LFDFT—A Practical Tool for Coordination Chemistry


Abstract
:1. Introduction
2. Theory
2.1. General
2.2. Computational Details
2.3. Methodology
3. Results and Discussion
3.1. Low-Lying Excited States of [Eu(NO)(phenanthroline)]
3.2. X-ray Absorption Spectra of Cerocene
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shibasaki, M.; Yoshikawa, N. Lanthanide Complexes in Multifunctional Asymmetric Catalysis. Chem. Rev. 2002, 102, 2187–2210. [Google Scholar] [CrossRef] [PubMed]
- Trummer, D.; Searles, K.; Algasov, A.; Guda, S.A.; Soldatov, A.V.; Ramanantoanina, H.; Safonova, O.V.; Guda, A.A.; Copéret, C. Deciphering the Phillips Catalyst by Orbital Analysis and Supervised Machine Learning from Cr Pre-edge XANES of Molecular Libraries. J. Am. Chem. Soc. 2021, 143, 7326–7341. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.C.G. Benefiting from the Unique Properties of Lanthanide Ions. Accounts Chem. Res. 2006, 39, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Poe, T.N.; Molinari, S.; Beltran-Leiva, M.J.; Celis-Barros, C.; Ramanantoanina, H.; Albrecht-Schönzart, T.E. Influence of Outer-Sphere Anions on the Photoluminescence from Samarium(II) Crown Complexes. Inorg. Chem. 2021, 60, 15196–15207. [Google Scholar] [CrossRef] [PubMed]
- Sessoli, R.; Powell, A.K. Strategies towards single molecule magnets based on lanthanide ions. Coord. Chem. Rev. 2009, 253, 2328–2341. [Google Scholar] [CrossRef]
- Dreiser, J. Molecular lanthanide single-ion magnets: From bulk to submonolayers. J. Phys. Condens. Matter 2015, 27, 183203. [Google Scholar] [CrossRef]
- Ramanantoanina, H.; Studniarek, M.; Daffé, N.; Dreiser, J. Non-empirical calculation of X-ray magnetic circular dichroism in lanthanide compounds. Chem. Commun. 2019, 55, 2988–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daul, C. Density functional theory applied to the excited states of coordination compounds. Int. J. Quantum Chem. 1994, 52, 867–877. [Google Scholar] [CrossRef]
- Chermette, H. Density functional theory: A powerful tool for theoretical studies in coordination chemistry. Coord. Chem. Rev. 1998, 178–180, 699–721. [Google Scholar] [CrossRef]
