
Computational Assessment of Xanthones from African Medicinal Plants as Aldose Reductase Inhibitors

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
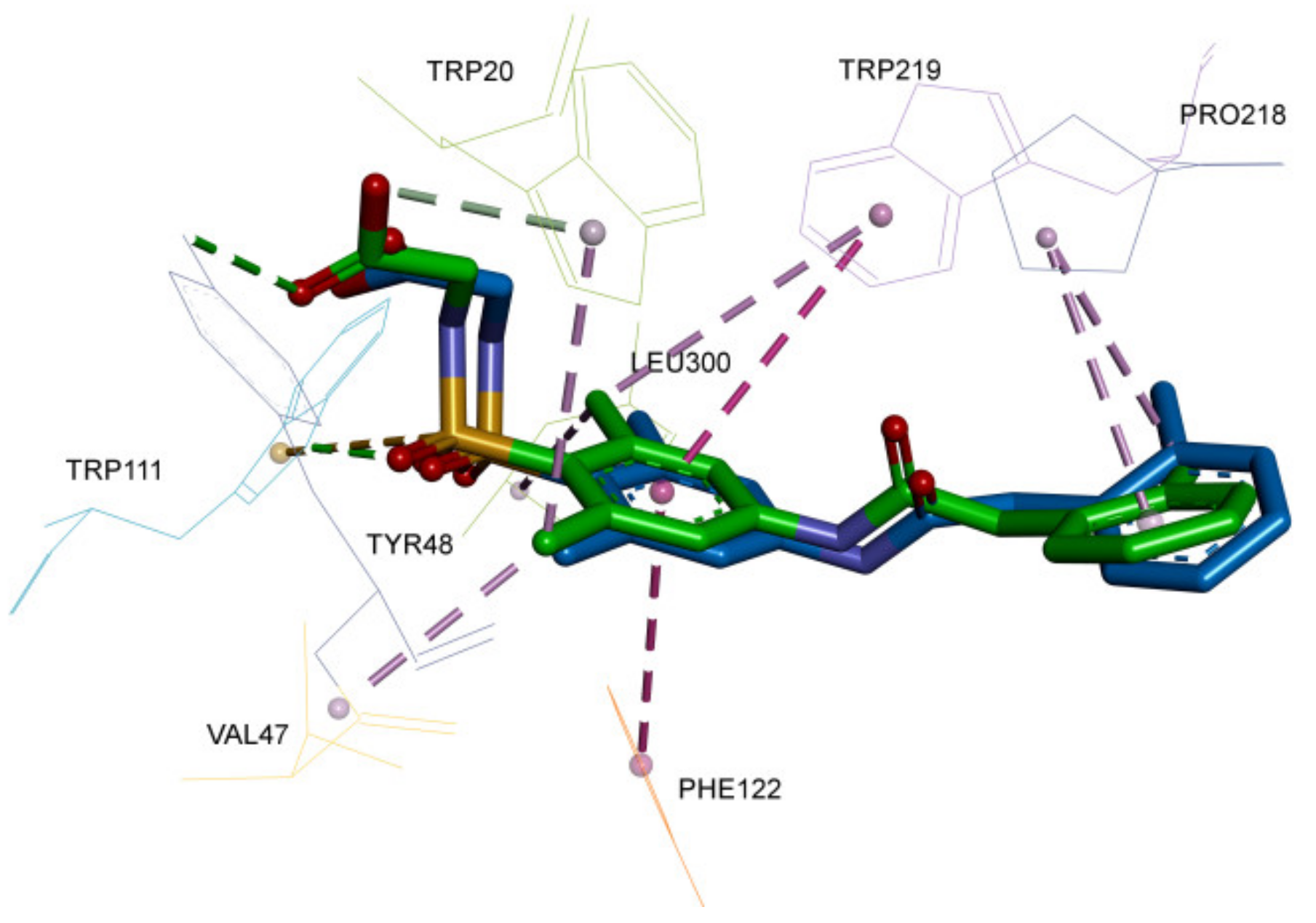
2.1. Validation of Docking Protocol
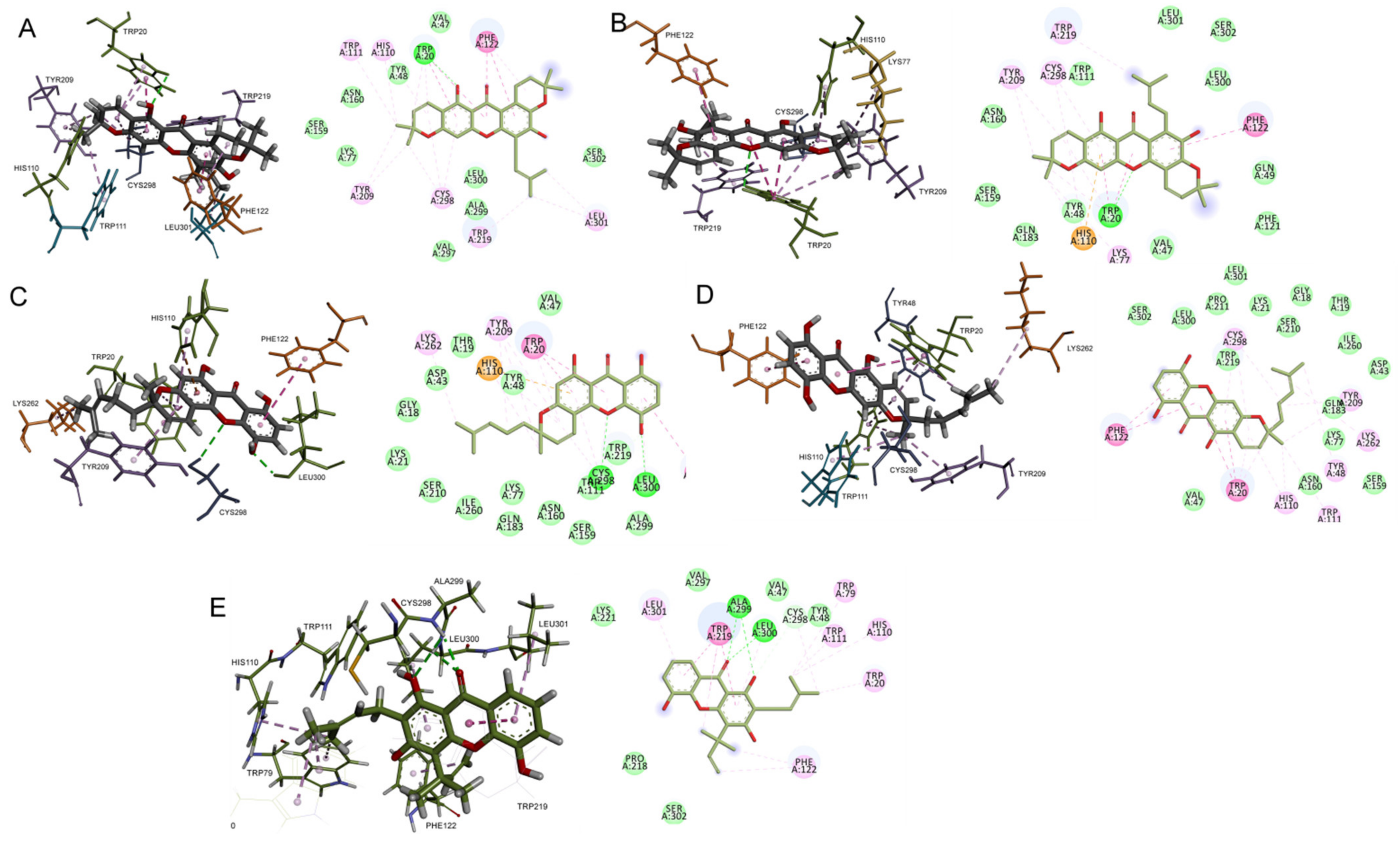
2.2. Molecular Docking Interaction of Xanthones with Aldose Reductase Enzyme
2.3. ADMET and Drug-Likeness Analysis
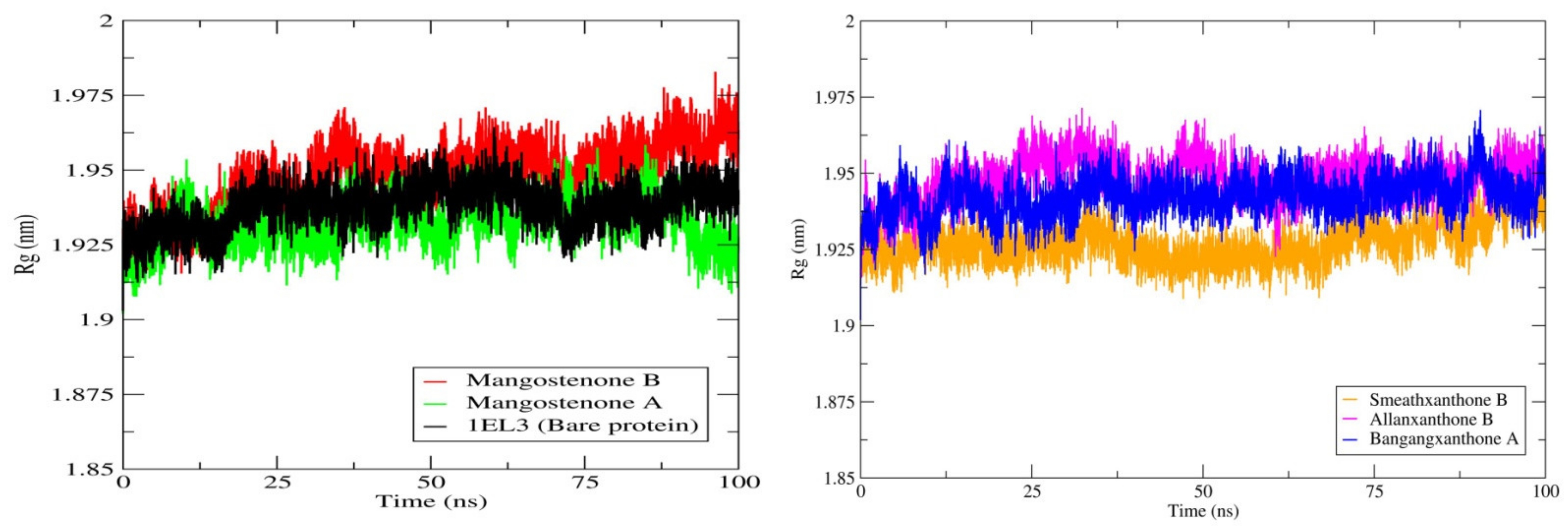
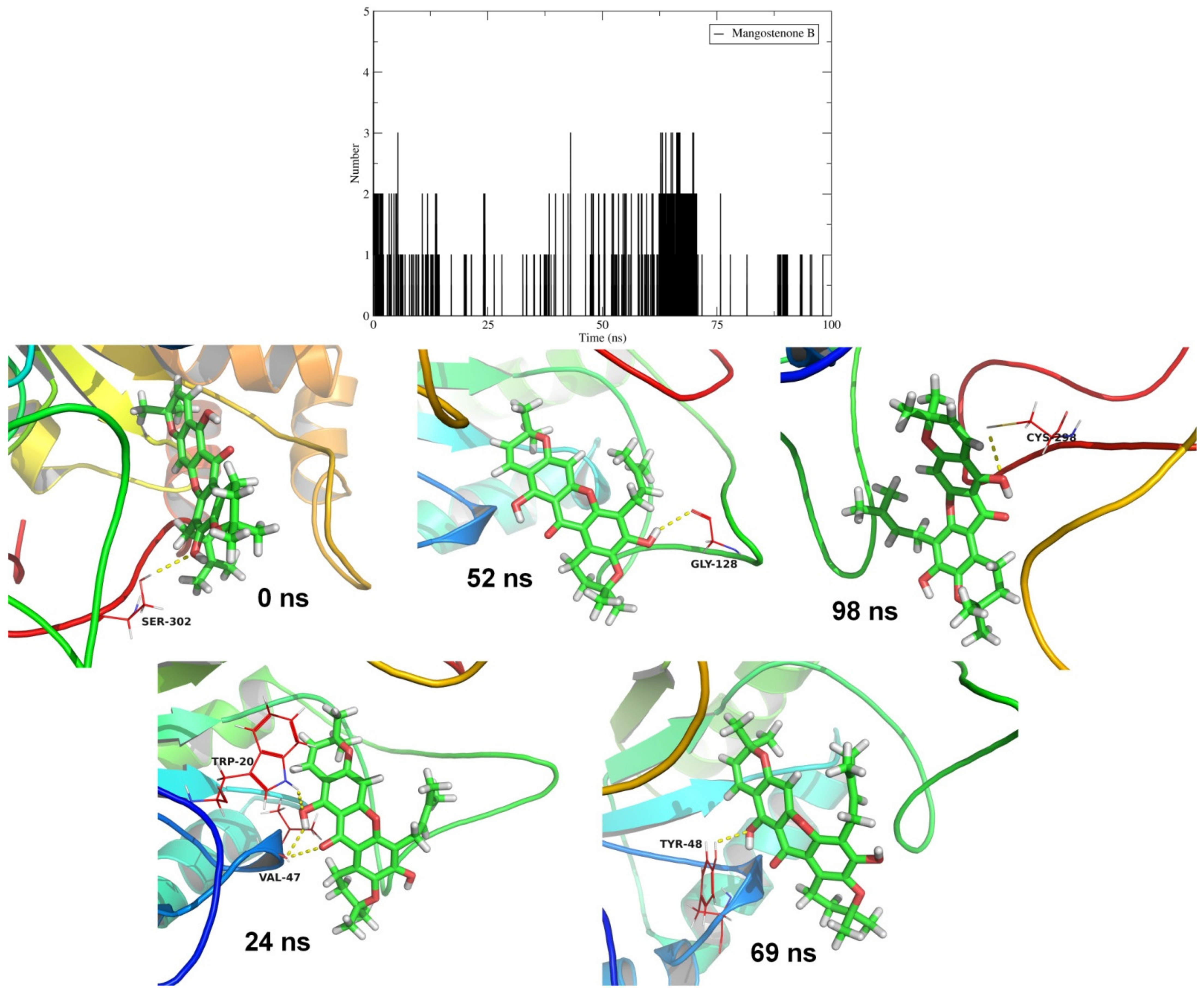
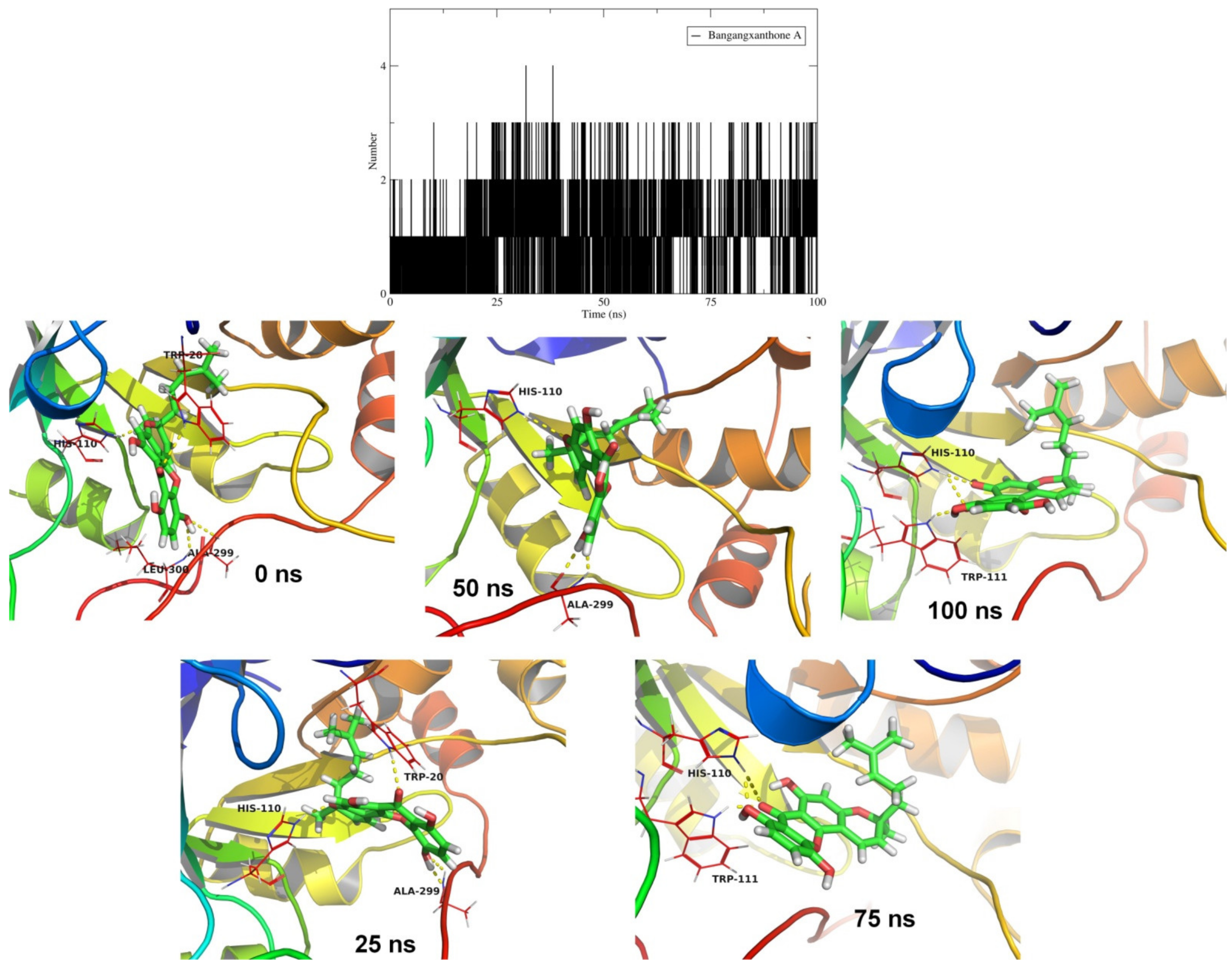
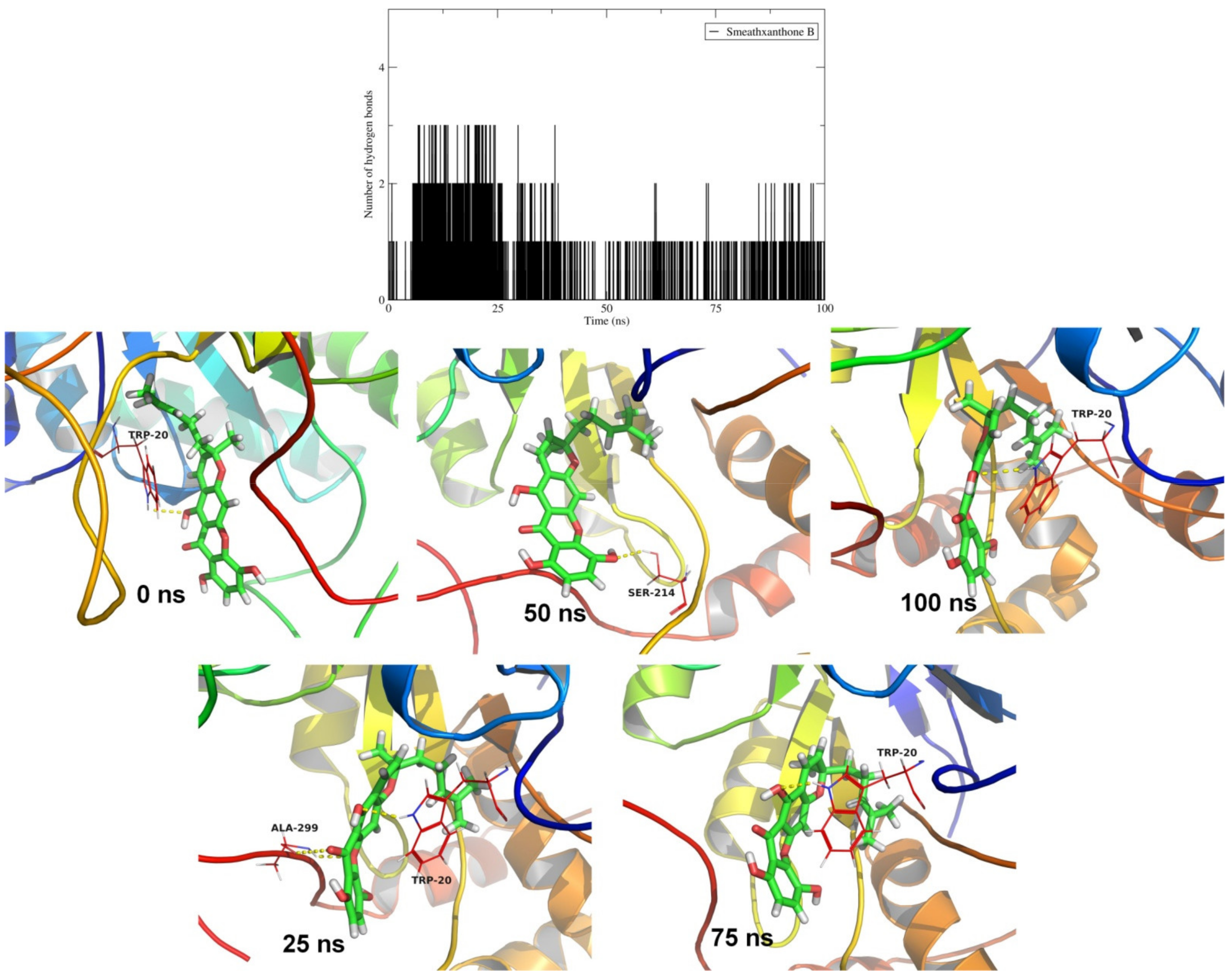
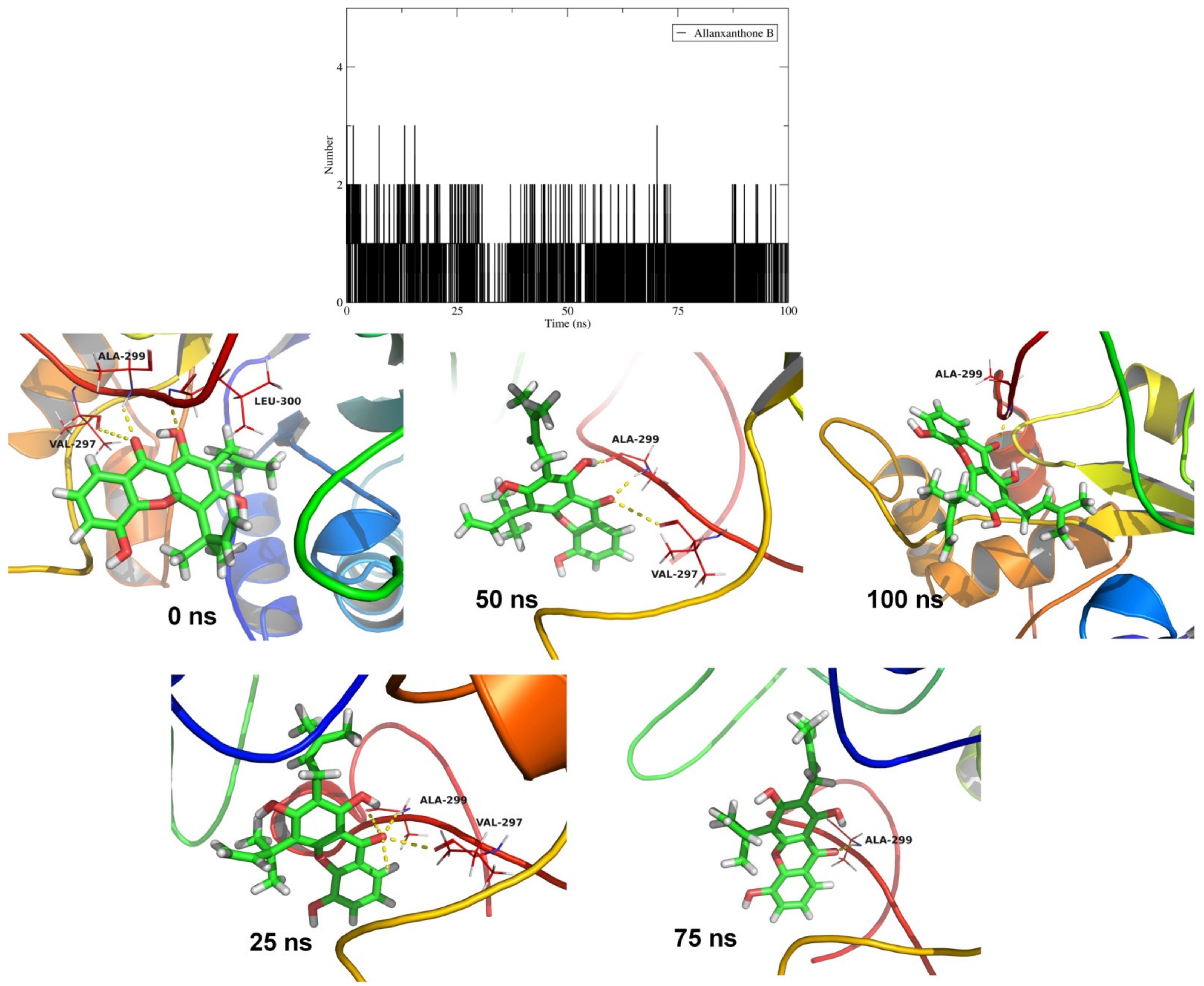
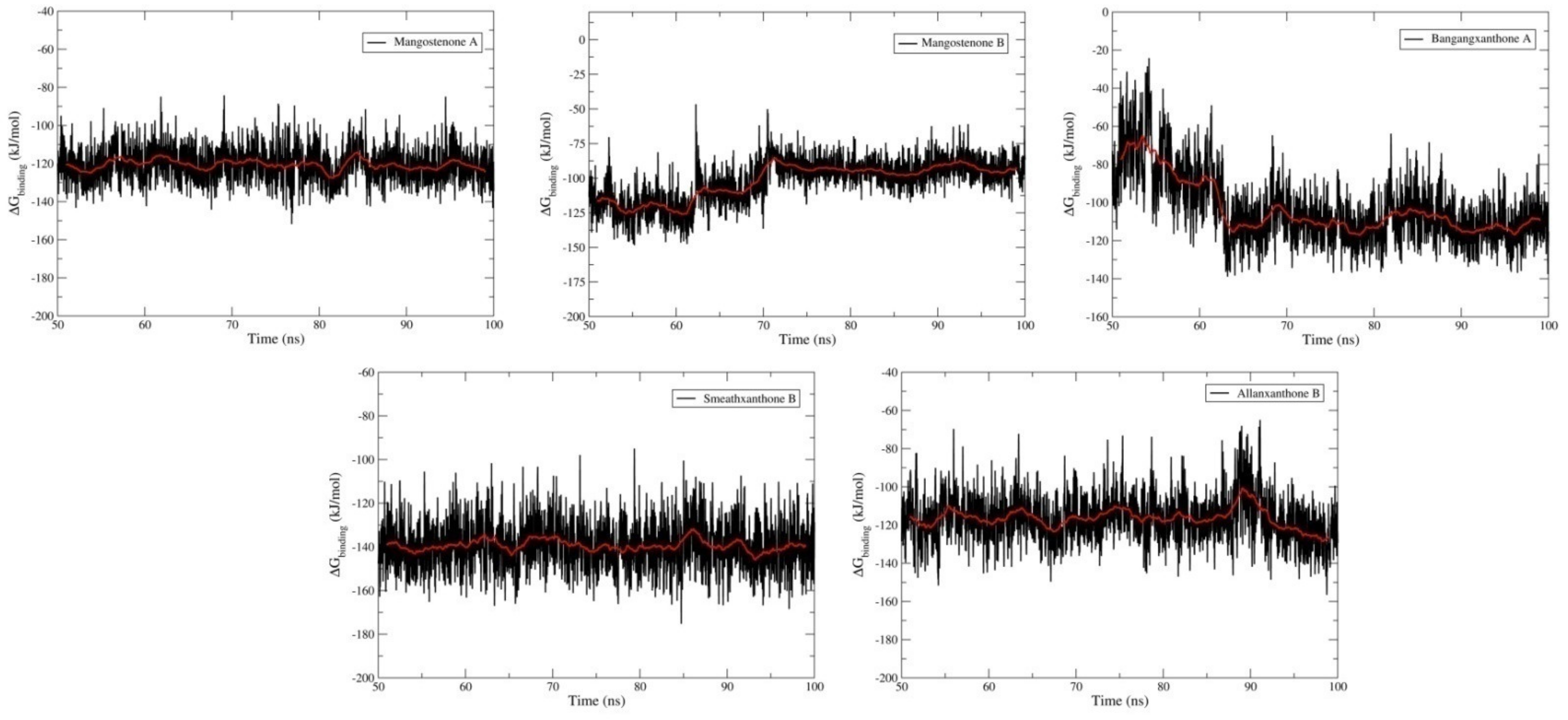
2.4. Molecular Dynamics Simulation Studies and MM-PBSA Calculations
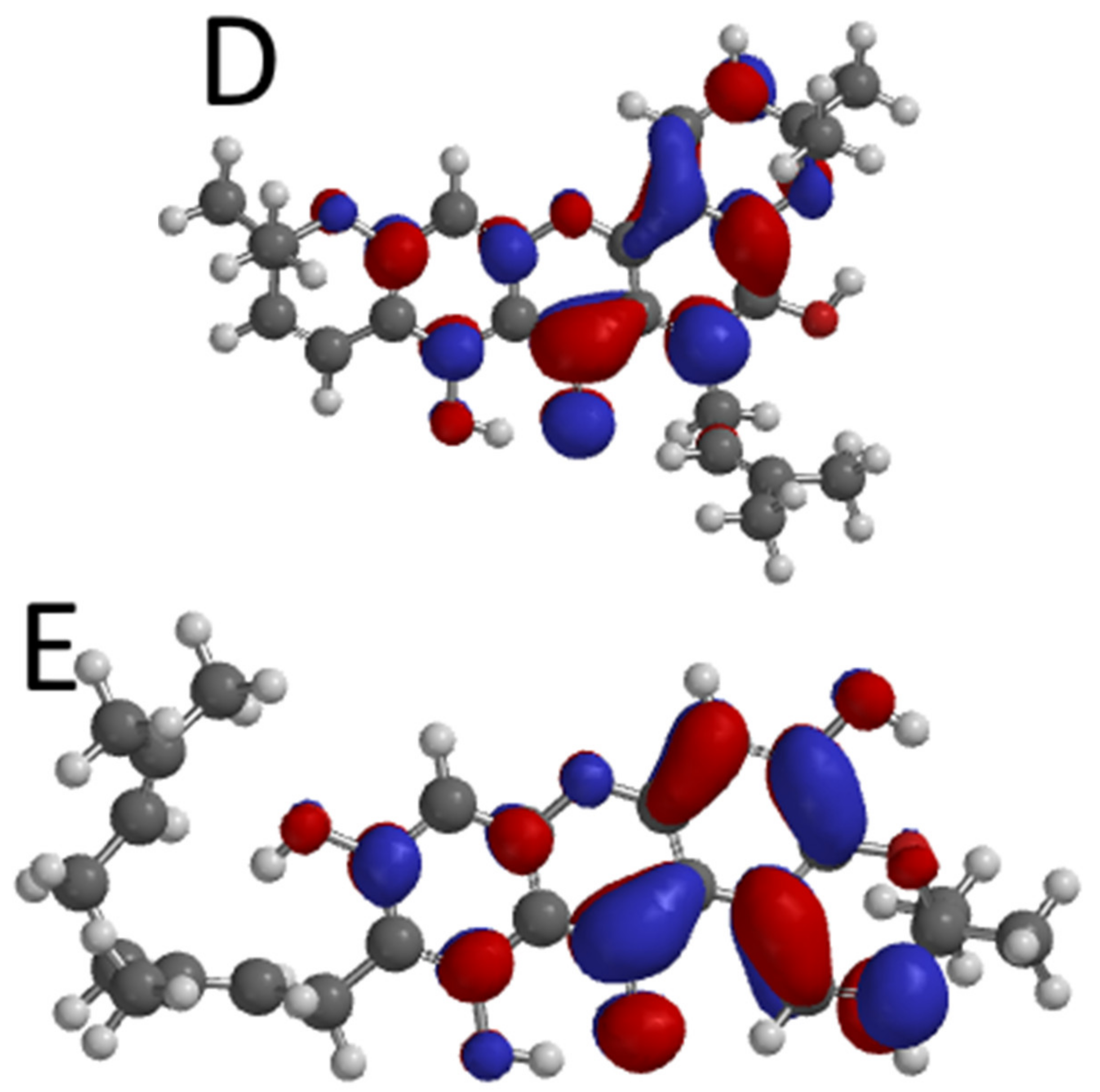
2.5. Global Reactivity Descriptor and Stability Properties of Hit Molecules
2.6. Molecular Electrostatic Potential of the Top-Ranked Xanthones
3. Materials and Methods
3.1. Receptor and Ligand Preparation
3.2. Validation of Docking Protocol and Molecular Docking Studies
3.3. ADME and Drug Likeness Predictions
3.4. Molecular Dynamics Simulation and MM-PBSA Calculations
3.5. DFT Studies of Top Ranked Aldose Reductase Inhibitors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yabe-Nishimura, C. Aldose Reductase in Glucose Toxicity: A Potential Target for the Prevention of Diabetic Complications. Pharmacol. Rev. 1998, 50, 21–34. [Google Scholar] [PubMed]
- Dréanic, M.-P.; Edge, C.M.; Tuttle, T. New Insights into the Catalytic Mechanism of Aldose Reductase: A QM/MM Study. ACS Omega 2017, 2, 5737–5747. [Google Scholar] [CrossRef] [PubMed]
- Grewal, A.S.; Sharma, N.; Singh, S.; Arora, S.J. Molecular Docking Evaluation of Some Natural Phenolic Compounds as Aldose Reductase Inhibitors for Diabetic Complications. J. Pharm. Technol. Res. Manag. 2017, 5, 135–148. [Google Scholar] [CrossRef]
- Wang, L.; Gu, Q.; Zheng, X.; Ye, J.; Liu, Z.; Li, J.; Hu, X.; Hagler, A.; Xu, J. Discovery of New Selective Human Aldose Reductase Inhibitors through Virtual Screening Multiple Binding Pocket Conformations. J. Chem. Inf. Model. 2013, 53, 2409–2422. [Google Scholar] [CrossRef]
- Syiem, D.; Majaw, S. Effect of Different Solvent Extracts of Potentilla Fulgens L. on Aldose Reductase and Sorbitol Dehydrogenase in Normoglycemic and Diabetic Mice. Pharmacologyonline 2011, 3, 63–72. [Google Scholar]
- Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and Mechanisms of Myocardial Lipotoxicity in Obesity. J. Intern. Med. 2018, 284, 478–491. [Google Scholar] [CrossRef]
- Kim, T.H.; Kim, J.K.; Kang, Y.-H.; Lee, J.-Y.; Kang, I.J.; Lim, S.S. Aldose Reductase Inhibitory Activity of Compounds from Zea Mays L. BioMed Res. Int. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Thakur, S.; Gupta, S.K.; Ali, V.; Singh, P.; Verma, M. Aldose Reductase: A Cause and a Potential Target for the Treatment of Diabetic Complications. Arch. Pharm. Res. 2021, 44, 655–667. [Google Scholar] [CrossRef]
- Quattrini, L.; La Motta, C. Aldose Reductase Inhibitors: 2013-Present. Expert Opin. Ther. Pat. 2019, 29, 199–213. [Google Scholar] [CrossRef]
- Miyamoto, S. Recent Advances in Aldose Reductase Inhibitors: Potential Agents for the Treatment of Diabetic Complications. Expert Opin. Ther. Pat. 2002, 12, 621–631. [Google Scholar] [CrossRef]
- Klebe, G.; Kramer, O.; Sotriffer, C. Strategies for the Design of Inhibitors of Aldose Reductase, an Enzyme Showing Pronounced Induced-Fit Adaptations. Cell. Mol. Life Sci. (CMLS) 2004, 61, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Sotriffer, C.A.; Krämer, O.; Klebe, G. Probing Flexibility and “Induced-Fit” Phenomena in Aldose Reductase by Comparative Crystal Structure Analysis and Molecular Dynamics Simulations. Proteins 2004, 56, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.V.; Bhadoriya, K.S.; Bari, S.B. QSAR and Flexible Docking Studies of Some Aldose Reductase Inhibitors Obtained from Natural Origin. Med. Chem. Res. 2012, 21, 1665–1676. [Google Scholar] [CrossRef]
- Pathania, S.; Randhawa, V.; Bagler, G. Prospecting for Novel Plant-Derived Molecules of Rauvolfia Serpentina as Inhibitors of Aldose Reductase, a Potent Drug Target for Diabetes and Its Complications. PLoS ONE 2013, 8, e61327. [Google Scholar] [CrossRef]
