Lates calcarifer)" Journal of Marine Science and Engineering 3, no. 2: 382-400. https://doi.org/10.3390/jmse3020382