- Schwarz, K. DFT calculations of solids with LAPW and WIEN2k. J. Solid State Chem. 2003, 176, 319–328. [Google Scholar] [CrossRef]
- Grimme, S.; Waletzke, M. A combination of Kohn–Sham density functional theory and multi-reference configuration interaction methods. J. Chem. Phys. 1999, 111, 5645–5655. [Google Scholar] [CrossRef]
- Szalay, P.G.; Müller, T.; Gidofalvi, G.; Lischka, H.; Shepard, R. Multiconfiguration Self-Consistent Field and Multireference Configuration Interaction Methods and Applications. Chem. Rev. 2012, 112, 108–181. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Verma, P.; Cramer, C.J.; Gagliardi, L.; Truhlar, D.G. Combining Wave Function Methods with Density Functional Theory for Excited States. Chem. Rev. 2018, 118, 7249–7292. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, W.; Calvello, S.; Soncini, A. Configuration-averaged 4f orbitals in ab initio calculations of low-lying crystal field levels in lanthanide(iii) complexes. Phys. Chem. Chem. Phys. 2016, 18, 15807–15814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, S.; Lang, L.; Neese, F. A dynamic correlation dressed complete active space method: Theory, implementation, and preliminary applications. J. Chem. Phys. 2017, 147, 234109. [Google Scholar] [CrossRef]
- Singh, S.K.; Atanasov, M.; Neese, F. Challenges in Multireference Perturbation Theory for the Calculations of the g-Tensor of First-Row Transition-Metal Complexes. J. Chem. Theory Comput. 2018, 14, 4662–4677. [Google Scholar] [CrossRef]
- Burke, K. Perspective on density functional theory. J. Chem. Phys. 2012, 136, 150901. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for Density Functional Theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Verma, P.; Truhlar, D.G. Status and Challenges of Density Functional Theory. Trends Chem. 2020, 2, 302–318. [Google Scholar] [CrossRef]
- Schwarz, K.; Blaha, P. Solid state calculations using WIEN2k. Comput. Mater. Sci. 2003, 28, 259–273. [Google Scholar] [CrossRef]
