Abstract
This study investigates the influences and environmental preferences of amide–π interactions, a relatively unexplored class of charge-free interactions, in oligomeric phycocyanins. In a data set of 20 proteins, we observed 2086 amide–π interactions, all of which were part of the protein backbone. Phe and Tyr residues were found to be involved in amide–π interactions more frequently than Trp or His. The most favorable amide–π interactions occurred within a pair distance range of 5–7 Å, with a distinct angle preference for T-shaped ring arrangements. Multiple interaction patterns suggest that approximately 76% of the total interacting residues participate in multiple amide–π interactions. Our ab initio calculations revealed that most amide–π interactions have energy from 0 to −2 kcal/mol. Stabilization centers of phycocyanins showed that all residues in amide–π interactions play a crucial role in locating one or more such centers. Around 78% of the total interacting residues in the dataset contribute to creating hot-spot regions. Notably, the amide–π interacting residues were found to be highly evolutionarily conserved. These findings enhance our understanding of the structural stability and potential for protein engineering of phycocyanins used as bioactive natural colorants in various industries, including food and pharmaceuticals.
1. Introduction
Phycobiliproteins (PBPs) are water-soluble, intensely fluorescent holoproteins of various colors, with covalently bound linear tetrapyrrole phycobilin chromophores, functioning as auxiliary components in the photosynthetic apparatus of cyanobacteria and certain algae [1]. The most abundant PBPs, differing in their protein structure, phycobilin content attached to conserved cysteine residues, absorption spectra, and fluorescent properties, are red phycoerythrins, with red phycoerythrobilin as the typical chromophore; and blue phycocyanins, most often C-phycocyanin (C-PC) and allophycocyanin (APC) with purple-blue phycocyanobilin pigment. Besides numerous health benefits, including antioxidant, anti-inflammatory, immunoregulatory, anticancer, and antidiabetic properties, PBPs have massive commercial potential as natural colorants in the nutraceutical, cosmetics, and pharmaceutical industries [2].
Phycocyanins share a typical PBP structure, which starts with assembling amino acid chain monomers, heterodimers composed of α and β subunits, and phycobilins linked via a thioether bond. Each subunit typically consists of eight α-helices separated by loops and a similar globulin-like fold. Water monomers spontaneously aggregate to form ring-shaped trimers (αβ)3, with a rotational symmetry and a central channel. Trimers aggregate in pairs to form hexamers (αβ)6, occasionally assisted with additional linker proteins [3]. The stability of phycocyanin oligomers mostly depends on their origin and amino acid composition, then pH (optimal stability between pH 5.5–6.0), temperature (up to 45 °C), light, and some exogenous substances [4]. The molecular forces responsible for the observed differences in thermal and chemical stability/instability of different phycocyanin aggregates still need to be fully understood [5]. Therefore, understanding the nature of non-covalent interactions is equally important to see what causes these property variations, limiting their even greater use in the food industry.
Amide groups are abundant in protein binding sites, and their less polar π-surface is often exposed for ligand interaction. Although amides are generally recognized for their importance in forming hydrogen bonding interactions in proteins and protein–ligand interactions [6,7,8], the amide–π interaction with the electron-rich face of an aromatic ring is less thoroughly studied [8,9,10]. The Protein Data Bank (PDB) analysis indicates that both intramolecular and intermolecular amide–π interactions are ubiquitous in proteins [11]. Previous reports have characterized amide–π interactions in a computational model [9,12], a peptide model [13], and a protein-small molecule interaction system [14]. The Diederich lab has taken the lead in increasing our understanding of these interactions in protein stability, and the knowledge gained from this structure–activity relationship study and the detection of the binding mode continues to inspire their use in rational design [15,16,17].
The present study expands on our previous work on the non-covalent interactions, cation–π and π–π interactions of phycocyanin crystal structures by analyzing the same protein group (dataset) concerning amide–π interactions to better understand their stabilizing role [18,19,20]. We have focused our study on the phycocyanin interfaces; therefore, the amide–π interactions within a protein are not considered. The results might contribute to understanding the structural stability of this class of essential evolutionary proteins with emerging practical applications and future designs of novel protein–bioactive compound interactions.
2. Materials and Methods
2.1. Dataset
For this study, we employed the PDB, accessed on 17 November 2022, listing 202,662 resolved structures [21]. The selection criteria for phycocyanin proteins to be included in the dataset were: (a) we accepted the structures of proteins containing the phycocyanin alpha or beta subunit domain (SCOP Classification, version 1.75) [22]; (b) theoretical model and NMR structures were excluded due to difficulties in defining their accuracy compared to X-ray diffraction studies. Only crystal structures with a resolution of 2.0 Å or better and a crystallographic R-factor of 25.0% or lower were accepted; (c) only representatives with at least 30% sequence identity were included in the analyses.
After assembling the dataset, several structures containing ligands and mutant amino acids were rejected (they can disrupt the native structure of the protein), leaving 20 phycocyanins in the dataset used for our bioinformatics analysis. Hydrogen atoms were added and optimized if needed using the REDUCE program [23] with its default settings. This software adds hydrogen atoms to macromolecule (protein and DNA) structures in standardized geometry, optimizing them to the orientations of OH, SH, NH3+, Met methyls, Asn and Gln side-chain amides, and His rings. The REDUCE software selects the best hydrogen positions by choosing the best overall score from all possible combinations, considering single scores assigned for each residue and groups containing movable protons partitioned in closed sets of local interacting networks.
