Abstract
Synechococcus is a dominant genus of the coastal phytoplankton with an effective contribution to primary productivity. Here, the phylogenetic and phenogenetic composition of Synechococcus in the coastal Yellow Sea was addressed by sequencing marker gene methods. Meanwhile, its co-occurrence pattern with bacterial and eukaryotic microbes was further investigated based on the construction of networks. The result revealed that Synechococcus abundance ranged from 9.8 × 102 cells mL−1 to 1.6 × 105 cells mL−1, which was significantly correlated to sampling depth and nutrient contents of nitrite, ammonia, and dissolved silicon. A total of eight Synechococcus phylogenetic lineages were detected, of which clade III was dominant in most of the samples. Meanwhile, clade I increased along the water column and even reached a maximum value of 76.13% at 20 m of station B. Phenogenetically, Synechococcus PT3 was always the predominant pigment type across the whole study zone. Only salinity was significantly correlated to the phenogenetic constitution. The networks revealed that Synechococcus co-occurred with 159 prokaryotes, as well as 102 eukaryotes including such possible grazers as Gymnodinium clades and Alveolata. Potential function prediction further showed that microbes co-occurring with Synechococcus were associated with diverse element cycles, but the exact mechanism needed further experimentation to verify. This research promotes exploring regularity in the genomic composition and niche position of Synechococcus in the coastal ecosystem and is significant to further discuss its potential participation in materials circulation and bottom-up effects in microbial food webs.
1. Introduction
Marine Synechococcus is a main group of photosynthetic picophytoplankton with the feature of self-assembly orange phycoerythrin for light capturing. Taxonomically, it belongs to Cyanobacteria and is demonstrated with rich phylogenetic diversity [1]. Through molecular detections, Synechococcus can be classified into three phylogenetic subclusters including subcluster 5.1 (S5.1), subcluster 5.2 (S5.2), and subcluster 5.3 (S5.3) [2], which at least consist of 20 clades, 2 clades, and 6 clades, respectively [3]. Furthermore, the different constitution of the main light-harvesting antennae for photosystem II, phycobilisomes (PBSs), leads to its rich phenogenetic diversity [4]. Three main pigment types (PTs), PT1, PT2, and PT3, can be identified [5]. Due to the independent evolution between phylogenetics and PTs, it is impossible to simultaneously identify these two assemblage characteristics of Synechococcus through bioinformation based on a single gene marker [6]. Barcode DNA genes, such as the 16S–23S internally transcribed spacer (ITS) [7], rpoC1 (subunit of RNA polymerase) [8] and petB (cytochrome b6) are frequently taken to identify the genotype diversity. Additionally, operons, including cpcBA [9], cpeBA [8], or multimarker [10] have already been utilized in the research of phenogenetic diversity.
Synechococcus exhibits a diverse distribution concerning abundance, phylogenetic composition, and PTs around the world’s oceans. It is widely distributed in the global marine ecosystems, from equatorial to polar sea waters [11]. In particular, it is most abundant in coastal zones and upwelling areas, with the highest abundance of 106 cells mL−1 [12]. The abundance distribution has been studied in Chinese surrounding sea areas, covering the South China Sea [13,14], the East China Sea [15], and some estuaries [16]. Followed the rise of the sequencing technique, researchers began to pay close attention to discuss the genotypes of Synechococcus as well as their niche division. The phylogenetic composition of Synechococcus has been revealed in the north Yellow Sea [17], East China Sea [18], the South China Sea [19], and the Hong Kong coastal water [20], respectively. However, it remains largely unrevealed in the south Yellow Sea featured with a temperate climate at higher latitude. In addition, the study of Synechococcus PTs is mostly confined to the estuary areas [9,16], but it is largely void in the north sea areas of China.
As a widespread and abundant phytoplankton, Synechococcus plays an essential role in carbon assimilation. In physiological experiments, Synechococcus can also participate in various metabolic activities and so be further involved in other element geochemical cycles, such as nitrogen [21], phosphorus [22], and silicon [23]. Whether Synechococcus maintains these functions in the field still needs to be confirmed from the ecological scale. Traditionally, co-culture experiments can infer and validate their interactions by constructing community assemblages [24]. However, they are limited by the poorly simulative “wild” environment and the status of uncultured microbes [25]. Furthermore, the mass of potentially interactive species complicates the determination of community-wide interactions using experimental research. With the development of high-throughput sequencing technology, network methods based on bioinformatics analysis and mathematical modeling has been used for investigating these interactions among taxa (co-occurrence patterns) across complex communities [26,27]. The study on the manipulation of grassland microbiome revealed that different types of microflora influenced multiple functions in nutrient cyclings, such as arbuscular mycorrhizal fungi and nitrogen-fixing bacteria limiting nutrients to plants [28]. Until now, co-occurrence network analysis has been applied in soil [29,30], fluvial [31], lake [32], and marine [33] ecosystems to explore the interactions between different microbiotas.
The Yellow Sea is a continental shelf sea with an average depth of 45 m in the Western Pacific Ocean. As it is semi-enclosed by the mainland, the Yellow Sea is affected by both anthropogenic activities and environmental changes, which have led to ecological problems of nutrient eutrophication, pollutant inflow, and biodiversity loss [34]. The presence of abundant nutrients and diverse substances is known to have affected the diversity of the coastal microorganisms and changed their metabolic pathways [35], but rare studies focused on their stresses on the composition and performance of Synechococcus assemblages. Along the latitude 36° N is a typical section of the Yellow Sea with gradually increasing land distance and deepening water depth. Numerous physicochemical [36,37] and biological [38] studies have been conducted here. It was also selected in our study with the goal to (i) reveal the distribution pattern of Synechococcus in terms of cell abundance and the impact of environmental variables on it along the Yellow Sea section; (ii) unveil both phylogenetic and phenogenetic diversity and structure of Synechococcus assemblages and their environmental constraints; and (iii) study the co-occurrence pattern of Synechococcus with multiple eukaryotic and prokaryotic taxa to explore its ecological functions associated with microbiome Synechococcus in the coastal ecosystem.
2. Materials and Methods
2.1. Sampling
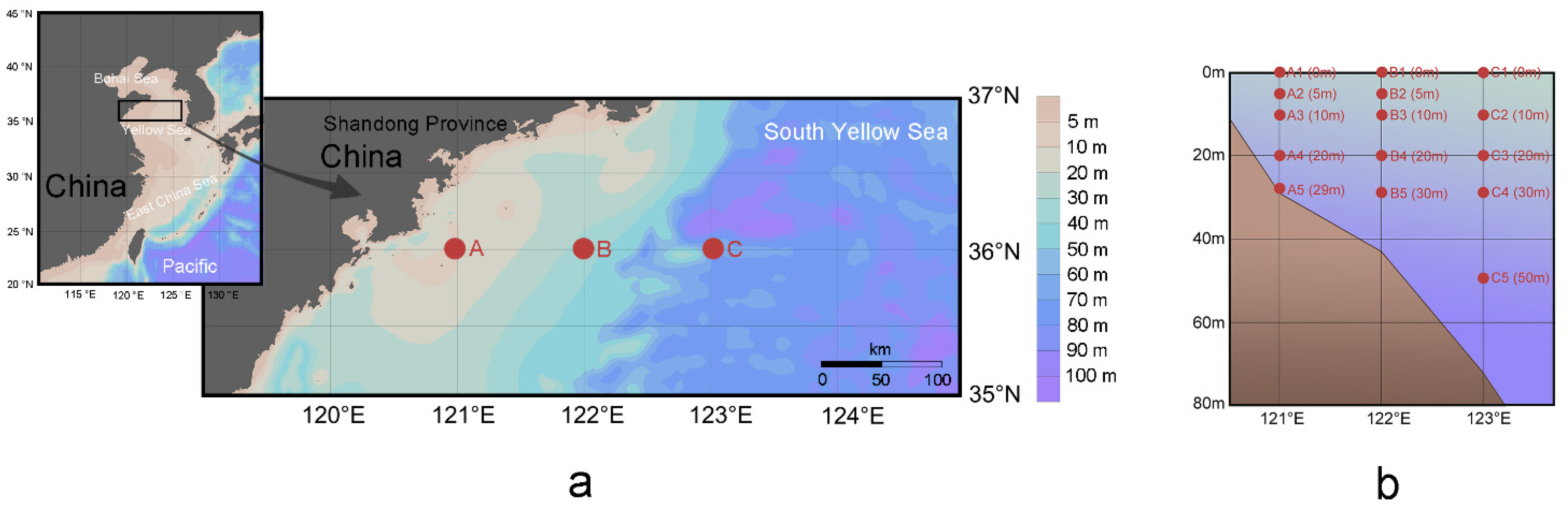
Field sampling was conducted in the South Yellow Sea by taking Research Vessel Kexue III from August 29 to September 6 in 2018 (Figure 1). Three sample stations on the same latitude of 36° N were designated according to their representatives at relatively shallow (29 m), average (43 m), and relatively deep (72 m) depths. Fifteen samples were collected from standard depths according to the specifications for the oceanographic survey (National Standard of China, GB/T 12763-2007). A total of 1.40 mL seawater was taken in sterile tubes in quintuplicate, and then 0.20 mL paraformaldehyde (final concentration 0.5%) was added to determine the count of Synechococcus [39]. The tubes were flash-frozen in liquid nitrogen, and then stored at −80 °C until analysis using flow cytometry. One liter seawater was filtered through 48 μm nylon mesh to remove microphytoplankton, and then through 0.22 μm polycarbonate membrane (Millipore Co., Bedford, MA, USA). Acquired membranes were preserved for high-throughput sequencing analysis by storing in liquid nitrogen.
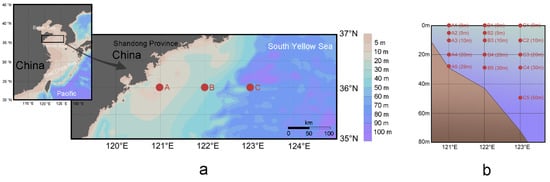
Figure 1.
Geographic (a) and vertical position with depth in the parentheses (b) of all samples in the coastal South Yellow Sea; backgrounds were visualized using Ocean Data View v.5.1.7.