- Casida, M.; Huix-Rotllant, M. Progress in Time-Dependent Density-Functional Theory. Annu. Rev. Phys. Chem. 2012, 63, 287–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Hannachi, D.; Haroun, M.F.; Khireddine, A.; Chermette, H. Optical and nonlinear optical properties of Ln(Tp)2, where Ln = La,…,Lu and Tp = tris(pyrazolyl)borate: A DFT + TD-DFT study. New J. Chem. 2019, 43, 14377–14389. [Google Scholar] [CrossRef]
- Gutierrez, F.; Rabbe, C.; Poteau, R.; Daudey, J.P. Theoretical Study of Ln(III) Complexes with Polyaza-Aromatic Ligands: Geometries of [LnL(H2O)n]3+ Complexes and Successes and Failures of TD-DFT. J. Phys. Chem. A 2005, 109, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Momeni, M.R.; Brown, A. Why Do TD-DFT Excitation Energies of BODIPY/Aza-BODIPY Families Largely Deviate from Experiment? Answers from Electron Correlated and Multireference Methods. J. Chem. Theory Comput. 2015, 11, 2619–2632. [Google Scholar] [CrossRef]
- Ramanantoanina, H.; Daul, C. A non-empirical calculation of 2p core-electron excitation in compounds with 3d transition metal ions using ligand-field and density functional theory (LFDFT). Phys. Chem. Chem. Phys. 2017, 19, 20919–20929. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, M.; Daul, C.; Rauzy, C. New insights into the effects of covalency on the ligand field parameters: A DFT study. Chem. Phys. Lett. 2003, 367, 737–746. [Google Scholar] [CrossRef]
- Ramanantoanina, H.; Sahnoun, M.; Barbiero, A.; Ferbinteanu, M.; Cimpoesu, F. Development and applications of the LFDFT: The non-empirical account of ligand field and the simulation of the f–d transitions by density functional theory. Phys. Chem. Chem. Phys. 2015, 17, 18547–18557. [Google Scholar] [CrossRef] [Green Version]
- Vlahović, F.; Perić, M.; Gruden-Pavlović, M.; Zlatar, M. Assessment of TD-DFT and LF-DFT for study of d– d transitions in first row transition metal hexaaqua complexes. J. Chem. Phys. 2015, 142, 214111. [Google Scholar] [CrossRef] [Green Version]