The PDB IDs of all selected phycocyanin chain structures were as follows: 1all (APC from Arthrospira platensis), 1b33 (APC from Mastigocladus laminosus), 1cpc (C-PC from Microchaete diplosiphon), 1f99 (R-PC from Polysiphonia urceolata), 1gh0 (C-PC from Arthrospira platensis), 1jbo (C-PC from Synechococcus elongatus), 1kn1 (APC from Neopyropia yezoensis), 1phn (PC from Cyanidium caldarium), 2bv8 (PC from Nostoc sp. R76DM), 2vjt (APC from Gloeobacter violaceus), 2vml (PC from Gloeobacter violaceus), 3dbj (APC from Thermosynechococcus vulcanus), 3o18 (C-PC from Themosynechococcus vulcanus), 4f0u (APC from Synechococcus elongatus PCC 7942), 4l1e (C-PC from Leptolyngbya sp. N62DM), 4lm6 (PC612 from Hemiselmis virescens M1635), 4lms (PC577 from Hemiselmis pacifica CCMP 706), 4po5 (APC from Synechocystis PCC 6803), 4rmp (APC from Phormidium sp. A09DM), and 4yjj (PC from Phormidium rubidum).
2.2. Amide–π Interaction Analysis in Phycocyanin Interfaces
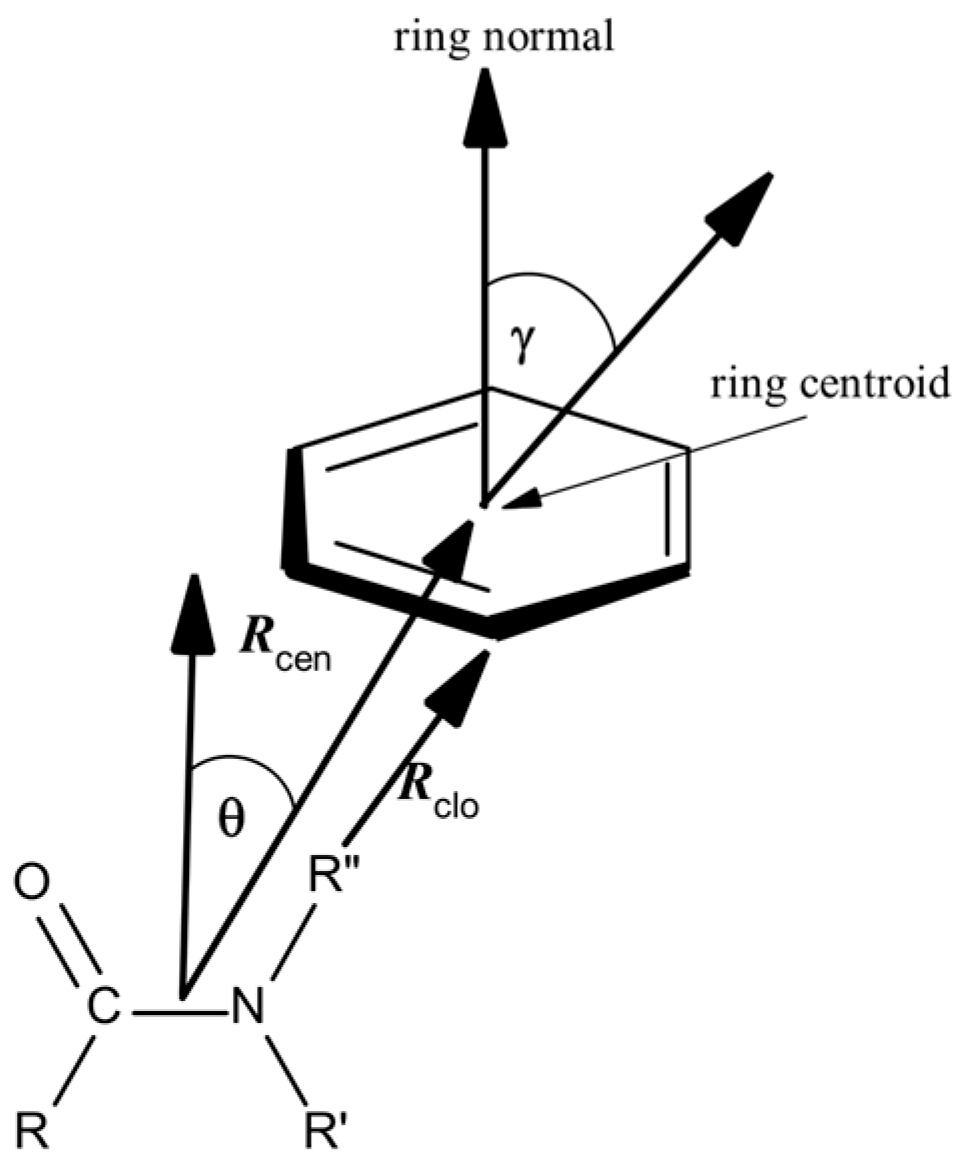
Protein structures selected containing various types of amide–π interactions were analyzed using Discovery Studio Visualizer 2021 (Dassault Systemes BIOVIA, San Diego, CA, USA) [24], with some specific criteria and geometrical feature settings. Amide–π stacked interactions happen between an amide group and a π ring if the subsequent criteria are met (Figure 1): (a) The distance between the centroid of the amide group and the π rings falls within the π–π centroid (max dist, Rcen; 7 Å); (b) An atom from each group should be within the π–π nearest atom (max dist, Rclo; 7 Å); (c) The angle θ between the normal of one or both groups and the centroid–centroid vector have to be between 0° and ± the stacked π–amide θ angle cut-off distance (90°), and the angle γ between the normal to each have to be between 0° and ± the stacked π–amide γ angle cut-off (90°). The centroid of the amide group plane was taken as the midpoint of the C and N atoms. Compared to the criteria applied in examinations of small molecules in the CSD (Cambridge Structural Database), these criteria were narrowly more relaxed. We opted for marginally looser criteria because the structural variations in the crystal structures of proteins are generally more significant than in the crystal structures of small molecules. Earlier studies proved π interactions as long-range interactions, containing notable binding forces even at intermolecular distances of 7 Å [25,26,27].