Physical parameters and chlorophyll a (chl. a) content were detected on boat through a CTD sensor (Sea-Bird Electronics Inc., Bellevue, WA, USA). The nutrient concentrations were determined by an AA3 segmented flow analyzer (Seal Analytical GmbH, Germany) [40].
2.2. Cell Abundance of Synechococcus
Synechococcus abundance was detected using a BD FACSAria™ flow cytometer. Yellow–green fluorescent beads (2.0 μm, Polysciences, Warrington, PA, USA) were added to each sample before the detection. The forward and right-angle light scattering and four fluorescence signals were collected and analyzed using WinMDI 2.9. Red fluorescence excited by a 488 nm laser indicates the presence of chl. a. Red fluorescence excited by 635 nm indicates the presence of phycocyanin (PC), while orange fluorescence indicates the presence of phycoerythrin (PE).
2.3. Structures of Synechococcus Assemblages and Microbial Communities
2.3.1. DNA Extraction
Total nucleic acids were extracted from the membrane bead-beaten with 500 mg of 0.10 mm silica/zirconia glass beads (BioSpec Products Inc., Bartlesville, OK, USA), 0.50 mL 120 mM (pH 8.0) K2HPO4 butter, and 0.50 mL phenol-chloroform-isoamyl alcohol (25:24:1) (pH 8.0) by Mini-BeadBeater (BioSpec, USA). The supernatants were collected to extract DNA using the Fast DNA spin kit (MP BIO, Santa Ana, CA, USA) following the instruction of the kit. Agarose gel electrophoresis and the Nano Drop 2000c spectrophotometer (Thermo Fisher, Waltham, MA, USA) were used to assess the quality and concentration of the extracted DNA. Then, extracted DNA was kept at −20 °C until further analysis.
2.3.2. PCR Reactions
Polymerase chain reaction (PCR) was performed in the thermocycler GeneAmp® PCR system 9700 (Perkin Elmer, Foster City, CA, USA) in triplicate [41]. The rpoC1 gene of Synechococcus was amplified by the primers rpoC1-39F (5′-GGNATYGTYTGYGAGCGYTG-3′) and rpoC1-462R (5′-CGYAGRCGCTTGRTCAGCTT-3′) with a unique 6 nt barcode attached at the end of the forward primer [42]. The PCR reactions for rpoC1 gene were conducted with a 2 × Rapid Taq Master Mix (Vazyme, Nanjing, China) in a 20 μL reaction volume containing 20 ng template DNA. The PCR process started with a 3-min pre-denaturation at 95 °C, followed with 37 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and elongation at 72 °C for 45 s, and ended with a final extension at 72 °C for 10 min. The cpeBA operon of Synechococcus was amplified in the same way but by the primers peBF (5′-GACCTACATCGCWCTGGGYG-3′) and peAR (5′-CCMACAACCARGCAGTAGTT-3′) with annealing at 53 °C [16]. The V4 regions of the 16S RNA gene and 18S RNA gene were amplified with the same 20 μL reaction volume by the universal prokaryotic primers 515F/806R and the specific eukaryotic planktonic primers 547F (5′-CCAGCASCYGCGGTAATTCC-3′)/952R (5′-ACTTTCGTTCTTGATYRA-3′), respectively [43,44]. The PCR process for 18S started with a 2-min pre-denaturation at 94 °C, followed by 10 cycles of denaturation at 94 °C for 30 s, annealing at 65 °C for 30 s, 18 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s and elongation at 72 °C for 30 s, and ended with a final extension at 72 °C for 10 min [44].
2.3.3. Sequencing
For the rpoC1, 16S, and 18S genes, high-throughput sequencing for the PCR products after being gel purified was conducted at Majorbio Co., Ltd. (Shanghai, China). Sequencing libraries were generated using NEB Next®Ultra™DNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) following the manufacturer’s recommendations, and index codes were added. The library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. At last, the library was sequenced on an Illumina MiSeq platform and 300 bp paired-end reads were generated. For the cpeBA gene, PCR products were purified by agarose gel electrophoresis with a TaKaRa MiniBEST DNA Fragment Purification Kit (TaKaLa, Kusatsu, Japan) and then cloned into a pMD-19T vector with a Cloning Kit (TaKaLa, Dalian, China). The recombinant products were subsequently transformed into Escherichia coli DH5α competent cells. After incubation, recombinant clones were picked up and sequenced by the RuiBo Sequencing Company (Qingdao, China) for constructing clone libraries.
2.3.4. Sequence Data Processing
The raw data were quality-controlled [45], chimeras-filtered [46], and redundancy-eliminated [47] to downstream analyses using QIIME2. Sequences of rpoC1 and cpeBA genes were aligned using Clustal W [48] and trimmed by BioEdit [49]. Sequences were grouped into OTUs (operational taxonomic units) at 95% similarity. Representative sequences of OTU were first identified using BLASTn against the nt database, and those not belonging to Cyanobacteria were picked out. The 15 most abundant OTUs sequenced based on the rpoC1 gene (covering 96.3% of total reads) and cpeBA operon (covering 94.2% of total reads) were aligned with the reference sequences (Tables S1 and S2). Maximum likelihood phylogenetic trees were constructed by Mega 7.014 with the model GTR+G+I and 200 bootstraps [9]. For 16S and 18S sequences, the OTU clustering was performed considering a similarity of 97% to obtain the representative OTU sequences [46]. The Greengenes v.135/16S database and the Silva v.132/18S eukaryota database were used to obtain the taxonomic information corresponding to each OTU. The function of 16S sequencing data was annotated using the script collapse_table.py based on the Functional Annotation of Prokaryotic Taxa (FAPROTAX) database in Python v.2.7 [41,50].
2.4. Construction of Co-Occurrence Network
Rare OTUs of 16S and 18S genes with a total relative abundance < 0.01% in all samples were filtered out prior to the construction of microbial community networks [29]. Spearman correlation matrixes among OTUs were calculated based on the relative abundance [51,52]. The connections in the network with the absolute value of Spearman coefficients > 0.6 and p values < 0.05 were considered significant [53]. Network properties were calculated using the psych package [54], and the networks were drawn using Gephi v0.9.2 [39,55]. Global genetic and functional correlation networks encompassing all genera and functions were constructed from the correlation matrix. Metabolic cycling networks were constructed using similar methods based on the relative abundance of metabolic functions annotated by the FAPROTAX database. To determine whether the proportion and type of metabolic cycles involving Synechococcus vary along the vertical water column, the samples were selected to represent the upper (A1, A2, B1, B2, and C1) and bottom (A5, B5, C4, and C5) layers according to the depths and photosynthetically active radiation (PAR) [56].
2.5. Statistical Analysis
Alpha diversity indexes and beta diversity distance were calculated using the core_diversity_analyses.py script. The environmental data were normalized and scaled to statistical analysis. The vegan package v.2.5-6 in R v3.6.1 was used to calculate the variance inflation factor (VIF), and then we picked nonredundant environmental variables to conduct redundancy analysis (RDA) with OTU tables [57]. Statistical parameters of co-occurrence networks, such as modularity and density, were calculated using the built-in program of Gephi v0.9.2 [58]. Correlationships among environmental variables and between environmental variables and Synechococcus cell abundance were examined by Spearman’s correlations using the corrplot package v.0.84 in the R programming language v3.6.1. Differences in Synechococcus cell abundance among three stations were tested using analysis of variance (ANOVA) with a post hoc test (Tukey test) in the software Statistical Product and Service Solutions (SPSS v.17.0). A p-value less than 0.05 denoted statistical significance. The software TBtools (Toolbox for Biologists) v1.055 [59], and the vegan package v.2.5-6 in R v3.6.1 was utilized to draw a heatmap.
3. Results
3.1. Environmental Variables
The environmental variables of each sample are shown in Table 1, and their Spearman’s correlations are shown in Table S3. The water depth increases from the shallow coastal station A to C. Positive correlations between longitude and oxygen (ρ = 0.89) and with salinity (ρ = 0.59) were found. Along the vertical water column with the increase in sampling depth, salinity (ρ = 0.84), the concentration of dissolved total phosphorus (DTP; ρ = 0.85), dissolved silicon (DSi; ρ = 0.78), and phosphate (PO43−; ρ = 0.73) gradually increased, whereas temperature (ρ = −0.91) and concentration of ammonia (NH4+; ρ = −0.75) continually decreased. In particular, the presence of YSCWM (Yellow Sea Cold Water Mass) caused a sharp drop in temperature in the lower water layers. Correlations between nutrient concentrations were also revealed (Table S3).

Table 1.
Physicochemical variables of each sample (a).