- Naveen, J.; Baskaran, V. Antidiabetic Plant-Derived Nutraceuticals: A Critical Review. Eur. J. Nutr. 2018, 57, 1275–1299. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Z.; Zuo, G.; Lim, S.S.; Yan, H. Defatted Seeds of Oenothera Biennis as a Potential Functional Food Ingredient for Diabetes. Foods 2021, 10, 538. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Yan, H. In Situ Net Fishing of α-Glucosidase Inhibitors from Evening Primrose (Oenothera Biennis) Defatted Seeds by Combination of LC-MS/MS, Molecular Networking, Affinity-Based Ultrafiltration, and Molecular Docking. Food Funct. 2022, 13, 2545–2558. [Google Scholar] [CrossRef]
- Mohammadhosseini, M.; Sarker, S.D.; Akbarzadeh, A. Chemical Composition of the Essential Oils and Extracts of Achillea Species and Their Biological Activities: A Review. J. Ethnopharmacol. 2017, 199, 257–315. [Google Scholar] [CrossRef]
- De la Fuente, J.Á.; Manzanaro, S. Aldose Reductase Inhibitors from Natural Sources. Nat. Prod. Rep. 2003, 20, 243–251. [Google Scholar] [CrossRef]
- Dineshkumar, B.; Mitra, A.; Manjunatha, M. Studies on the Anti-Diabetic and Hypolipidemic Potentials of Mangiferin (Xanthone Glucoside) in Streptozotocin-Induced Type 1 and Type 2 Diabetic Model Rats. Int. J. Adv. Pharm. Sci. 2010, 1, 75–85. [Google Scholar]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New Inhibitor of the TAp73 Interaction with MDM2 and Mutant P53 with Promising Antitumor Activity against Neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Lemos, A.; Gomes, A.S.; Loureiro, J.B.; Brandão, P.; Palmeira, A.; Pinto, M.M.M.; Saraiva, L.; Sousa, M.E. Synthesis, Biological Evaluation, and In Silico Studies of Novel Aminated Xanthones as Potential P53-Activating Agents. Molecules 2019, 24, 1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-P.; Zhao, W.; Xia, Z.-Y.; Kong, G.-H.; Lu, X.-P.; Hu, Q.-F.; Gao, X.-M. Three Novel Xanthones from Garcinia Paucinervis and Their Anti-TMV Activity. Molecules 2013, 18, 9663–9669. [Google Scholar] [CrossRef]
- Luo, C.-T.; Mao, S.-S.; Liu, F.-L.; Yang, M.; Chen, H.; Kurihara, H.; Li, Y. Antioxidant Xanthones from Swertia Mussotii, a High Altitude Plant. Fitoterapia 2013, 91, 140–147. [Google Scholar] [CrossRef]
- Leiro, J.; Arranz, J.A.; Yáñez, M.; Ubeira, F.M.; Sanmartín, M.L.; Orallo, F. Expression Profiles of Genes Involved in the Mouse Nuclear Factor-Kappa B Signal Transduction Pathway Are Modulated by Mangiferin. Int. Immunopharmacol. 2004, 4, 763–778. [Google Scholar] [CrossRef]