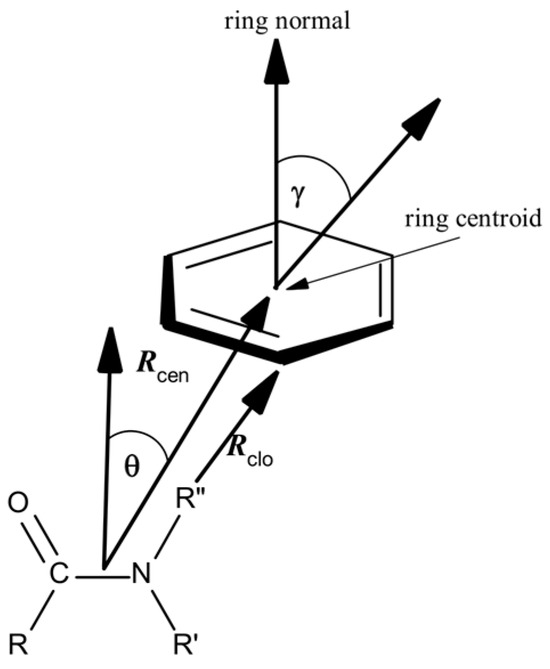
Figure 1.
Parameters for amide–π interactions: Rcen is the distance between the centroid of the amide group and the π ring; Rclo is the distance between π–π the closest atoms from each group; θ is the angle between the amide–centroid vector and a chosen vector on principle axis of the aromatic ring; and γ is the angle between the normal of each ring.
2.3. Computation of Amide–π Interaction Energy in Phycocyanin Interfaces
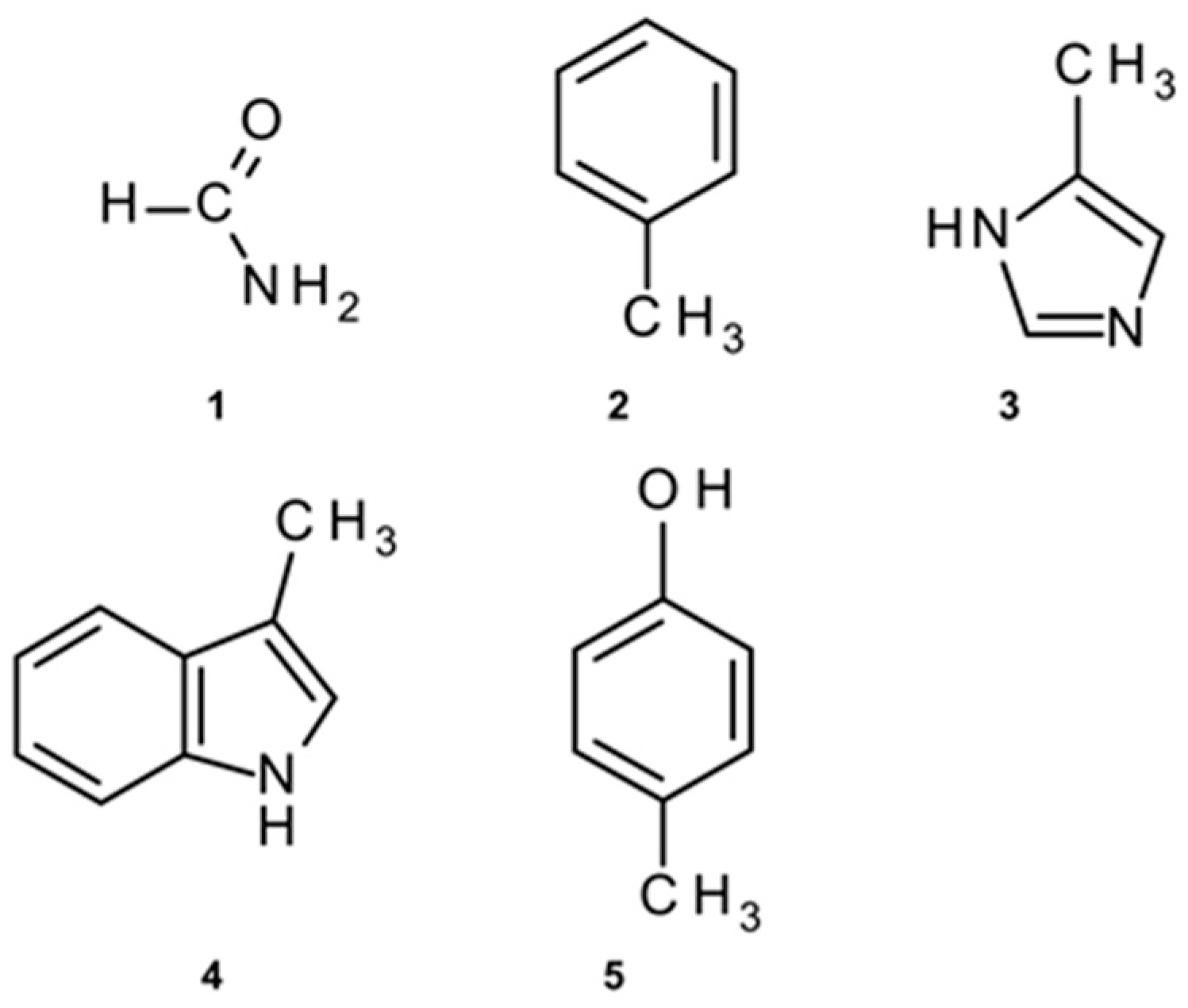
To perform ab initio methods in determining the energies of amide–π pairs on the desired level of theory, with adequate accuracy and yet in a satisfactory time frame, calculations were conducted on structurally reduced model systems [27,28]. We employed formamide (1) as mimics for amide groups. Phenylalanine was simplified to methylbenzene (2), His to 5-methyl-1H-imidazole (3), while Trp and Tyr were reduced to 3-methyl-1H-indole (4) and 4-methylphenol (5), respectively (Figure 2).
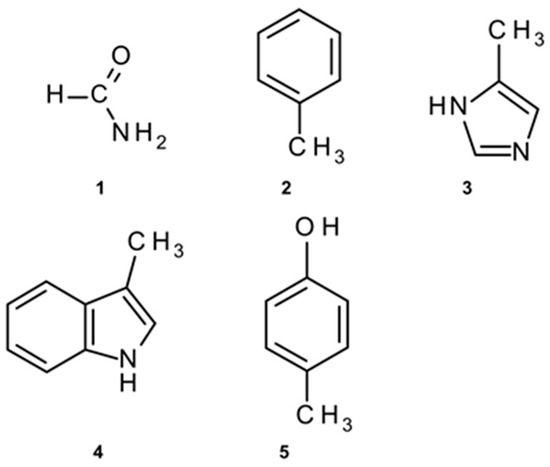
Figure 2.
Structurally simplified structures used for amide–π interaction energy calculations: (1) instead of amide; (2) instead of Phe; (3) instead of His; (4) instead of Trp; (5) instead of Tyr.
Ab initio calculations were performed in a vacuum using Jaguar from Schrödinger Suite 2018-1 (Schrödinger, Inc., New York, NY, USA) [29], utilizing the LMP2 method with triple zeta Dunning’s correlation consistent basis set [30] and aug diffuse functions [31]. The LMP2 method applied to studying amide–π interactions was considerably faster than the MP2 method. In contrast, both methods’ calculated interaction energies and equilibrium distances are almost identical [32]. Several authors found that LMP2 is an excellent method for calculating protein interaction energies [33,34].