- Ramanantoanina, H. A DFT-based theoretical model for the calculation of spectral profiles of lanthanide M4,5-edge X-ray absorption. J. Chem. Phys. 2018, 149, 054104. [Google Scholar] [CrossRef] [Green Version]
- Vanquickenborne, L.; Ceulemans, A. Ligand-field models and the photochemistry of coordination compounds. Coord. Chem. Rev. 1983, 48, 157–202. [Google Scholar] [CrossRef]
- Gerloch, M.; Harding, J.H.; Woolley, R.G. The context and application of ligand field theory. In Inorganic Chemistry; Springer: Berlin/Heidelberg, Germany, 1981; pp. 1–46. [Google Scholar]
- Woolley, R.G. Ligand-field analysis of transition-metal complexes. Int. Rev. Phys. Chem. 1987, 6, 93–141. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alvarez, S.; Mealli, C.; Falceto, A.; Cahill, T.J.; Zeng, T.; Manca, G. From Widely Accepted Concepts in Coordination Chemistry to Inverted Ligand Fields. Chem. Rev. 2016, 116, 8173–8192. [Google Scholar] [CrossRef] [PubMed]
- Baerends, E.; Ziegler, T.; Atkins, A.; Autschbach, J.; Baseggio, O.; Bashford, D.; Bérces, A.; Bickelhaupt, F.; Bo, C.; Boerrigter, P.; et al. ADF 2021.1, SCM; Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2021. [Google Scholar]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.K.; Musgrave, C.B. Prediction of transition state barriers and enthalpies of reaction by a new hybrid density-functional approximation. J. Chem. Phys. 2001, 115, 11040–11051. [Google Scholar] [CrossRef]
- Lenthe, E.V.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Lenthe, E.V.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.P.; Goel, N.; Singh, G.; Srivastava, P. Syntheses, structural and thermal studies of Eu(III) and Gd(III) complexes with 2, 6-dinitrophenol and 1, 10-phenanthroline/2, 2′-bipyridine ligands. Inorganica Chim. Acta 2012, 387, 294–307. [Google Scholar] [CrossRef]
- Walter, M.D.; Booth, C.H.; Lukens, W.W.; Andersen, R.A. Cerocene Revisited: The Electronic Structure of and Interconversion Between Ce2(C8H8)3 and Ce(C8H8)2. Organometallics 2009, 28, 698–707. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, I.; Trendafilova, N.; Zahariev, T.; Danchova, N.; Gutzov, S. Theoretical insight in highly luminescent properties of Eu(III) complex with phenanthroline. J. Lumin. 2018, 202, 192–205. [Google Scholar] [CrossRef]