- Braga, E.J.; Corpe, B.T.; Marinho, M.M.; Marinho, E.S. Molecular Electrostatic Potential Surface, HOMO–LUMO, and Computational Analysis of Synthetic Drug Rilpivirine. Int. J. Sci. Eng. Res. 2016, 7, 315–319. [Google Scholar]
- Ripphausen, P.; Nisius, B.; Peltason, L.; Bajorath, J. Quo Vadis, Virtual Screening? A Comprehensive Survey of Prospective Applications. J. Med. Chem. 2010, 53, 8461–8467. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Oyedele, A.-Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular Modeling in Drug Discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Hong, S.; Liow, C.H.; Yuk, J.M.; Byon, H.R.; Yang, Y.; Cho, E.; Yeom, J.; Park, G.; Kang, H.; Kim, S.; et al. Reducing Time to Discovery: Materials and Molecular Modeling, Imaging, Informatics, and Integration. ACS Nano 2021, 15, 3971–3995. [Google Scholar] [CrossRef]
- Faloye, K.O.; Bekono, B.D.; Fakola, E.G.; Ayoola, M.D.; Bello, O.I.; Olajubutu, O.G.; Owoseeni, O.D.; Mahmud, S.; Alqarni, M.; Al Awadh, A.A.; et al. Elucidating the Glucokinase Activating Potentials of Naturally Occurring Prenylated Flavonoids: An Explicit Computational Approach. Molecules 2021, 26, 7211. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Motta, S.; Bonati, L. Modeling Binding with Large Conformational Changes: Key Points in Ensemble-Docking Approaches. J. Chem. Inf. Model. 2017, 57, 1563–1578. [Google Scholar] [CrossRef]
- Haroon, H.B.; Perumalsamy, V.; Nair, G.; Anand, D.K.; Kolli, R.; Monichen, J.; Prabha, K. Repression of Polyol Pathway Activity by Hemidesmus Indicus Var. Pubescens R.Br. Linn Root Extract, an Aldose Reductase Inhibitor: An In Silico and Ex Vivo Study. Nat. Prod. Bioprospect. 2021, 11, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Madeswaran, A.; Umamaheswari, M.; Asokkumar, K.; Sivashanmugam, T.; Subhadradevi, V.; Jagannath, P. In Silico Docking Studies of Aldose Reductase Inhibitory Activity of Commercially Available Flavonoids. Bangladesh J. Pharmacol. 2012, 7, 266–271. [Google Scholar] [CrossRef]
- Vellai, R.D.; Pillai, S.S. Aldose Reductase Inhibitiory Effect of Gymnemic Acid, Trigonelline and Ferulic Acid-An In Silico Approach. Int. J. Pharmacogn. Phytochem. Res. 2017, 9, 31–39. [Google Scholar] [CrossRef]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.-P.; Fenart, L. Modelling of the Blood–Brain Barrier in Drug Discovery and Development. Nat. Rev. Drug Discov. 2007, 6, 650–661. [Google Scholar] [CrossRef]