3.2. Synechococcus Abundance and Its Environmental Constraints
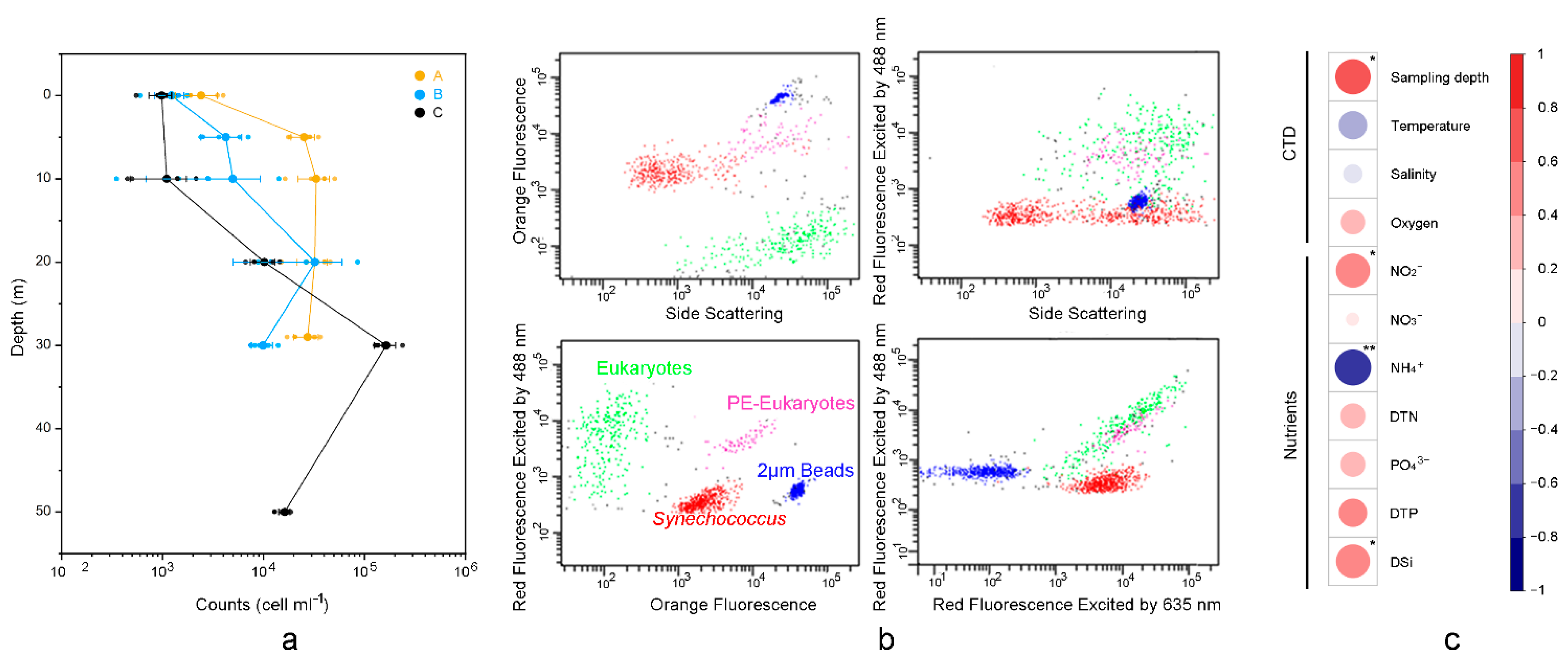
In the studied zone, Synechococcus counts ranged from 9.8 × 102 cells mL−1 (C1) to 1.6 × 105 cells mL−1 (C4), with an average of 2.4 × 104 cells mL−1 (Figure 2). The cell abundance of Synechococcus was highest at C4 (1.6 × 105 cells mL−1), followed by A3 (3.3 × 104 cells mL−1) and B4, A4 (3.2 × 104 cells mL−1).
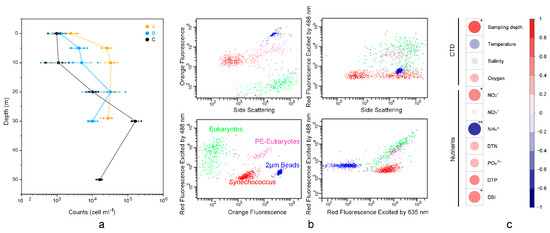
Figure 2.
Cell abundance of Synechococcus measured using flow cytometry at each station. (a) Line chart showing the relationship between cell abundance of Synechococcus and depth. (b) Flow cytometric signatures of Synechococcus, eukaryotes, and PE-eukaryotes. (c) Correlationships between Synechococcus count and physicochemical variables; The color scale on the right represents their Spearman correlation coefficient. “**” denotes p < 0.01, “*” denotes p < 0.05.
Synechococcus counts showed no significant difference among the three stations. However, they correlated with the vertically sampled water layer significantly (ρ = 0.62; Figure 2). The concentrations of nitrite (NO2−; ρ = 0.58) and DSi (ρ = 0.57) were positively correlative to the cell abundance of Synechococcus, while NH4+ (ρ = −0.66) was negatively correlative to the Synechococcus counts.
3.3. Phylogenetic and Phenogenetic Diversity and Structure of Synechococcus Assemblage
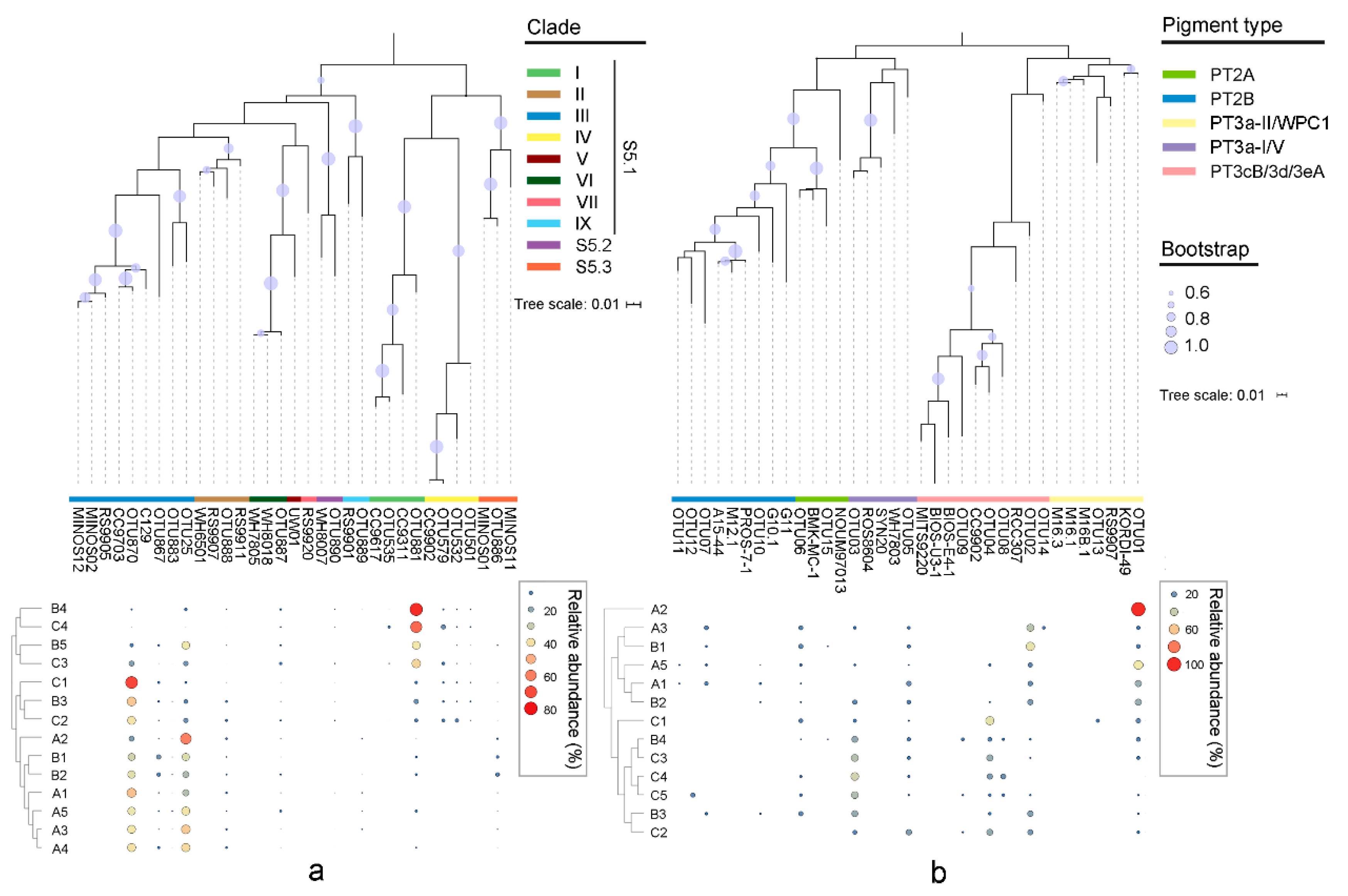
The rpoC1 gene generated 216,077 quality sequences from 14 samples, with an average of 15,434 sequences per sample (Table S4). The good coverage of all rpoC1 sequencing samples was >99.9%. The genomic diversities of Synechococcus assemblages were estimated by the NP-Shannon index and the Chao1 index (predicted OTUs) (Table S4). The high phylogenetic diversity usually appeared in the middle or bottom layers along the water column of stations B and C, while the surface layer of station A had higher diversity indexes. In the phylogenetic tree, a total of eight Synechococcus lineages were identified from 14 samples based on the rpoC1 gene (Figure 3a). Three subclusters of Synechococcus, S5.1, S5.2, and S5.3, all appeared in the studied area. Among them, S5.1 accounted for the highest proportion, with an average of 90.92%, followed by S5.3 (2.09%), while S5.2 as a rare subcluster accounted for only 0.54%. Both S5.1 and S5.2 were more abundant in station C, while S5.3 had a higher relative abundance in station B. Synechococcus clade III of S5.1 was the dominant clade, which possesses a high proportion of up to 60.82%, on average. In particular, it occupied more than 80% of the total clades in station A, which was about twice that in stations B and C. The proportion of clade I increased with the decrease in water depth and reached a maximum value of 76.13% at B4. Clade IV in station A was significantly less than that of station C and the lower water areas of station B. A higher relative abundance of clades II and IX was detected in station A, while that of clade VI was in station C. The proportion of Synechococcus S5.2 was not obviously different among samples, while that of S5.3 was relatively high in the surface area. The hierarchical clustering based on Bray–Curtis demonstrated that the phylogenetic composition of Synechococcus in samples from lower water layers of stations B and C was different from others (Figure 3a).
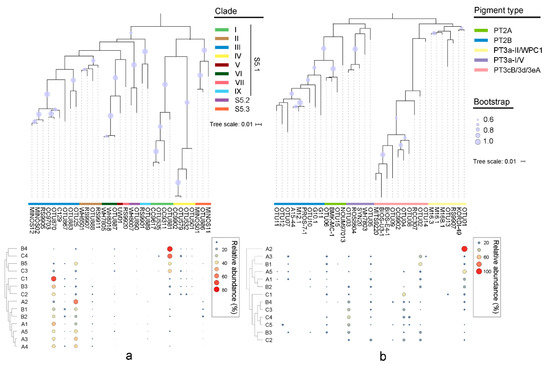
Figure 3.
Maximum likelihood phylogenetic tree of the 15 most abundant OTUs based on rpoC1 gene (a) and cpeBA operon (b). Heatmaps on the bottom show the relative abundance of OTUs in each library.