- Bünzli, J.C.G.; Piguet, C. Taking advantage of luminescent lanthanide ions. Chem. Soc. Rev. 2005, 34, 1048. [Google Scholar] [CrossRef]
- Thejo Kalyani, N.; Dhoble, S. Organic light emitting diodes: Energy saving lighting technology—A review. Renew. Sustain. Energy Rev. 2012, 16, 2696–2723. [Google Scholar] [CrossRef]
- Binnemans, K. Interpretation of europium(III) spectra. Coord. Chem. Rev. 2015, 295, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Jia, G.; Duan, C.K.; Wong, W.Y.; Wong, W.T.; Tanner, P.A. Crystal Structure, Spectroscopy and Crystal Field Analysis of Substituted 1, 10-Phenanthroline–Europium Complexes. Eur. J. Inorg. Chem. 2011, 2011, 637–646. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W. A challenge for density functionals: Self-interaction error increases for systems with a noninteger number of electrons. J. Chem. Phys. 1998, 109, 2604–2608. [Google Scholar] [CrossRef]
- de Silva, P.; Corminboeuf, C. Local hybrid functionals with orbital-free mixing functions and balanced elimination of self-interaction error. J. Chem. Phys. 2015, 142, 074112. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, K. Challenges for Theory and Computation. Computation 2017, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Hüfner. Optical Spectra of Transparent Rare Earth Compounds; Academic Press: Cambridge, MA, USA, 1978. [Google Scholar]
- Newman, D.J.; Ng, B. (Eds.) Crystal Field Handbook; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar] [CrossRef]
- Aravena, D.; Atanasov, M.; Neese, F. Periodic Trends in Lanthanide Compounds through the Eyes of Multireference ab Initio Theory. Inorg. Chem. 2016, 55, 4457–4469. [Google Scholar] [CrossRef]
- Tchougréeff, A.L.; Dronskowski, R. Nephelauxetic effect revisited. Int. J. Quantum Chem. 2009, 109, 2606–2621. [Google Scholar] [CrossRef]
- Tanner, P.A.; Yeung, Y.Y.; Ning, L. What Factors Affect the 5D0 Energy of Eu3+? An Investigation of Nephelauxetic Effects. J. Phys. Chem. A 2013, 117, 2771–2781. [Google Scholar] [CrossRef]