The geometries of interacting structures were optimized using the LMP2/aug-pVTZ(-f) level of theory, and their single point energies were calculated at the LMP2/aug-pVTZ level. The optimized geometries were placed in space to match the corresponding complexes by superimposing heavy atoms onto their respective coordinates from crystal structures, and then the energies of the dimeric structures that were produced this way were calculated.
The amide–π interaction energies in dimers (amide–π pairs) were calculated as the difference between the energy of the complex and the sum of the energies of the monomers in their optimized geometries.
2.4. Computation of Hot-Spot Residues
Hot-spot residues in the examined interfaces are predicted with the HotRegion web server using accessible surface area (ASA) and knowledge-based pair energies of each residue [35]. Two residues are specified to be in contact if the distance between any two atoms of the two residues from different chains is smaller than the sum of their corresponding van der Waals radii plus 0.5 Å [36,37]. Interface residues with observed binding free energies of ≥2.0 kcal/mol were deemed hot spots, while interfacing residues with <0.4 kcal/mol were marked non-hot spots. A network of hot spots is constructed to define hot regions. In the network, the nodes are the hot-spot residues, and the edges are connected between nodes if the two hot-spot residues are in contact. Two hot-spot residues are defined as being in contact if the distance between their Cα atoms is smaller than 6.5 Å [38]. Thereafter, connected network elements are found. Consider that the nodes in a connected component are equal to or greater than three. In that case, the connected component is termed a hot region, and the hot-spot residues in this connected component are termed the members of this particular hot region. The ASA of each residue in the monomer state and the complex form were calculated using the NACCESS program [39].