The cpeBA gene generated 497 quality sequences from 13 samples, with an average of 38 sequences per sample, through the method of clone library (Table S4). The good coverage of the cpeBA gene was >73.0%, except in A2 (only 1 OTU). In the tree based on the cpeBA operon, five well-separated clusters were formed (Figure 3b). PT2 and 3a Synechococcus could be classified by the sequencing of the cpeBA sequence, while Synechococcus PT3cB, 3d, and 3eA could not be distinguished from each other. PT3a sequences from clade I and V formed a cluster (hereafter PT3a-I/V) and were separated from the cluster formed by those from clade II and WPC1 (hereafter PT3a-II/WPC1). The sample A2 only contained 1 OTU, so it was removed from statistical analyses. Synechococcus PT3, including PT3a-I/V, PT3a-II/WPC1, and PT3cB/3d/3eA, was dominant in the studied area with a mean proportion of 73.04% in all samples, while that of PT2, including PT2A and PT2B, decreased to 13.42%. Among PT3, PT3cB/3d/3eA had the highest relative abundance in the studied section, with a peak of 52.94% in C2. PT3a-I/V had a higher proportion in station C, while PT3a-II/WPC1 was more abundant in station A. For PT2, the relative abundance of PT2A was relatively higher than PT2B. In samples C2 and C3, both PT2A and PT2B were not detected. Furthermore, PT2B was not found in B4 and most water layers of station C. The clustering analysis showed that stations A and C each formed a separated cluster, while samples from station B alternatively clustered in them (Figure 3b).
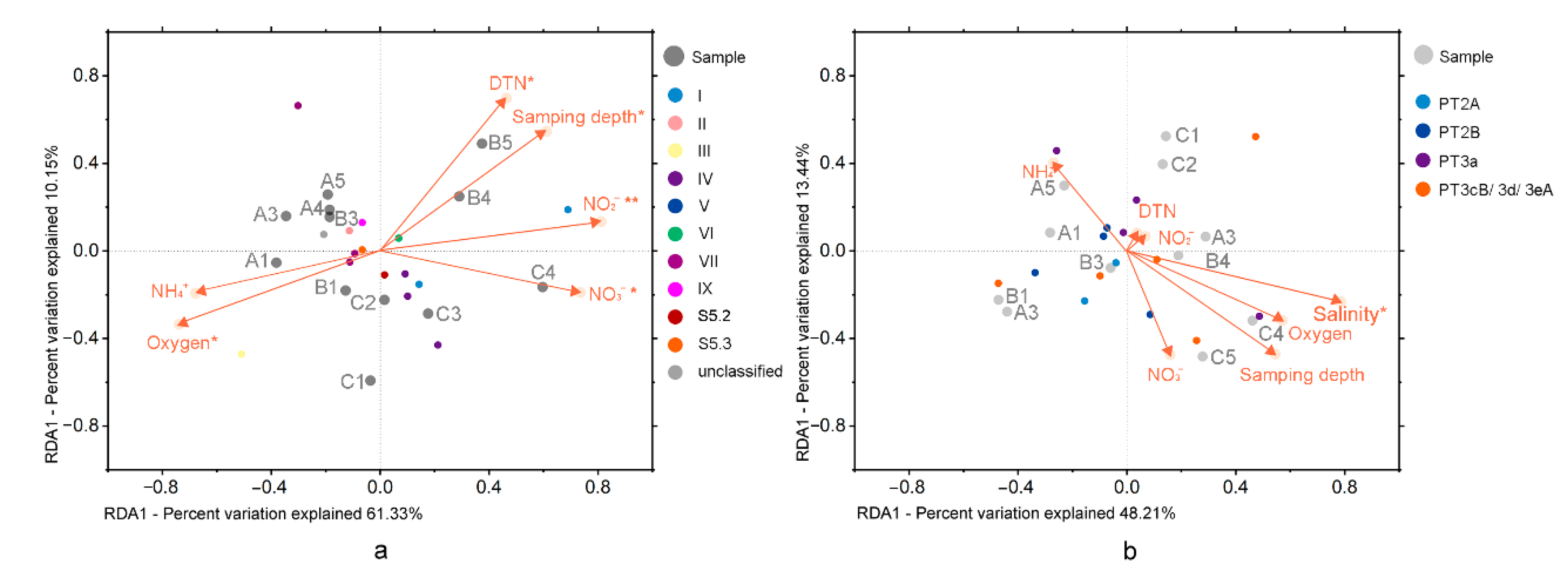
The first axis and the second axis of the RDA analysis based on the rpoC1 gene explained 61.33% and 10.15% of the variance in Synechococcus assemblages, respectively (Figure 4). Five environmental variables, NO2− (ρ = 0.60), oxygen (ρ = 0.58), sampling depth (ρ = 0.56), DTN (ρ = 0.54), and NO3− (ρ = 0.49), significantly correlated to the community. Clade I and IV relative abundances were associated with nitrite/nitrate-N, while clade III showed correlations with oxygen. Clade VI was associated with sampling depth. The first axis and the second axis of the RDA analysis based on the cpeBA gene totally explained 61.65% of the variance of Synechococcus assemblage. Only salinity (ρ = 0.64) was significantly correlated to the phenotypic constitution.
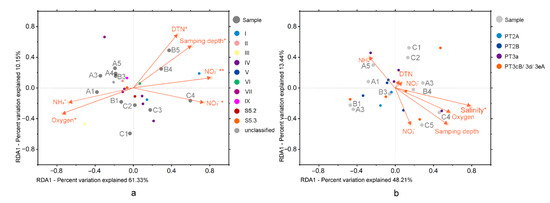
Figure 4.
Correlation plots based on a redundancy analysis (RDA), revealing the relationship between samples, environmental factors, and the 15 most abundant OTUs based on rpoC1 gene (a) and cpeBA operon (b). “*” indicates p < 0.05, “**” indicates p < 0.01.
The Spearman correlation analysis was performed to examine the links between Synechococcus clades and PTs (Figure S1). It showed that S5.1 clade I and IV as well as S5.2 positively correlated with PT3a-I/V (low phycourobilin: phycoerythrobilin), while clade III negatively correlated with it. No other significant correlation between Synechococcus clades and PTs was detected. The result supported the independent evolution between phylogenetic and phenogenetic diversity of Synechococcus.
3.4. Microbial Co-Occurrence with Synechococcus
The 16S rRNA gene generated 840,321 quality sequences from 15 samples, with an average of 56,021 sequences per sample. The 18S rRNA gene generated 850,827 quality sequences from 15 samples, with an average of 56,722 sequences per sample. Good coverages of all sequencing samples were >99.9%. The average value of the NP-Shannon in prokaryotic communities was 3.70, while the Chao1 index was 741 (Table S5). For eukaryotic communities, the average value of the NP-Shannon index was 3.65, and the Chao1 index was 466. The composition of the prokaryotic and eukaryotic communities was shown in Figure S2. The relative abundance of Synechococcus in the total prokaryotic community was high, maximally reaching 47.94% in A3 (Figure S3). Six OTUs were detected belonging to the genus Synechococcus, which dominated the phylum Cyanobacteria with 46.72–99.22% occupation (Figure S4). No OTU was categorized in Prochlorococcus.
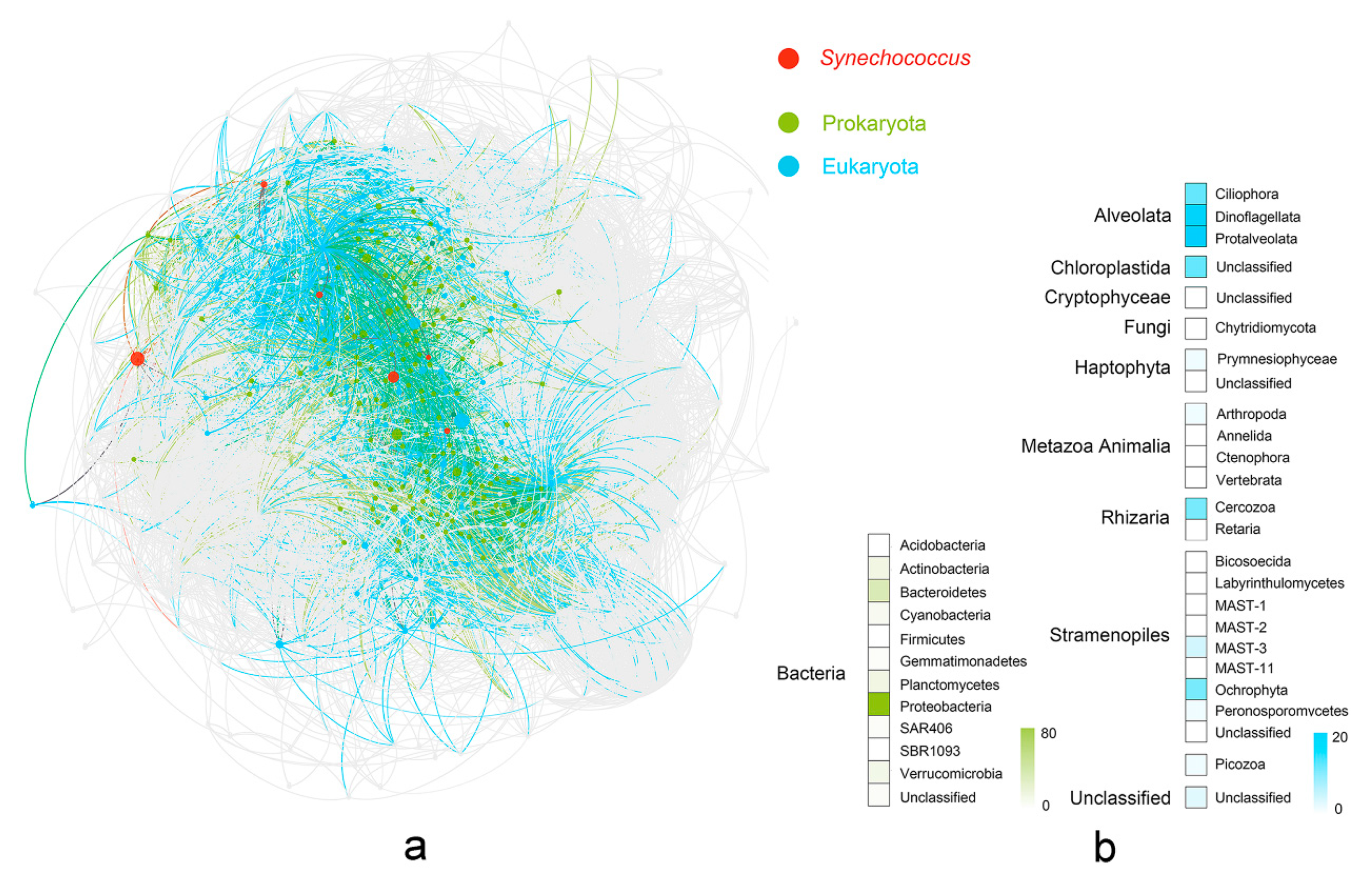
In the co-occurrence network, genetic correlation linked 869 nodes via 37,072 connections, corresponding to 30,707 positive and 6365 negative correlations (Figure S5). Synechococcus was connected to 159 prokaryotic OTU nodes, including 5 archaea and 154 bacteria (Figure 5). Proteobacteria was the most abundant prokaryotic group (76 OTUs), followed by Bacteroidetes (27), Actinobacteria (11), and Planctomycetes (11). A total of 102 of the 18S OTUs were associated with Synechococcus, which can be classified into eight known kingdoms (Alveolata, Chloroplastida, Cryptophyceae, Fungi, Haptophyta, Metazoa Animalia, Rhizaria, and Stramenopiles; Figure 5; Table S6). Among these kingdoms, Alveolata was represented the most (46), which included three phyla of Protalveolata (19), Dinoflagellata (17), and Ciliophora (10). The second-most abundant kingdom was Stramenopiles (21), which contained eight known phyla associated with Synechococcus.