- Dorenbos, P. Lanthanide 4f-electron binding energies and the nephelauxetic effect in wide band gap compounds. J. Lumin. 2013, 136, 122–129. [Google Scholar] [CrossRef]
- Cowan, R.D. The Theory of Atomic Structure and Spectra; University of California Press: Oakland, CA, USA, 1981. [Google Scholar]
- de Groot, F. Multiplet effects in X-ray spectroscopy. Coord. Chem. Rev. 2005, 249, 31–63. [Google Scholar] [CrossRef]
- de Groot, F.; Kotani, A. Core Level Spectroscopy of Solids; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar] [CrossRef] [Green Version]
- Thole, B.T.; van der Laan, G.; Fuggle, J.C.; Sawatzky, G.A.; Karnatak, R.C.; Esteva, J.M. 3d X-ray-absorption lines and the 3d94fn+1 multiplets of the lanthanides. Phys. Rev. B 1985, 32, 5107–5118. [Google Scholar] [CrossRef]
- Edelstein, N.M.; Allen, P.G.; Bucher, J.J.; Shuh, D.K.; Sofield, C.D.; Kaltsoyannis, N.; Maunder, G.H.; Russo, M.R.; Sella, A. The Oxidation State of Ce in the Sandwich Molecule Cerocene. J. Am. Chem. Soc. 1996, 118, 13115–13116. [Google Scholar] [CrossRef] [Green Version]
- Mooßen, O.; Dolg, M. Two interpretations of the cerocene electronic ground state. Chem. Phys. Lett. 2014, 594, 47–50. [Google Scholar] [CrossRef]
- Smiles, D.E.; Batista, E.R.; Booth, C.H.; Clark, D.L.; Keith, J.M.; Kozimor, S.A.; Martin, R.L.; Minasian, S.G.; Shuh, D.K.; Stieber, S.C.E.; et al. The duality of electron localization and covalency in lanthanide and actinide metallocenes. Chem. Sci. 2020, 11, 2796–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerridge, A.; Coates, R.; Kaltsoyannis, N. Is Cerocene Really a Ce(III) Compound? All-Electron Spin–Orbit Coupled CASPT2 Calculations on M(η8-C8H8)2 (M = Th, Pa, Ce). J. Phys. Chem. A 2009, 113, 2896–2905. [Google Scholar] [CrossRef] [PubMed]
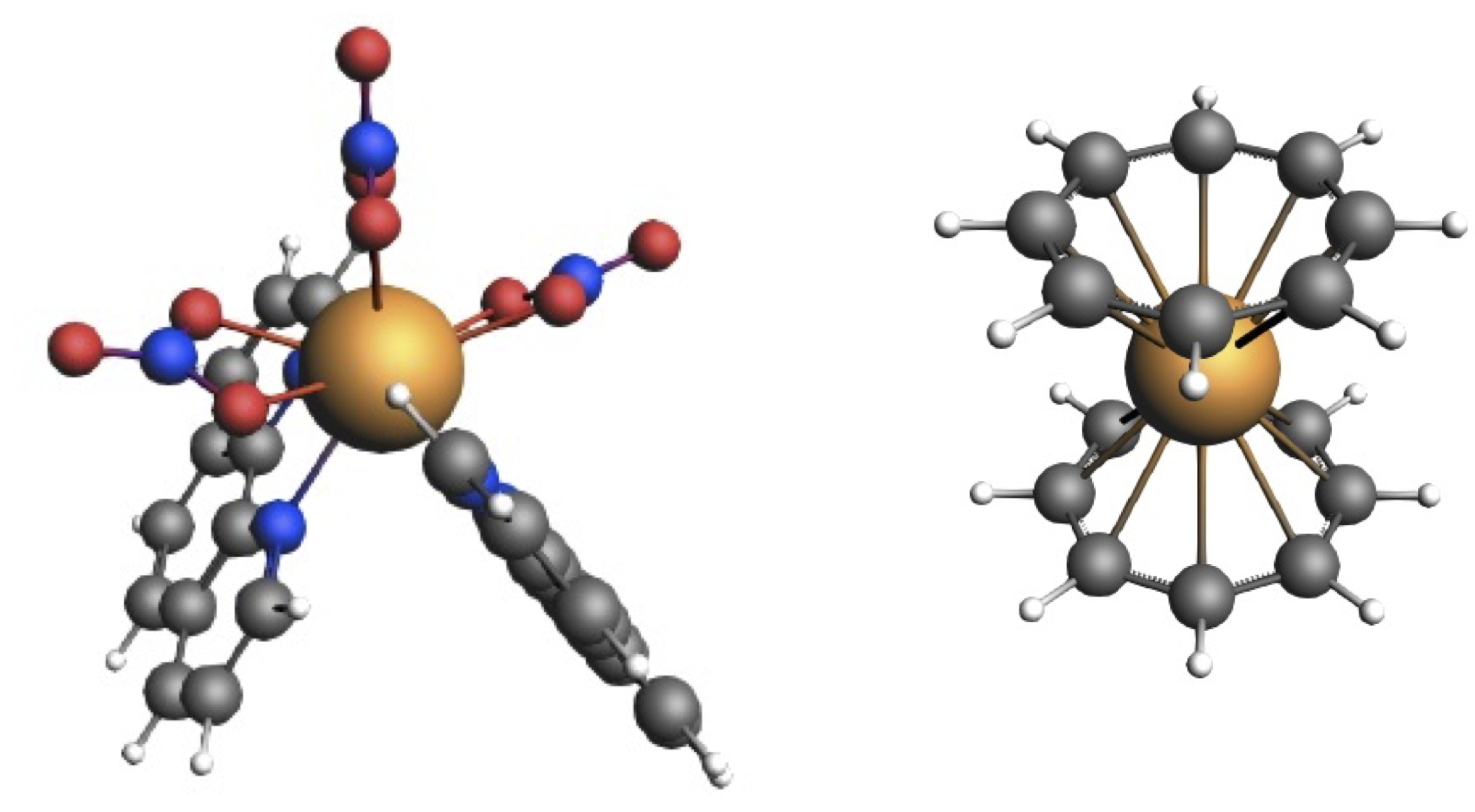



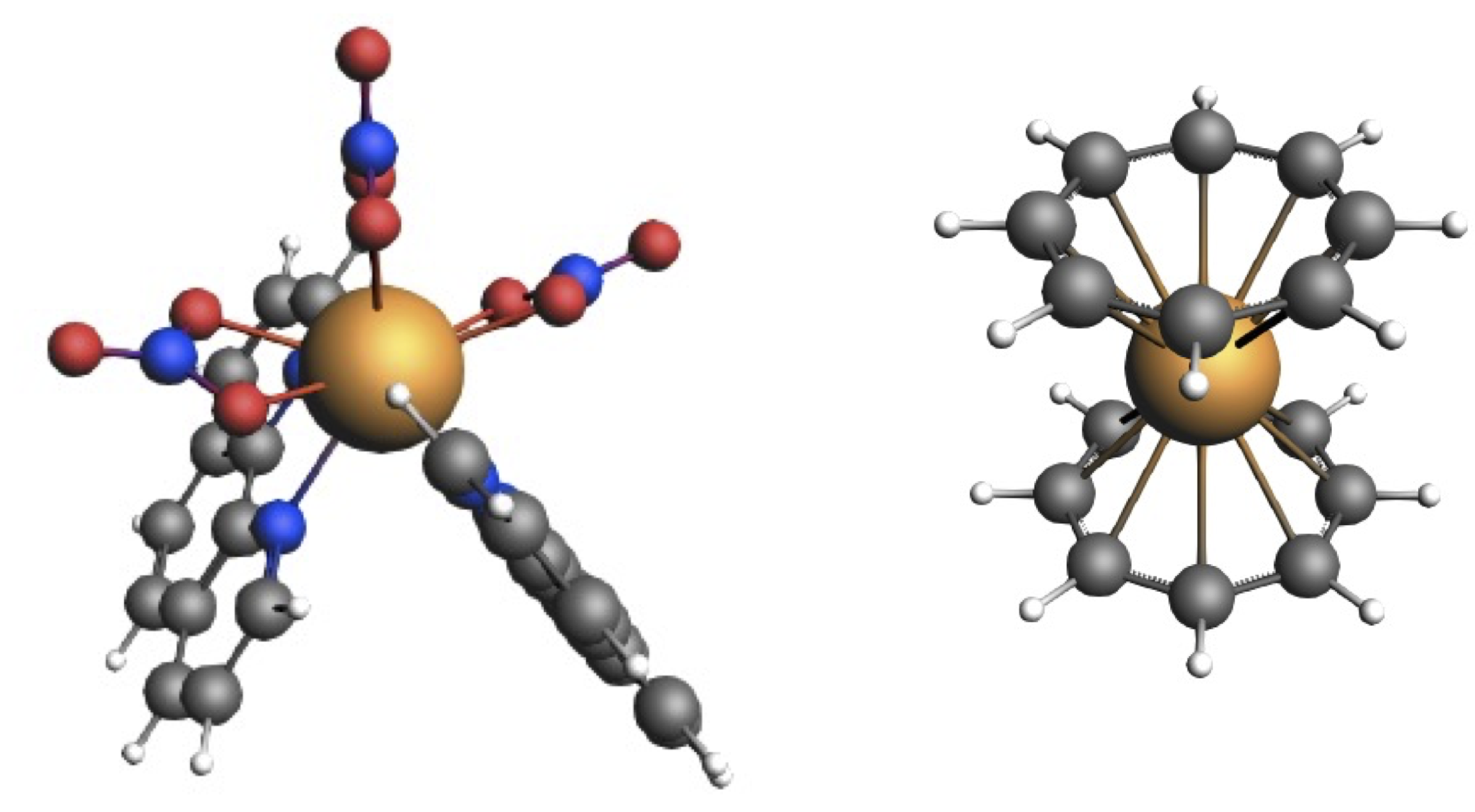{kind=link}
{kind=link}
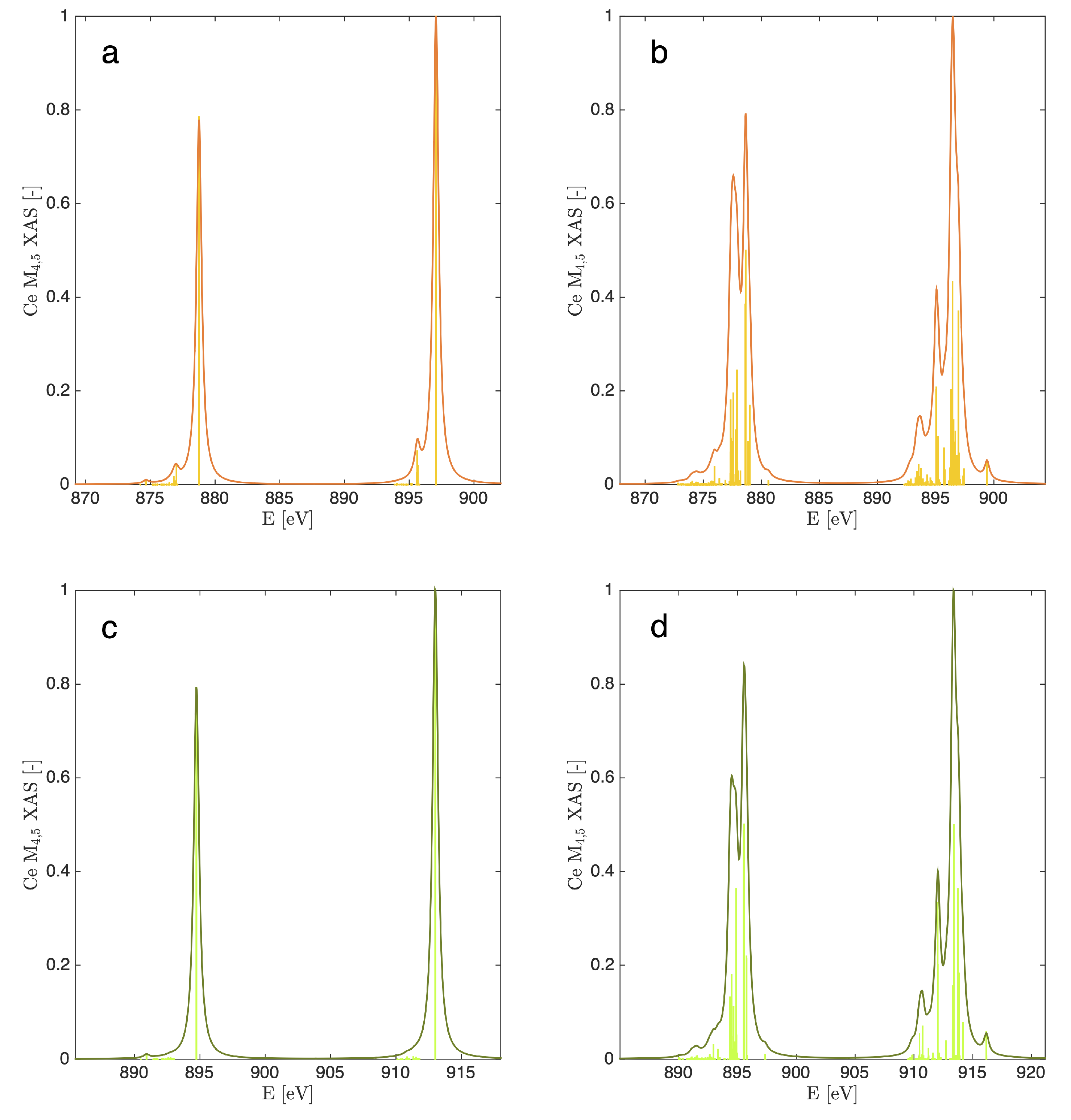{kind=link}