- James, P.; Davis, S.P.; Ravisankar, V.; Nazeem, P.A.; Mathew, D. Novel Antidiabetic Molecules from the Medicinal Plants of Western Ghats of India, Identified Through Wide-Spectrum in Silico Analyses. J. Herbs Spices Med. Plants 2017, 23, 249–262. [Google Scholar] [CrossRef]
- Sharma, P.; Joshi, T.; Joshi, T.; Chandra, S.; Tamta, S. In Silico Screening of Potential Antidiabetic Phytochemicals from Phyllanthus Emblica against Therapeutic Targets of Type 2 Diabetes. J. Ethnopharmacol. 2020, 248, 112268. [Google Scholar] [CrossRef]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An Aldose Reductase Inhibitor for the Treatment of Diabetic Neuropathy. Pharmacotherapy 2008, 28, 646–655. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein Structure-Based Drug Design: From Docking to Molecular Dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra Precision Docking, Free Energy Calculation and Molecular Dynamics Simulation Studies of CDK2 Inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Monajjemi, M.; Oliaey, A. Gyration Radius and Energy Study at Different Temperatures for Acetylcholine Receptor Protein in Gas Phase by Monte Carlo, Molecular and Langevin Dynamics Simulations. J. Phys. Theor. Chem. 2009, 5, 19–26. [Google Scholar]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Stein, S.A.M.; Loccisano, A.E.; Firestine, S.M.; Evanseck, J.D. Principal Components Analysis: A Review of Its Application on Molecular Dynamics Data. In Annual Reports in Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2006; Volume 2, pp. 233–261. ISBN 978-0-444-52822-3. [Google Scholar]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Subramanian, N.; Sundaraganesan, N.; Jayabharathi, J. Molecular Structure, Spectroscopic (FT-IR, FT-Raman, NMR, UV) Studies and First-Order Molecular Hyperpolarizabilities of 1,2-Bis(3-Methoxy-4-Hydroxybenzylidene)Hydrazine by Density Functional Method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 76, 259–269. [Google Scholar] [CrossRef]
- Domingo, L.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef]