2.5. Computation of Stabilization Centers
Protein stabilization centers (SC) are clusters of amino acid residues creating cooperative, non-covalent, long-range interactions [40]. Measured as individual interactions, stabilization forces from non-covalent long-range interactions are weak. Nevertheless, since they are inherently cooperative in regions where they act in a group of SC, they could play an essential role in maintaining the overall stability of protein structures. To examine the SC of interaction-forming residues, we employed the SCide program [41]. The criteria SCide operates to determine SC are as follows: (a) two residues are in contact if there is at least one heavy atom–atom distance smaller than the sum of their van der Waals radii plus 1 Å; (b) a contact is identified as a “long-range” interaction if the interacting residues are at least ten amino acids separated; (c) two residues build stabilization centers if they are in long-range interaction and if it is feasible to select one–one residue from both flanking tetrapeptides of these two residues that make at least seven contacts between these two triplets [42].
2.6. Computation of Conservation of Amino Acid Residues
The conservation score of amino acid residues in each phycocyanin protein was computed using the ConSurf web server [42]. This bioinformatics tool calculates the conservation by comparing the given PDB polypeptide chain sequence with the proteins deposited in the Swiss-Prot database [43] and discovers the ones that are homologous to the PDB sequence. The number of PSI-BLAST iterations and the E-value cut-off used in all similarity searches were 1 and 0.001, respectively. The subsequent multiple alignments used all the sequences evolutionarily related to each protein in the analyzed dataset. These protein sequence alignments classified the residues into nine categories, from highly variable to highly conserved. Residues with a score of 1 were considered highly variable, and those with a score of 9 were classified as highly conserved.
3. Results and Discussion
This paper aims to study the role of amide–π interactions in interfaces of phycocyanin proteins and their environmental preferences. We conducted a computational analysis of the 20 X-ray crystallographic structures of phycocyanin-containing proteins (APC and C/R-PC from various species) and summarized amide–π interactions to better understand intrinsic phycocyanin oligomers’ high stability. Furthermore, the relative preference of amide–π interacting amino acids in interfaces, interaction geometries, and energetic contribution of amide–π interactions, hot-spot residues, stabilization centers, and the conservation score of amino acid residues were examined.
3.1. Distribution of Amide–π Interactions