| Levels | LFDFT | Exp. | Percent Error | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| State | I | (1) | (2) | (3) | (4) | (1) | (2) | (3) | (4) | |
| F | A | 0 | 0 | 0 | 0 | 0 | - | - | - | - |
| F | A | 214 | 261 | 273 | 349 | 295 | −27.46 | −11.53 | −7.46 | 18.31 |
| B | 393 | 396 | 392 | 368 | 367 | 7.08 | 7.90 | 6.81 | 0.27 | |
| B | 700 | 523 | 498 | 389 | 444 | 57.66 | 17.79 | 12.16 | −12.39 | |
| F | B | 946 | 971 | 974 | 986 | 947 | −0.11 | 2.53 | 2.85 | 4.12 |
| B | 1020 | 975 | 980 | 1022 | 981 | 3.98 | −0.61 | -0.10 | 4.18 | |
| A | 1047 | 1117 | 1108 | 1023 | 1016 | 3.05 | 9.94 | 9.06 | 0.69 | |
| A | 1288 | 1134 | 1109 | 1027 | 1080 | 19.26 | 5.00 | 2.69 | −4.91 | |
| A | 1323 | 1135 | 1112 | 1039 | 1111 | 19.08 | 2.16 | 0.09 | −6.48 | |
| F | B | 1882 | 1884 | 1874 | 1839 | - | - | - | - | |
| A | 1909 | 1894 | 1882 | 1852 | - | - | - | - | ||
| B | 1952 | 1919 | 1905 | 1859 | 1808 | 7.96 | 6.14 | 5.37 | 2.82 | |
| A | 2011 | 1926 | 1910 | 1865 | 1846 | 8.94 | 4.33 | 3.47 | 1.03 | |
| B | 2027 | 1932 | 1913 | 1870 | 1857 | 9.15 | 4.04 | 3.02 | 0.70 | |
| B | 2032 | 1932 | 1921 | 1873 | 1893 | 7.34 | 2.06 | 1.48 | −1.06 | |
| A | 2137 | 1985 | 1962 | 1874 | - | - | - | - | ||
| F | B | 2244 | 2742 | 2764 | 2771 | 2587 | −13.26 | 5.99 | 6.84 | 7.11 |
| A | 2473 | 2812 | 2818 | 2780 | 2603 | −4.99 | 8.03 | 8.26 | 6.80 | |
| A | 2698 | 2834 | 2834 | 2799 | 2633 | 2.47 | 7.63 | 7.63 | 6.30 | |
| B | 2790 | 2890 | 2876 | 2801 | 2648 | 5.36 | 9.14 | 8.61 | 5.78 | |
| A | 2866 | 2897 | 2885 | 2812 | 2735 | 4.79 | 5.92 | 5.48 | 2.82 | |
| A | 2945 | 2913 | 2888 | 2838 | 2872 | 2.54 | 1.43 | 0.56 | −1.18 | |
| A | 3072 | 2915 | 2898 | 2843 | 2946 | 4.28 | −1.05 | −1.63 | −3.50 | |
| B | 3179 | 2983 | 2945 | 2850 | 2967 | 7.15 | 0.54 | −0.74 | −3.94 | |
| B | 3245 | 2987 | 2950 | 2886 | 3086 | 5.15 | −3.21 | −4.41 | −6.48 | |
| D | A | 16,081 | 16,517 | 16,535 | 16,874 | 17,241 | −6.73 | −4.20 | −4.09 | −2.13 |
| D | A | 17,705 | 18,128 | 18,143 | 18,485 | 18,945 | −6.55 | −4.31 | −4.23 | −2.43 |
| B | 17,716 | 18,164 | 18,176 | 18,488 | - | - | - | - | ||
| B | 17,806 | 18,199 | 18,206 | 18,493 | - | - | - | - | ||