- Stefaniu, A.; Pintilie, L. Molecular Descriptors and Properties of Organic Molecules; InTech: Rijeka, Croatia, 2018. [Google Scholar]
- Srivastava, K.; Shimpi, M.R.; Srivastava, A.; Tandon, P.; Sinha, K.; Velaga, S.P. Vibrational Analysis and Chemical Activity of Paracetamol–Oxalic Acid Cocrystal Based on Monomer and Dimer Calculations: DFT and AIM Approach. RSC Adv. 2016, 6, 10024–10037. [Google Scholar] [CrossRef]
- Ayeni, A.O.; Akinyele, O.F.; Hosten, E.C.; Fakola, E.G.; Olalere, J.T.; Egharevba, G.O.; Watkins, G.M. Synthesis, Crystal Structure, Experimental and Theoretical Studies of Corrosion Inhibition of 2-((4-(2-Hydroxy-4-Methylbenzyl)Piperazin-1-Yl)Methyl)-5-Methylphenol – A Mannich Base. J. Mol. Struct. 2020, 1219, 128539. [Google Scholar] [CrossRef]
- Kawsar, S.; Bulbul, M.; Hosen, M.; Ferdous, J.; Chowdhury, T.; Misbah, M. DFT Study, Physicochemical, Molecular Docking, and ADMET Predictions of Some Modified Uridine Derivatives. Int. J. New Chem. 2020, 8, 88–110. [Google Scholar] [CrossRef]
- Alhosseini, A.H.; Shahab, S.; Lopatik, D.; Kuvaeva, Z.; Karankevich, H.; Kaviani, S.; Sheikhi, M. Synthesis, DFT Study and Bioactivity Evaluation of New Butanoic Acid Derivatives as Antiviral Agents. Biointerface Res. Appl. Chem. 2022, 12, 3522–3539. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology; Hempel, J.E., Williams, C.H., Hong, C.C., Eds.; Springer: New York, NY, USA, 2015; Volume 1263, pp. 243–250. ISBN 978-1-4939-2268-0. [Google Scholar]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today: Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain Χ1 and Χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Viet, M.H.; Li, M.S. Effects of Water Models on Binding Affinity: Evidence from All-Atom Simulation of Binding of Tamiflu to A/H5N1 Neuraminidase. Sci. World J. 2014, 2014, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Pan, D.; Li, J.; Zhang, L.; Shao, X. Application of Berendsen Barostat in Dissipative Particle Dynamics for Nonequilibrium Dynamic Simulation. J. Chem. Phys. 2017, 146, 124108. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and Efficiency of the Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand (PubChem ID) | Chemical Structure | Binding Energy (kcal/mol) | Hydrogen Bond Interaction | Hydrophobic Interaction | Pi-Interaction | Electrostatic Interaction - | |