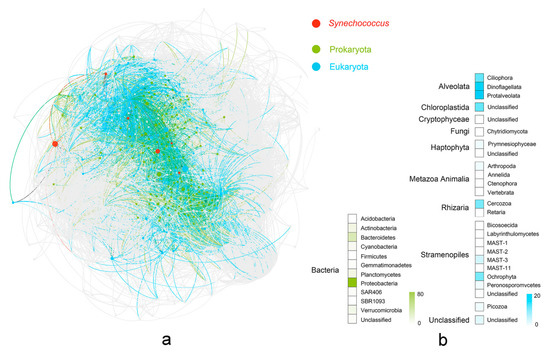
Figure 5.
Microbial community co-occurrence network associated with Synechococcus. (a) The network of microbial community relationships encompassing all prokaryotic and eukaryotic OTUs was constructed from the correlation matrix using a force-directed layout algorithm; red nodes represent six OTUs belong to Synechococcus; gray nodes represent OTUs with the absolute value of Spearman coefficients < 0.6 and p > 0.05 with Synechococcus; green nodes represent prokaryotic OTUs with the absolute value of Spearman coefficients > 0.6 and p < 0.05 with Synechococcus; blue nodes represent eukaryotic OTUs with the absolute value of Spearman coefficients > 0.6 and p < 0.05 with Synechococcus. (b) Heatmap showing the number of predominant kingdoms/phyla associated with Synechococcus using color legend; two domains were represented with different colors (prokaryota: green; eukaryota: blue).
4. Discussion
4.1. Distribution Features of Synechococcus Cell Abundance
In the studied Yellow Sea area, Synechococcus counts maximally reach 1.6 × 105 cells mL−1, with an average of 2.4 × 104 cells mL−1 (Figure 2). Along the seawater column, the counts of Synechococcus increased first and then decreased with the maximum cell abundance appearing in the middle layer as A3 (10 m, 3.3 × 104 cells mL−1), B4 (20 m, 3.2 × 104 cells mL−1), and C4 (30 m, 1.6 × 105 cells mL−1). The count of Synechococcus in the studied area generally accorded with that in other coastal seas of the world: using the parametric model, the global quantitative distribution of Synechococcus reached 104 cells mL−1 except for the Arctic Ocean and the Southern Ocean [60]. The similar distribution characteristics of Synechococcus abundance that ranged from 102 to 104 cells mL−1 in the coastal Yellow Sea waters were also observed [61]. The YSCWM—a bulk of cold water below the seasonal thermocline with a lower temperature (5–12 °C) and relatively constant salinity (31.5–32.5 PSU)—is an important phenomenon in the Yellow Sea, which has an impact on the summer strategy and vertical distribution of zooplankton and phytoplankton [62,63]. The marked decrease in Synechococcus counts in lower layers, particularly in station C, may attribute to the existence of YSCWM. Sequencing results also demonstrated that Synechococcus was dominant in the prokaryotic community with the absence of Prochlorococcus. Consequently, as the only highly abundant picocyanobacteria, Synechococcus is an important prokaryotic contributor to primary production in the studied coastal Yellow Sea.
The distribution of Synechococcus was significantly correlated to the sampling depth and contents of nitrite, ammonia, and dissolved silicon in the studied area. Consistent with the result of previous studies, the distribution of Synechococcus was affected by the sampling depth (Table 1) [64,65]. It could be closely related to the vertical attenuation in PAR, which is known to significantly affect the physiological activity of Synechococcus, including the amount of chl. a, total carotenoids, and D1 protein in the photosystem II reaction center [65,66]. The correlation between Synechococcus and nitrogen source (NO2− and NH4+) was also worthy of investigation. Marine Synechococcus isolates have been reported to utilize NH4+, NO2−, NO3−, urea, and amino acids as nitrogen sources via systems facilitating the active uptake of these compounds [67]. This effect of the nitrogen source was considered to be related to the light-harvesting pigment protein, phycoerythrin, of Synechococcus [68]. Nitrogen shortage was another factor affecting the vertical distribution of Synechococcus, as the low light levels in the deep euphotic zone cannot provide sufficient energy for assimilating NO2− and NH4+ for their N-rich phycobilisomes [69]. However, planktons prefer to incorporate NH4+ rather than nitrate in the coastal ecosystem [70]. The enrichment of NH4+ may attract relatively larger planktons such as diatoms, which may compete for nutrients and niches with Synechococcus. Some of these larger planktons may even prey on Synechococcus, which may explain the negative correlation between NH4+ concentration and Synechococcus counts. Our results also identified a correlation between Synechococcus and another material, DSi, which has been shown to favor its biomass [23,71,72].
4.2. Structure and Niche Occupation of Synechococcus Assemblages
Despite the ubiquitous and cosmopolitan distribution of the genus Synechococcus, there might be unseen barriers among the world’s oceans, and thus diverse genotypes of Synechococcus evolve. Some genotypes widely distribute, and others might restrict their habitat to narrow areas of marine environments, resulting in the current world distribution pattern of Synechococcus. Consistent with the results of Xia, Synechococcus S5.1 was the most abundant and widespread subcluster in the coastal Yellow Sea [8]. S5.2 and S5.3 were also widespread but occurred at low abundance. Generally, I, II, and IV are the most abundant in the Western Pacific Ocean, and the global ocean in general, in which clades I and IV appear to be the dominant type in temperate, coastal environments, while clade II widely dominated in subtropical/tropical and off-shore, oligotrophic environments [73,74]. Clade III also occurred in warm waters, but with a lower abundance and a narrower distribution [8]. However, in our study, clade III dominated in most samples, while clade I, II, and IV added up to one third of clade III on average. Another study by Xia showed that clade III is highly abundant in samples when the salinity is intermediate to high [9]. Tai and Palenik deemed the presence of clade III in this coastal environment to be more reflective of physical oceanographic patterns, such as stratification and the Inshore Countercurrent [75]. Synechococcus clade I was generally typical in cold, mesotrophic, and eutrophic waters [74]. Our research also exhibited the increase in clade I along with the vertical water column, especially at the bottom area located in the YSCWM zone.
The co-occurrence of pigment types in the studied coastal Yellow Sea was observed, and PT3 was always the dominant PT across the whole study area. This result is consistent with the opinion that one PT usually predominates in a sea area, although different PTs can co-occur [76]. The clone library method may not detect the rare taxa, while it is available to respond to the dominant species [16]. Attempts were made to detect the cpcBA operon, which encodes the phycocyanobilin (PCB). However, these attempts were not successful in any samples using the primer set of SyncpcB-Fw and SyncpcA-Rev [77]. Our gene marker cannot identify PC-only Synechococcus without the cpcBA operon, so cannot study the niche occupation of PT1 in the studied area. It was accepted that PT1 Synechococcus was found in turbid estuarine waters [78], while it was almost absent from the oceanic waters [10]. PT2 thrives in turbid coastal or continental shelf waters [79], whereas PT3 would mainly occur in mesotrophic or oligotrophic oceanic waters [9]. Consistently, it was ensured that from station A to C, the proportion of Synechococcus containing phycourobilin (PUB) increased gradually, while that containing no PUB decreased and was even absent in some water layers, demonstrating that underwater light spectral properties have a strong selective pressure on Synechococcus populations.