| LFDFT | Reference | ||||||
|---|---|---|---|---|---|---|---|
| (1) | (2) | (3) | (4) | (a) | (b) | (c) | |
| F(4f,4f) | 11.4244 | 11.7216 | 11.7334 | 11.9464 | 8.7164 | 10.2648 | 10.7841 |
| F(4f,4f) | 7.1200 | 7.3052 | 7.3126 | 7.4444 | 7.3652 | 7.4879 | |
| F(4f,4f) | 5.1085 | 5.2413 | 5.2466 | 5.3410 | 5.2875 | 5.6741 | |
| (4f) | 0.1604 | 0.1610 | 0.1607 | 0.1600 | 0.1652 | 0.1731 | |
| B(4f,4f) | −0.0519 | −0.0245 | −0.0188 | 0.0172 | −0.0196 | ||
| B(4f,4f) | −0.2515 | −0.1332 | −0.1147 | −0.0217 | −0.0471 | ||
| LFDFT | Reference | |||||
|---|---|---|---|---|---|---|
| [Ce(COT)] | [Ce(COT)] | |||||
| (1) | (2) | (1) | (2) | (a) | (b) | |
| F(4f,4f) | - | - | 9.0259 | 8.5968 | - | 10.01 |
| F(4f,4f) | - | - | 5.6126 | 5.3400 | - | 6.35 |
| F(4f,4f) | - | - | 4.0234 | 3.8265 | - | 4.57 |
| G(3d,4f) | 3.4783 | 3.2930 | 3.7890 | 3.6082 | 3.78 | 4.06 |
| G(3d,4f) | 2.0595 | 1.9495 | 2.2432 | 2.1359 | 2.21 | 2.37 |
| G(3d,4f) | 1.4287 | 1.3524 | 1.5560 | 1.4816 | 1.52 | 1.64 |
| F(3d,4f) | 5.4411 | 5.1962 | 5.9528 | 5.7140 | 5.65 | 5.99 |
| F(3d,4f) | 2.4219 | 2.3010 | 2.6421 | 2.5243 | 2.53 | 2.71 |
| (3d) | 7.5344 | 7.5331 | 7.5357 | 7.5343 | 6.80 | 7.45 |
| (4f) | 0.0781 | 0.0742 | 0.0852 | 0.0814 | 0.086 | 0.106 |
| B(4f,4f) | −0.0523 | −0.0460 | −0.0350 | −0.0058 | ||
| B(4f,4f) | −3.0090 | 0.9311 | −1.5888 | −0.3282 | ||
| B(4f,4f) | 0.3738 | −0.9137 | 0.1651 | −0.2111 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramanantoanina, H. LFDFT—A Practical Tool for Coordination Chemistry. Computation 2022, 10, 70. https://doi.org/10.3390/computation10050070
Ramanantoanina H. LFDFT—A Practical Tool for Coordination Chemistry. Computation. 2022; 10(5):70. https://doi.org/10.3390/computation10050070
Chicago/Turabian StyleRamanantoanina, Harry. 2022. "LFDFT—A Practical Tool for Coordination Chemistry" Computation 10, no. 5: 70. https://doi.org/10.3390/computation10050070
APA StyleRamanantoanina, H. (2022). LFDFT—A Practical Tool for Coordination Chemistry. Computation, 10(5), 70. https://doi.org/10.3390/computation10050070