|---|---|---|---|---|---|---|---|
| Amino Acid Residue | Distance (Å) | ||||||
| Mangostenone B (CID 21672078) | 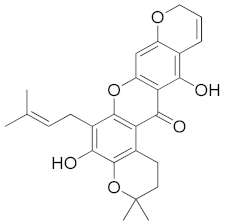 | −11.2 | Trp20 | 2.75 | Trp20, His110, Trp111, Phe122, Tyr209, Trp219, Cys298, Leu301 | Trp20, His110, Trp111, Phe122, Tyr209, Trp219, | - |
| Bangangxanthone A (CID 11524043) | 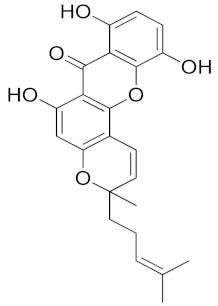 | −10.8 | Cys298 Leu300 | 3.07 2.16 | Trp20, His110, Phe122, Tyr209, Lys262, Cys298, Leu300 | Trp20, His110, Phe122, Tyr209, Leu300 | His110 |
| Smeathxanthone B (CID 11625362) | 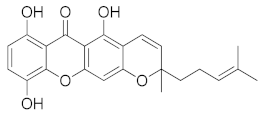 | −10.8 | - | - | Trp20, Tyr48, His110, Trp111, Phe122, Tyr209, Lys262, Cys298 | Trp20, Tyr48, His110, Trp111, Phe122, Tyr209 | - |
| Mangostenone A (CID 509267) | 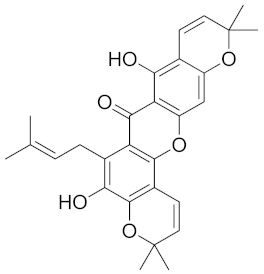 | −10.7 | Trp20 | 2.86 | Trp20, Lys77 His110, Phe122, Tyr209, Trp219, Cys298 | Trp20, His110, Phe122, Tyr209, Trp219 | His110 |
| Allanxanthone B (CID 11328706) |  | −10.6 | Ala299 Leu300 | 2.10 3.02 2.52 | Trp20, Trp79, His110, Trp111, Phe122, Trp219, Cys298, Leu300, Leu301 | Trp20, Trp79, His110, Trp111, Phe122, Trp219, Leu300, Leu301 | |
| Ligand | Lipinski Rule | HIA | BBB | Solubility | Carcinogenicity | Acute Oral Toxicity | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Log P | TPSA (Å) | HBA | HDD | MW (Da) | Rotatable Bond | ||||||
| Mangostenone B | 5.38 | 89.13 | 6 | 2 | 462.53 | 2 | + | + | −4.025 | - | 2.332 |
| Bangangxanthone A | 4.10 | 100.13 | 6 | 3 | 394.42 | 3 | + | + | −4.364 | - | 2.099 |
| Smeathxanthone B | 4.03 | 100.13 | 6 | 3 | 394.42 | 3 | + | + | −4.364 | - | 2.468 |
| Mangostenone A | 5.28 | 89.13 | 6 | 2 | 460.52 | 2 | + | + | −3.759 | - | 2.791 |
| Allanxanthone B | 4.68 | 100.13 | 6 | 3 | 462.53 | 5 | + | + | −4.270 | - | 2.765 |
| Epalrestat | 2.46 | 115.00 | 3 | 1 | 319.40 | 4 | + | - | −3.512 | - | 3.025 |
| Compounds | van der Waal Energy (kJ.mol−1) | Electrostatic Energy (kJ.mol−1) | Polar Solvation Energy (kJ.mol−1) | SASA Energy (kJ.mol−1) | Binding Energy (kJ.mol−1) |
|---|---|---|---|---|---|
| Mangostenone A | −148.646 (0.172) | −4.996 (0.047) | 48.007 (0.131) | −15.039 (0.018) | −120.672 (0.163) |