4.3. Potential Ecological Functions of Synechococcus in the Coastal Ecosystem
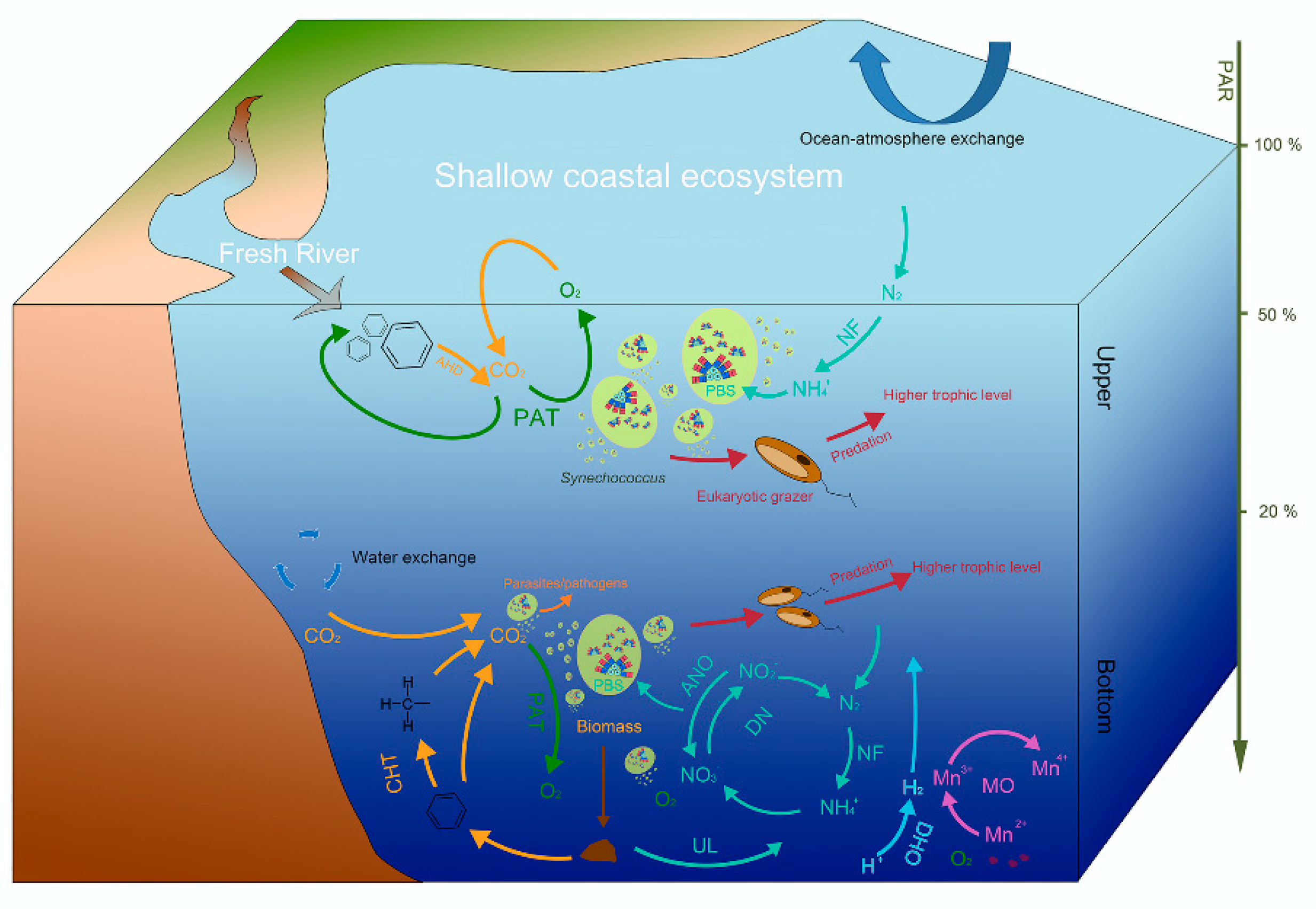
Synechococcus may play roles in multiple metabolic cycles in ecosystems via interactions with various microbial assemblages. From network results, Synechococcus was directly correlated with 159 prokaryotes and 102 eukaryotes (Figure 5). Correlations between Synechococcus and other prokaryotes could be explained by a similar adaptability to the external environment, reciprocity on metabolite supply, competition for nutrients, and the antergic effect of pathogenic bacteria. Microbiota roles in biochemical cycles can be revealed from genomics-based analyses [50]. From genomic data, Synechococcus is found to be possibly involved with some metabolic activities, such as photoautotrophy [80], nitrogen fixation [81], and extracellular degradation of hydrocarbons (Figure S6) [82]. The connection of Synechococcus with other metabolic activities, such as manganese oxidation and dark hydrogen oxidation, may result in the exchange of metabolic products (oxygen, carbon dioxide, and ammonia). Based on these multiple metabolic activities, Synechococcus appears to participate in carbon, nitrogen, hydrogen, and manganese cycling, outlining its feedback to the ecosystem. In addition, predation-based interactions can contribute to the transfer of materials and energy assimilated by Synechococcus to higher trophic levels.
In coastal areas, the environmental factors and biogeochemical cycles in different water layers vary, caused by the attenuation of light, exchanges between the atmosphere and ocean, and an inflow of freshwater rivers [56,83,84]. Using the function prediction of FAPROTAX, Synechococcus was found to associate with microbiota that are involved in different metabolic pathways and element cycles in upper and bottom water layers (Figure 6 and Figure S6). In addition to photosynthesis and nitrogen fixation, Synechococcus was associated with the degradation of certain hydrocarbons carried by fresh rivers in upper layers. Comparatively, in the bottom layer, Synechococcus was related to more diverse microbial metabolic cycles, including carbon, nitrogen, hydrogen, and manganese cycles. Among these, 77.85% of the metabolic activities were aerobic chemoheterotrophy, which was on account of the supply of oxygen, and the decomposition of its biomass. In addition, previous studies have suggested that the prasinophyte Tetraselmis sp. (belonging to the Alveolata kingdom) and Gymnodinium-like dinoflagellates (belonging to the Chloroplastida kingdom) were Synechococcus grazers under certain conditions, and play important roles in determining the fluctuation of the Synechococcus population [85]. Here, significant correlations between 102 eukaryotes and Synechococcus were revealed, which included five Gymnodinium clades. No significant correlation was observed between Synechococcus and Tetraselmis sp., while the association with 46 extra Alveolata was observed. These eukaryotes may be potential grazers of Synechococcus, which could transfer biomass assimilated by Synechococcus to upper trophic levels of the food chain for maintaining the stability and coordination of the ecosystem (bottom-up effects on the food web; Figure 6).
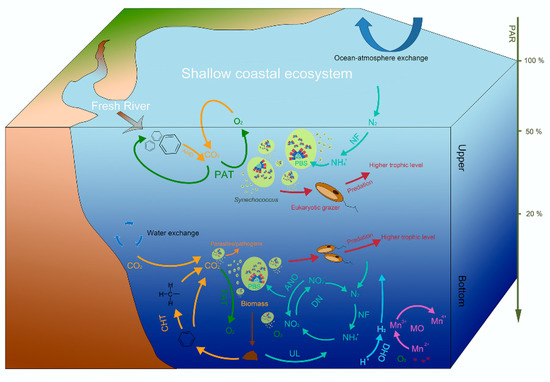
Figure 6.
Schematic diagram illustrating the coupling of the biochemical cycles associated with Synechococcus and mediated by microorganisms in the coastal ecosystem. PBS, phycobilisomes; PAR, photosynthetically active radiation; AHD, aromatic hydrocarbon degradation; PAT, photoautotrophy; NF, nitrogen fixation; CHT, chemoheterotrophy; ANO, aerobic nitrite oxidation; UL, ureolysis; MO, manganese oxidation. NO3−, nitrate; NO2−, nitrite; NH4+, ammonium; N2, nitrogen; H2, hydrogen; CO2, carbon dioxide; O2, oxygen; Mn, manganese.
5. Conclusions
The distribution of Synechococcus was studied in a typical section of the Yellow Sea. The cell abundance in the studied area was 2.4 × 104 cells mL−1 on average, which was significantly correlated to the sampling depth, nitrogen source (NO2− and NH4+), and DSi. The intense decrease in the count in the lower layer probably attributes to the YSCWM. Synechococcus clade III was dominant in most samples. Clade I increased along the water column, and its proportion exceeded that of clade III in some bottom layers. Phenogeneticly, PT3 was always the dominant PT across the whole study area. From station A to C, the proportion of PT3 increased gradually, while PT2 decreased and was even absent in some water layers. RDA showed that multiple environmental variables were significantly correlated to the Synechococcus phylogenetic structure and salinity was significantly correlated to the Synechococcus phenogenetic structure. Cross-correlation co-occurrence networks revealed that Synechococcus significantly correlated with various microbial taxa, including 159 prokaryotes and 102 eukaryotes. Metabolic cycling networks showed that Synechococcus may be related to more diverse microbial metabolic cycles, including the carbon, nitrogen, hydrogen, and manganese cycles in the bottom layer of the photic zone. Furthermore, various in situ grazers of Synechococcus were predicted, including Gymnodinium clades and 46 extra Alveolata.
Due to the unseen barriers among the world’s oceans, diverse genotypes of Synechococcus evolved and occupied different areas of the global ocean. To further explore the current world distribution pattern of Synechococcus, additional studies that examine the distribution of Synechococcus species and their evolution are required. Additionally, functional roles and grazer observations based on amplicon sequencing in this study tend to be preliminarily sketched, and mesocosm experiments combined with isotopic tracer and molecular omics methods should be further applied to prove the conclusions in the future.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jmse9091018/s1, Figures S1 –S6, Tables S1–S6. Figure S1: Spearman rank correlation analysis between Synechococcus clades as determined using rpoC1 and pigment types as indirectly assessed using the cpeBA operon; Figure S2: The relative abundances of predominant taxa in the prokaryotic community at the phylum level (a) and the eukaryotic community at the kingdom level (b); Figure S3: The relative abundance of taxa in the prokaryotic community at the genus level. (a) Community bar map showing the relative abundance of the genus; The unclassified does not represent a single taxon, but the sum of all taxa not classified to genus level; (b) Chord diagram reveals the relationship between samples and Synechococcus abundance. The length of the left semicircle represents the relative abundance of the Synechococcus in the corresponding samples. The right semicircle represents the distribution proportion of Synechococcus in different samples; Figure S4: The relative abundances of OTUs belonging to phyla Cyanobacteria; Figure S5: Network of microbial community encompassing all prokaryotic and eukaryotic OTUs constructed from the correlation matrix. OTUs with Spearman coefficients > 0.6 and p < 0.05 were connected and graphed using a force-directed layout algorithm; Figure S6: Network of microbial metabolic function correlations encompassing metabolic functions constructed from the correlation matrix of upper and bottom samples. (a) Metabolic functions with the absolute value of Spearman coefficients > 0.6 and p < 0.05 were connected and graphed using a force-directed layout algorithm. Red nodes represent six OTUs belonging to Synechococcus. (b) Metabolic heat map associated with Synechococcus showing the relative abundance of predominant metabolic functions classified based on the main metabolic elements. (c) Metabolic heat map associated with Synechococcus showing the relative abundance of predominant metabolic functions classified based on respiratory types; Table S1: Reference sequences of rpoC1 gene; Table S2: Reference sequences of cpeBA operon; Table S3: Spearman correlation coefficients between environmental variables; Table S4: Sequencing information and alpha diversity indices of rpoC1 and cpeBA gene; Table S5: Sequencing information and alpha diversity indices of 16S and 18S gene; Table S6: Correlative eukaryotic taxa of Synechococcus as potential in-situ grazers in the ecosystem.
Author Contributions
Conceptualization, S.Q. and J.L.; methodology, J.L. and T.W.; investigation, J.L. and T.W.; data curation, T.W. and X.C.; writing—original draft preparation, T.W.; writing—review and editing, S.Q. and J.L.; visualization, T.W. and X.C.; supervision, S.Q.; project administration, J.L.; funding acquisition, J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financially supported by the Key Deployment Project of the Centre for Ocean Mega-Science, Chinese Academy of Sciences (COMS2020Q09), the National Key Research and Development Program of China (No. 2018YFD0901102).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequence data of this study were deposited in the sequence read archive of NCBI (US National Center for Biotechnology Information; https://www.ncbi.nlm.nih.gov/, accessed on 13 September 2021) under the accession number PRJNA595432.