Using the geometrical criteria defined in the Methods section, we have analyzed the frequency of aromatic amino acid residues involved in amide–π interactions. The results are presented in Table 1. There are, in total, 2086 amide–π interactions in phycocyanin protein interfaces in our data set, and it is interesting to note that there is an average of 104 interactions per protein.

Table 1.
Frequency of occurrence of amide–π interaction-forming residues in interfaces of phycocyanin proteins.
Although all protein structures have side-chain amide groups (Asn, Gln, and Arg) in interfaces, in our dataset, there are no amide–π interactions involving such groups. We detected that all interactions were of backbone amide–π interactions type. It is well known that this interaction class stabilizes tertiary and local structures and strengthens protein–ligand interactions [44]. It was found that Phe and Tyr had a higher occurrence of forming amide–π interactions than His and Trp out of the aromatic residues (Table 1). A plausible explanation might be that Phe and Tyr occur most frequently, while less than 1% of our database’s His and Trp residues are in phycocyanin interfaces [18]. In addition, the higher acceptor efficiency of the His and Trp side chains compared to Tyr and Phe is due to the conjugate nature of two planar rings containing a heteroatom.
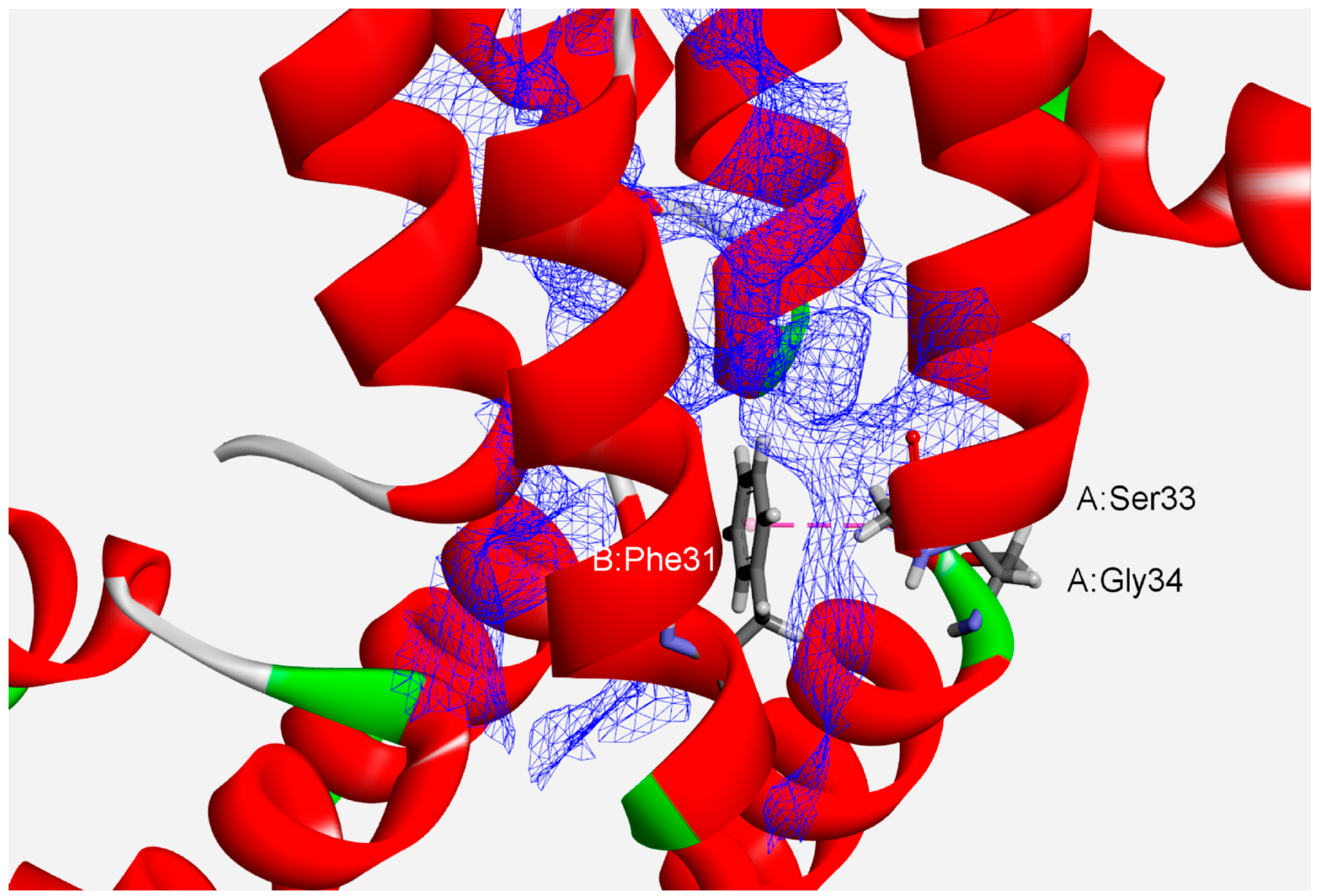
Analyses of protein interfaces have revealed that binding energies are not distributed uniformly. Rather, specific critical residues called hot spots comprise only a small fraction of the interface, but account for most of the total binding energy [45,46]. Thus, we analyzed the interface hot-spot residues in phycocyanin protein subunits to comprehend their associations’ high stability. We have found 1628 (78%) hot-spot residues forming amide–π interactions (Table 1). Phe and Tyr are the only common hot-spot amino acids implicated in such interactions. The composition of the hot spots reveals that certain amino acids are preferred in the high-energy interactions between protein chains in protein interfaces. A typical interface with some of the amide–π interactions involving planar backbone residues (A:Ser33:C,O;Gly34:N–B:Phe31) is shown in Figure 3 (APC from the cyanobacterium Spirulina platensis, PDB ID code 1all). An amide group from A:Ser33 and Gly34 interacts with the π-system of B:Phe31. The hot-spot residues (A:Arg37, A:Val38, A:Tyr97, A:Val100, B:Ile5, B:Ile9, B:Tyr18, B:Leu19, B:Ile24, B:Leu27, B:Tyr30 and B:Phe31) are denoted by a blue wire-meshed surface. These residues are part of hot-region 0.
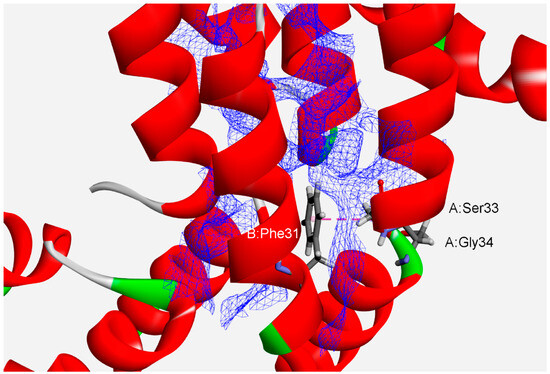
Figure 3.
Details of amide–π interaction involving more hot-spot residues (blue wire-meshed surface) at the interface of APC from the cyanobacterium Spirulina platensis (PDB ID code 1all). The amide–π interaction is labeled with a pink dashed line (A:Ser33:C,O;Gly34:N–B:Phe31).