| Mangostenone B | −126.43 (0.311) | −3.401 (0.048) | 44.466 (0.174) | −16.651 (0.029) | −102.039 (0.307) |
| Bangangxanthone A | −166.372 (0.276) | −8.469 (0.119) | 90.841 (0.300) | −19.222 (0.022) | −103.238 (0.347) |
| Smeathxanthone B | −218.585 (0.216) | −2.271 (0.064) | 102.529 (0.193) | −21.078 (0.017) | −139.426 (0.212) |
| Allanxanthone B | −148.569 (0.249) | −10.725 (0.080) | 59.471 (0.230) | −16.813 (0.024) | −116.627 (0.239) |
| Ligands | EHOMO (eV) | ELUMO (eV) | ΔEgap (eV) | µ (eV) | η (eV) | S (eV−1) | χ (eV) | ω (eV) |
|---|---|---|---|---|---|---|---|---|
| Mangostenone B | −5.31 | −1.38 | 3.93 | −3.35 | 1.97 | 0.51 | 3.35 | 2.86 |
| Bangangxanthone A | −5.57 | −1.93 | 3.86 | −3.75 | 1.82 | 0.55 | 3.75 | 3.86 |
| Smeathxanthone B | −5.41 | −1.91 | 3.58 | −3.70 | 1.79 | 0.56 | 3.70 | 3.82 |
| Mangostenone A | −5.34 | −1.60 | 3.74 | −3.47 | 1.87 | 0.53 | 3.47 | 3.22 |
| Allanxanthone B | −5.48 | −1.69 | 3.79 | −3.59 | 1.90 | 0.53 | 3.59 | 3.40 |
| Enzyme (PDB ID) | Native Ligand | Residues within 5 Å |
|---|---|---|
| Aldose reductase (1EL3) | [2,6-dimethyl-4-(2-O-tolyl-acetylamino)-benzenesulfonyl]-glycine | Trp20, Val47, Tyr48, Val77, Trp79, His110, Trp111, Phe122, Pro218, Trp219, Cys298, Leu300 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owoseeni, O.D.; Patil, R.B.; Phage, P.M.; Ogboye, R.M.; Ayoola, M.D.; Famuyiwa, S.O.; Gboyero, F.O.; Ndinteh, D.T.; Faloye, K.O. Computational Assessment of Xanthones from African Medicinal Plants as Aldose Reductase Inhibitors. Computation 2022, 10, 146. https://doi.org/10.3390/computation10090146
Owoseeni OD, Patil RB, Phage PM, Ogboye RM, Ayoola MD, Famuyiwa SO, Gboyero FO, Ndinteh DT, Faloye KO. Computational Assessment of Xanthones from African Medicinal Plants as Aldose Reductase Inhibitors. Computation. 2022; 10(9):146. https://doi.org/10.3390/computation10090146
Chicago/Turabian StyleOwoseeni, Onikepe Deborah, Rajesh B. Patil, Prajakta M. Phage, Ruth Mosunmola Ogboye, Marcus Durojaye Ayoola, Samson Oluwaseyi Famuyiwa, Felix Olusegun Gboyero, Derek Tantoh Ndinteh, and Kolade Olatubosun Faloye. 2022. "Computational Assessment of Xanthones from African Medicinal Plants as Aldose Reductase Inhibitors" Computation 10, no. 9: 146. https://doi.org/10.3390/computation10090146