Acknowledgments
The data and samples were collected by Research Vessel Kexue III. We acknowledge the assistance from the Engineering and Technical Service, Institute of Oceanology, Chinese Academy of Sciences for organizing the research voyage and sharing open data. We extend our gratitude to the journal reviewers for their comments and suggestions, which helped in significantly improving the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scanlan, D.J.; Ostrowski, M.; Mazard, S.; Dufresne, A.; Garczarek, L.; Hess, W.R.; Post, A.F.; Hagemann, M.; Paulsen, I.; Partensky, F. Ecological genomics of marine Picocyanobacteria. Microbiol. Mol. Biol. Rev. 2009, 73, 249–299. [Google Scholar] [CrossRef] [Green Version]
- Ahlgren, N.A.; Rocap, G. Diversity and distribution of marine Synechococcus: Multiple gene phylogenies for consensus classification and development of qPCR assays for sensitive measurement of clades in the ocean. Front Microbiol. 2012, 3, 213. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.H.; Noh, J.H.; Shim, J. Seasonal changes in picocyanobacterial diversity as revealed by pyrosequencing in temperate waters of the East China Sea and the East Sea. Aquat. Microb. Ecol. 2013, 71, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Dufresne, A.; Ostrowski, M.; Scanlan, D.J.; Garczarek, L.; Mazard, S.; Palenik, B.P.; Paulsen, I.T.; De Marsac, N.T.; Wincker, P.; Dossat, C.; et al. Unravelling the genomic mosaic of a ubiquitous genus of marine cyanobacteria. Genome Biol. 2008, 9, R90. [Google Scholar] [CrossRef] [PubMed]
- Six, C.; Thomas, J.-C.; Garczarek, L.; Ostrowski, M.; Dufresne, A.; Blot, N.; Scanlan, D.J.; Partensky, F. Diversity and evolution of phycobilisomes in marine Synechococcus spp.: A comparative genomics study. Genome Biol. 2007, 8, R259. [Google Scholar] [CrossRef] [PubMed]
- Humily, F.; Farrant, G.; Marie, D.; Partensky, F.; Mazard, S.; Perennou, M.; Labadie, K.; Aury, J.-M.; Wincker, P.; Segui, A.N.; et al. Development of a targeted metagenomic approach to study a genomic region involved in light harvesting in marine Synechococcus. FEMS Microbiol. Ecol. 2014, 88, 231–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Wilhelm, S.; Harvey, H.R.; Taylor, K.; Jiao, N.; Chen, F. Novel lineages of Prochlorococcus and Synechococcus in the global oceans. ISME J. 2011, 6, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Partensky, F.; Garczarek, L.; Suzuki, K.; Guo, C.; Cheung, S.Y.; Liu, H. Phylogeography and pigment type diversity of Synechococcus cyanobacteria in surface waters of the Northwestern Pacific Ocean. Environ. Microbiol. 2016, 19, 142–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X.; Guo, W.; Tan, S.; Liu, H. Synechococcus assemblages across the salinity gradient in a salt wedge estuary. Front. Microbiol. 2017, 8, 1254. [Google Scholar] [CrossRef] [Green Version]
- Grébert, T.; Doré, H.; Partensky, F.; Farrant, G.K.; Boss, E.; Picheral, M.; Guidi, L.; Pesant, S.; Scanlan, D.; Wincker, P.; et al. Light color acclimation is a key process in the global ocean distribution of Synechococcus cyanobacteria. Proc. Natl. Acad. Sci. USA 2018, 115, E2010–E2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, M.L.; Doré, H.; Garczarek, L.; Seuthe, L.; Müller, O.; Sandaa, R.-A.; Bratbak, G.; Larsen, A. Synechococcus in the Atlantic Gateway to the Arctic Ocean. Front. Mar. Sci. 2016, 3, 191. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.A.; Rocap, G.; Moffett, J.W. Production of cobalt binding ligands in a Synechococcus feature at the Costa Rica upwelling dome. Limnol. Oceanogr. 2005, 50, 279–290. [Google Scholar] [CrossRef]
- Agawin, N.; Duarte, C.; Agustıí, S.; McManus, L. Abundance, biomass and growth rates of Synechococcus sp. in a tropical coastal ecosystem (Philippines, South China Sea). Estuar. Coast. Shelf Sci. 2003, 56, 493–502. [Google Scholar] [CrossRef]
- Morozov, E.; Tang, D. Satellite Ocean colour algorithm for Prochlorococcus, Synechococcus, and Picoeukaryotes concentration retrieval in the South China Sea. Adv. Space Res. 2018, 63, 16–31. [Google Scholar] [CrossRef]
- Chiang, K.-P.; Kuo, M.-C.; Chang, J.; Wang, R.-H.; Gong, G.-C. Spatial and temporal variation of the Synechococcus population in the East China Sea and its contribution to phytoplankton biomass. Cont. Shelf Res. 2002, 22, 3–13. [Google Scholar] [CrossRef]
- Xia, X.; Liu, H.; Choi, D.; Noh, J.H. Variation of Synechococcus pigment genetic diversity along two turbidity gradients in the China Seas. Microb. Ecol. 2018, 75, 10–21. [Google Scholar] [CrossRef]
- Li, G.; Song, Q.; Zheng, P.; Zhang, X.; Zou, S.; Li, Y.; Gao, X.; Zhao, Z.; Gong, J. Dynamics and distribution of marine Synechococcus abundance and genotypes during seasonal hypoxia in a Coastal Marine Ranch. J. Mar. Sci. Eng. 2021, 9, 549. [Google Scholar] [CrossRef]
- Choi, D.H.; Noh, J.H. Phylogenetic diversity of Synechococcus strains isolated from the East China Sea and the East Sea. FEMS Microbiol. Ecol. 2009, 69, 439–448. [Google Scholar] [CrossRef]
- Jing, H.; Liu, H. Phylogenetic composition of Prochlorococcus and Synechococcus in cold eddies of the South China Sea. Aquat. Microb. Ecol. 2012, 65, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Vidyarathna, N.K.; Palenik, B.; Lee, P.; Liu, H. Comparison of the seasonal variations of Synechococcus assemblage structures in estuarine waters and coastal waters of Hong Kong. Appl. Environ. Microbiol. 2015, 81, 7644–7655. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.-C.; Lin, R.-F.; Chu, M.-K.; Chen, H.-M. Organization and expression of nitrogen-fixation genes in the aerobic nitrogen-fixing unicellular cyanobacterium Synechococcus sp. strain RF-1. Microbiology 1999, 145, 743–753. [Google Scholar] [CrossRef]
- Hung, S.-H.; Chung, C.-C.; Liao, C.-W.; Gong, G.-C.; Chang, J. Sequence diversity and expression levels of Synechococcus phosphate transporter gene in the East China Sea. J. Exp. Mar. Biol. Ecol. 2013, 440, 90–99. [Google Scholar] [CrossRef]
- Brzezinski, M.A.; Krause, J.W.; Baines, S.B.; Collier, J.L.; Ohnemus, D.C.; Twining, B.S. Patterns and regulation of silicon accumulation in Synechococcus spp. J. Phycol. 2017, 53, 746–761. [Google Scholar] [CrossRef]
- Trosvik, P.; Rudi, K.; Straetkvern, K.O.; Jakobsen, K.S.; Naes, T.; Stenseth, N.C. Web of ecological interactions in an experimental gut microbiota. Environ. Microbiol. 2010, 12, 2677–2687. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [Green Version]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2011, 6, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Dai, T.; Su, Z.; Hasegawa, K.; Tian, J.; Chen, L.; Wen, D. A tripartite microbial-environment network indicates how crucial microbes influence the microbial community ecology. Microb. Ecol. 2019, 79, 342–356. [Google Scholar] [CrossRef]
- Wagg, C.; Schlaeppi, K.; Banerjee, S.; Kuramae, E.E.; Van der Heijden, M.G.A. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 2019, 10, 4841. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wang, H.; Dsouza, M.; Lou, J.; He, Y.; Dai, Z.; Brookes, P.C.; Xu, J.; Gilbert, J.A. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 2016, 10, 1891–1901. [Google Scholar] [CrossRef]
- Shi, S.; Nuccio, E.E.; Shi, Z.J.; He, Z.; Zhou, J.; Firestone, M.K. The interconnected rhizosphere: High network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016, 19, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Widder, S.; Besemer, K.; Singer, G.; Ceola, S.; Bertuzzo, E.; Quince, C.; Sloan, W.T.; Rinaldo, A.; Battin, T.J. Fluvial network organization imprints on microbial co-occurrence networks. Proc. Natl. Acad. Sci. USA 2014, 111, 12799–12804. [Google Scholar] [CrossRef] [Green Version]
- Mikhailov, I.S.; Zakharova, Y.R.; Bukin, Y.S.; Galachyants, Y.P.; Petrova, D.; Sakirko, M.V.; Likhoshway, Y.V. Co-occurrence networks among bacteria and microbial eukaryotes of Lake Baikal during a spring phytoplankton bloom. Microb. Ecol. 2019, 77, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.A.; Countway, P.D.; Xia, C.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.-E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.B.C.; Kirby, M.X.; Berger, W.H.; Bjorndal, K.A.; Botsford, L.W.; Bourque, B.J.; Bradbury, R.; Cooke, R.H.; Erlandson, J.; Estes, J.A.; et al. Historical overfishing and the recent collapse of coastal ecosystems. Science 2001, 293, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubert, M.; Grob, C.; Howat, A.M.; Burns, O.J.; Pratscher, J.; Jehmlich, N.; Von Bergen, M.; Richnow, H.H.; Chen, Y.; Murrell, J.C. Methylamine as a nitrogen source for microorganisms from a coastal marine environment. Environ. Microbiol. 2017, 19, 2246–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Ning, X.; Su, J.; Lin, Y.; Xu, B. Environmental changes and the responses of the ecosystems of the Yellow Sea during 1976–2000. J. Mar. Syst. 2005, 55, 223–234. [Google Scholar] [CrossRef]
- Zhao, J.; Ran, W.; Teng, J.; Liu, Y.; Liu, H.; Yin, X.; Cao, R.; Wang, Q. Microplastic pollution in sediments from the Bohai Sea and the Yellow Sea, China. Sci. Total. Environ. 2018, 640–641, 637–645. [Google Scholar] [CrossRef]
- Zhou, F.; Xuan, J.; Huang, D.; Liu, C.; Sun, J. The timing and the magnitude of spring phytoplankton blooms and their relationship with physical forcing in the central Yellow Sea in 2009. Deep. Sea Res. Part II Top. Stud. Oceanogr. 2013, 97, 4–15. [Google Scholar] [CrossRef]
- Wang, T.; Chen, X.; Li, J.; Qin, S. Distribution and phenogenetic diversity of Synechococcus in the Bohai Sea. China. J. Oceanol. Limnol. 2021, in press. [Google Scholar] [CrossRef]
- Dafner, E.V. Segmented continuous-flow analyses of nutrient in seawater: Intralaboratory comparison of technicon autoanalyzer II and bran+luebbe continuous flow autoanalyzer III. Limnol. Oceanogr. Methods 2015, 13, 511–520. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Yu, S.; Bai, J.; Qin, S. Community characteristics and ecological roles of bacterial biofilms associated with various algal settlements on coastal reefs. J. Environ. Manag. 2019, 250, 109459. [Google Scholar] [CrossRef]
- Mühling, M.; Fuller, N.; Somerfield, P.; Post, A.; Wilson, W.; Scanlan, D.; Joint, I.; Mann, N. High resolution genetic diversity studies of marine Synechococcus isolates using rpoC1-based restriction fragment length polymorphism. Aquat. Microb. Ecol. 2006, 45, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.; et al. Improved bacterial 16S rRNA Gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 2016, 1, e00009–e00015. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Richlen, M.L.; Sehein, T.R.; Kulis, D.M.; Anderson, D.M.; Cai, Z. Microbial community structure and associations during a marine dinoflagellate bloom. Front. Microbiol. 2018, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.W.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Feizi, S.; Marbach, D.; Médard, M.; Kellis, M. Network deconvolution as a general method to distinguish direct dependencies in networks. Nat. Biotechnol. 2013, 31, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Krieger, A.M.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]
- Revelle, W.; Revelle, M.W. Package ‘psych’. 2015. Available online: https://cran.r-project.org/web/packages/psych/psych.pdf (accessed on 13 September 2021).