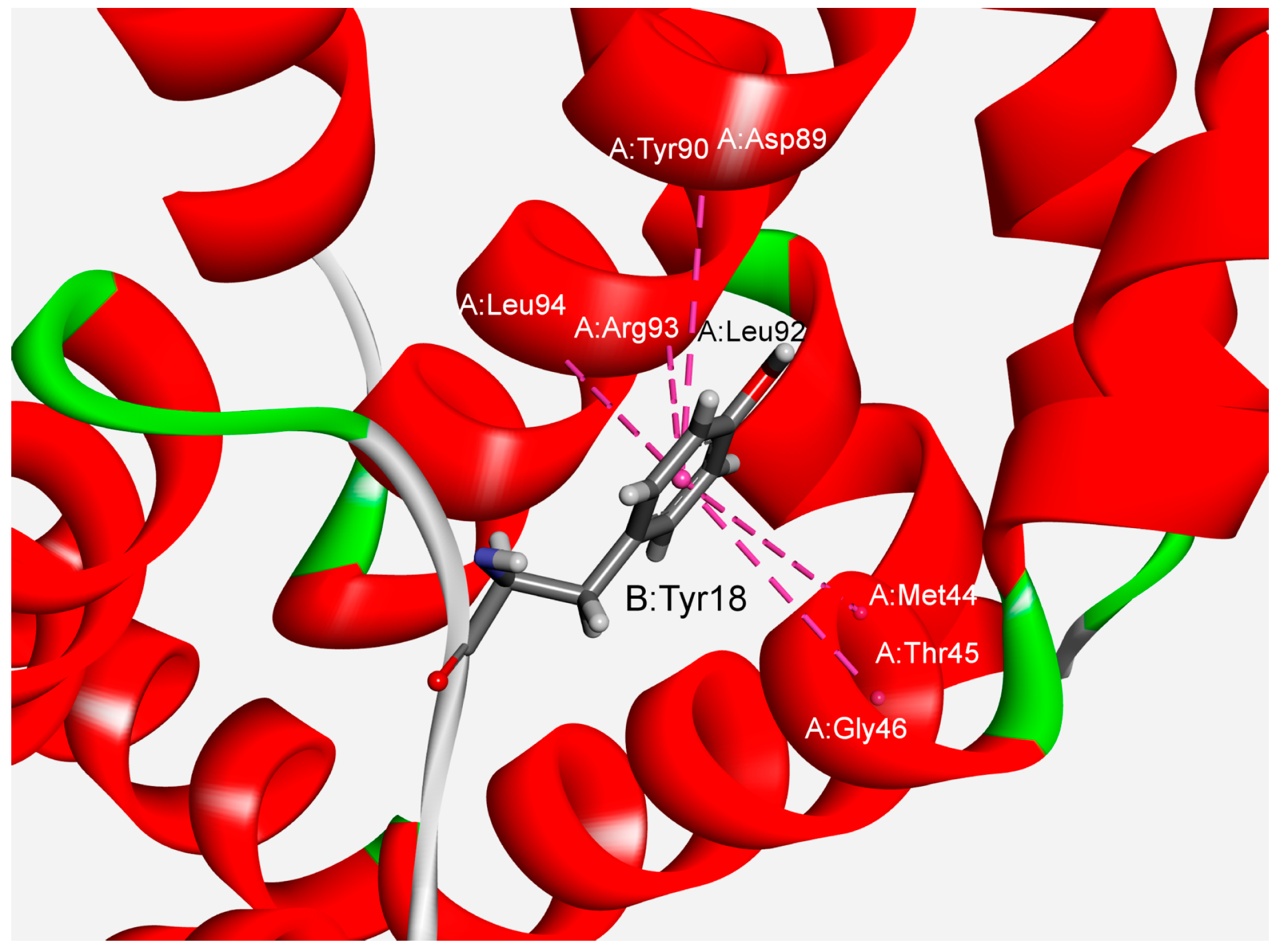
We have also studied the multiple amide–π interactions in phycocyanins. The analysis revealed that about 76% of the dataset’s total amide–π interactions are involved in forming multiple interactions. In many crystal structures of phycocyanins, a backbone amide can bind with several aromatic residues, and this type of interaction is marked as furcation. An illustrative example of amide–π interactions involving the presence of five amide groups surrounding one aromatic group is shown in Figure 4. Tyr18 from the B chain can interact with five amide groups from the A chain (Met44–Thr45, Thr45–Gly46, Asp89–Tyr90, Leu92–Arg93, Arg93–Leu94) simultaneously. The significance of multiple non-covalent weak interactions for cooperativity and controlling multicomponent supramolecular assemblies has been reported [47]. Another additional feature is observed as an additive property of these interactions, demonstrating an effect on the strength of the host–guest system.
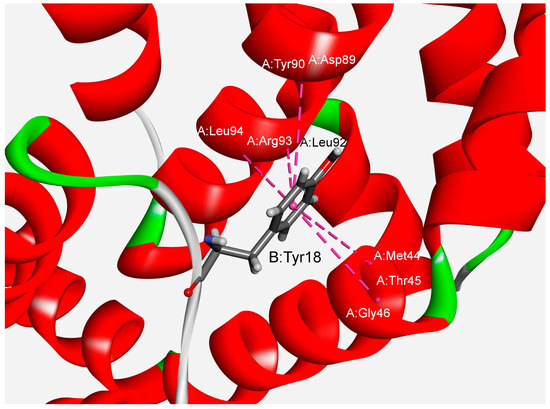
Figure 4.
Example of multiple amide–π interactions for the APC protein from the cyanobacterium Spirulina platensis (PDB ID code 1all). The interactions are marked with pink dashed lines.