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009; Available online: https://gephi.org/publications/gephi-bastian-feb09.pdf (accessed on 13 September 2021).
- Lin, S.; Zou, T.; Gao, H.; Guo, X. The vertical attenuation of irradiance as a function of turbidity: A case of the Huanghai (Yellow) Sea in spring. Acta Oceanol. Sin. 2009, 28, 66–75. [Google Scholar]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Flombaum, P.; Gallegos, J.L.; Gordillo, R.A.; Rincón, J.; Zabala, L.L.; Jiao, N.; Karl, D.M.; Li, W.K.W.; Lomas, M.; Veneziano, D.; et al. Present and future global distributions of the marine Cyanobacteria prochlorococcus and Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 9824–9829. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhao, L.; Xiao, T.; Zhao, S.; Xuan, J.; Li, C.; Ning, X. Spatial and temporal variation of picoplankton distribution in the Yellow Sea, China. Chin. J. Oceanol. Limnol. 2011, 29, 150–162. [Google Scholar] [CrossRef]
- Li, H.; Xiao, T.; Ding, T.; Lü, R. Effect of the Yellow Sea cold water mass (YSCWM) on distribution of bacterioplankton. Acta Ecol. Sin. 2006, 26, 1012–1019. [Google Scholar] [CrossRef]
- Mei, X.; Li, R.; Zhang, X.; Liu, Q.; Liu, J.; Wang, Z.; Lan, X.; Liu, J.; Sun, R. Evolution of the Yellow Sea warm current and the Yellow Sea cold water mass since the middle pleistocene. Palaeogeogr. Palaeoclim. Palaeoecol. 2016, 442, 48–60. [Google Scholar] [CrossRef]
- Bouman, H.A.; Ulloa, O.; Barlow, R.; Li, W.K.W.; Platt, T.; Zwirglmaier, K.; Scanlan, D.J.; Sathyendranath, S. Water-column stratification governs the community structure of subtropical marine picophytoplankton. Environ. Microbiol. Rep. 2011, 3, 473–482. [Google Scholar] [CrossRef]
- Lohscheider, J.N.; Strittmatter, M.; Küpper, H.; Adamska, I. Vertical distribution of epibenthic freshwater Cyanobacterial Synechococcus spp. strains depends on their ability for photoprotection. PLoS ONE 2011, 6, e20134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postius, C.; Kenter, U.; Wacker, A.; Ernst, A.; Böger, P. Light causes selection among two phycoerythrin-rich Synechococcus isolates from Lake Constance. FEMS Microbiol. Ecol. 1998, 25, 171–178. [Google Scholar] [CrossRef]
- Moore, L.R.; Post, A.F.; Rocap, G.; Chisholm, S.W. Utilization of different nitrogen sources by the marine cyanobacteria Prochlorococcus and Synechococcus. Limnol. Oceanogr. 2002, 47, 989–996. [Google Scholar] [CrossRef]
- Wyman, M.; Gregory, R.P.F.; Carr, N.G. Novel role for Phycoerythrin in a marine Cyanobacterium, Synechococcus strain DC2. Science 1985, 230, 818–820. [Google Scholar] [CrossRef]
- Carr, N.G.; Mann, N.H. The oceanic cyanobacterial picoplankton. In The Molecular Biology of Cyanobacteria; Springer: Berlin/Heidelberg, Germany, 1994; pp. 27–48. [Google Scholar]
- Rodrigues, R.M.; Williams, P.J.L.B. Inorganic nitrogen assimilation by picoplankton and whole plankton in a coastal ecosystem. Limnol. Oceanogr. 2002, 47, 1608–1616. [Google Scholar] [CrossRef]
- Baines, S.B.; Twining, B.; Brzezinski, M.A.; Krause, J.W.; Vogt, S.; Assael, D.; McDaniel, H. Significant silicon accumulation by marine Picocyanobacteria. Nat. Geosci. 2012, 5, 886–891. [Google Scholar] [CrossRef]
- Wei, Y.; Sun, J.; Zhang, X.; Wang, J.; Huang, K. Picophytoplankton size and biomass around equatorial eastern Indian Ocean. Microbiology 2019, 8, e00629. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, N.A.; Rocap, G. Culture isolation and culture-independent clone libraries reveal new marine Synechococcus ecotypes with distinctive light and N physiologies. Appl. Environ. Microbiol. 2006, 72, 7193–7204. [Google Scholar] [CrossRef] [Green Version]
- Zwirglmaier, K.; Jardillier, L.; Ostrowski, M.; Mazard, S.; Garczarek, L.; Vaulot, D.; Not, F.; Massana, R.; Ulloa, O.; Scanlan, D.J. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ. Microbiol. 2007, 10, 147–161. [Google Scholar] [CrossRef]
- Tai, V.; Palenik, B. Temporal variation of Synechococcus clades at a coastal Pacific Ocean monitoring site. ISME J. 2009, 3, 903–915. [Google Scholar] [CrossRef] [Green Version]
- Haverkamp, T.H.; Schouten, D.; Doeleman, M.; Wollenzien, U.; Huisman, J.; Stal, L.J. Colorful microdiversity of Synechococcus strains (Picocyanobacteria) isolated from the Baltic Sea. ISME J. 2008, 3, 397–408. [Google Scholar] [CrossRef]
- Haverkamp, T.H.; Acinas, S.G.; Doeleman, M.; Stomp, M.; Huisman, J.; Stal, L.J. Diversity and phylogeny of Baltic Sea picocyanobacteria inferred from their ITS and phycobiliprotein operons. Environ. Microbiol. 2007, 10, 174–188. [Google Scholar] [CrossRef]
- Liu, H.; Jing, H.; Wong, T.H.; Chen, B. Co-occurrence of phycocyanin-and phycoerythrin-rich Synechococcus in subtropical estuarine and coastal waters of Hong Kong. Environ. Microbiol. Rep. 2014, 6, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.; Phinney, D.; Yentsch, C. Water column transparency and the distribution of spectrally distinct forms of phycoerythrin-containing organisms. Mar. Ecol. Prog. Ser. 1998, 162, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Putt, M.; Glover, H.E. Diurnal patterns in photosynthetic capacity and depth-dependent photosynthesis-irradiance relationships in Synechococcus spp. and larger phytoplankton in three water masses in the Northwest Atlantic Ocean. Mar. Biol. 1986, 91, 205–217. [Google Scholar] [CrossRef]
- Spiller, H.; Shanmugam, K.T. Physiological conditions for nitrogen fixation in a unicellular marine Cyanobacterium, Synechococcus sp. strain SF1. J. Bacteriol. 1987, 169, 5379–5384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurster, M.; Mundt, S.; Hammer, E.; Schauer, F.; Lindequist, U. Extracellular degradation of phenol by the Cyanobacterium Synechococcus PCC 7002. Environ. Boil. Fishes 2003, 15, 171–176. [Google Scholar] [CrossRef]
- Barbier, E.B.; Hacker, S.D.; Kennedy, C.; Koch, E.W.; Stier, A.C.; Silliman, B.R. The value of estuarine and coastal ecosystem services. Ecol. Monogr. 2011, 81, 169–193. [Google Scholar] [CrossRef]
- Li, Y.; Tang, K.; Zhang, L.; Zhao, Z.; Xie, X.; Chen, C.-T.A.; Wang, D.; Jiao, N.; Zhang, Y. Coupled carbon, sulfur, and nitrogen cycles mediated by microorganisms in the water column of a shallow-water hydrothermal ecosystem. Front. Microbiol. 2018, 9, 2718. [Google Scholar] [CrossRef] [Green Version]
- Nagarkar, M.; Countway, P.D.; Du Yoo, Y.; Daniels, E.; Poulton, N.; Palenik, B. Temporal dynamics of eukaryotic microbial diversity at a coastal Pacific site. ISME J. 2018, 12, 2278–2291. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).