3.2. Interaction Pattern and Energetic Contribution of Amide–π Interactions
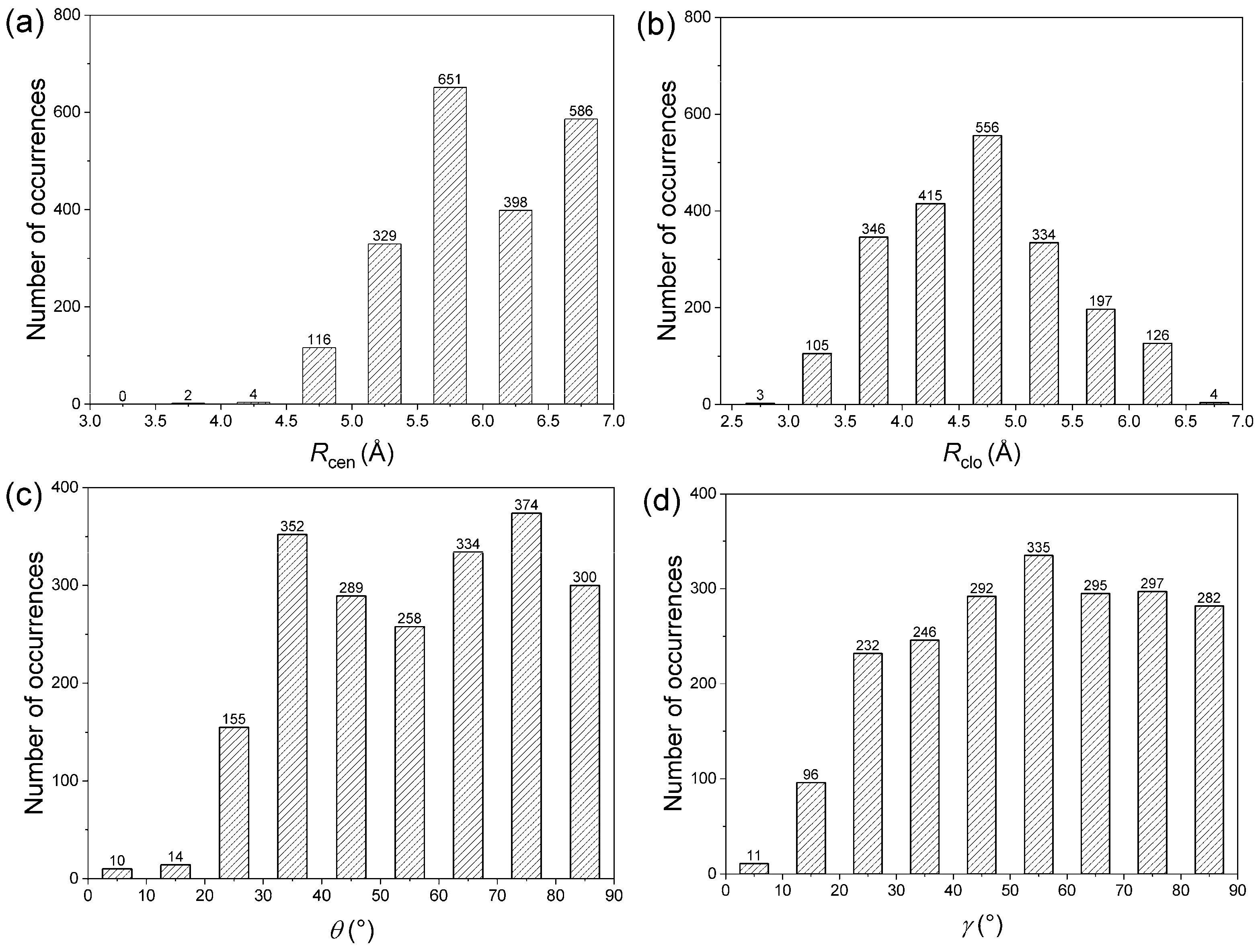
The native structure compromises many non-covalent interactions in proteins, and the geometrical features relating to the two residue types are expected to be rather broad. However, based on the distribution of interplanar angles, it was indicated that there is non-randomness in the packing of side chains [48]. Based on the orientation of the aromatic ring and amide group, the amide–π interactions have been broadly classified into edge-to-face (T-shaped), parallel displaced, and parallel stacked. The frequency distribution of the distance and angle parameters of amide–π interaction pairs are dissected (Figure 5).
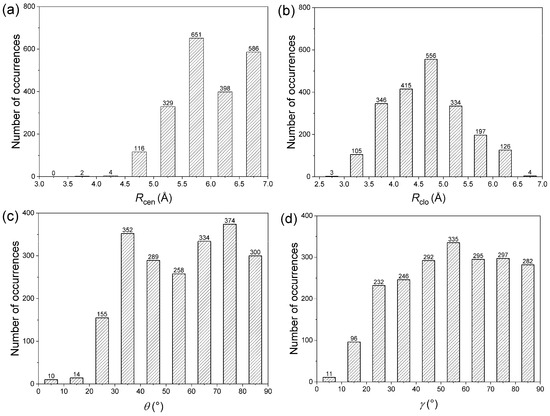
Figure 5.
Interaction pattern of amide–π interactions in phycocyanin interfaces: (a) Rcen distance distribution, (b) Rclo distance distribution, (c) θ angle distribution, (d) γ angle distribution.
The distribution of Rcen, the centroid–centroid distance, for amide–π interactions was found to be bimodal with a prominent minimum between 6.0 and 6.5 Å (Figure 5a). Outside the minimum, there are two distinct maxima with a narrow range of linearity, corresponding to parallel-stacked and T-shaped orientation, respectively, because parallel orientations have a shorter Rcen than T-shaped orientations. Due to evident physical constraints, interaction pairs rarely occur at separation distances below 4.5 Å. The distribution of Rclo for amide–π interactions was found to be a narrow peak at 4.5 Å (Figure 5b), the optimal average distance between two aromatic rings in a T-shaped orientation. This is because T-shaped orientations have shorter Rclo than parallel orientations. Regardless of the angle distribution, the interacting pairs are oriented to minimize Rclo between the two rings and thus maximize the van der Waals attraction. The plot of θ angle distribution derived from amide–π interaction pairs (Figure 5c) shows two prominent peaks centered at 35° and 75°. Regarding the plane–plane angle (γ), the angles were distributed between all angles (0°–90° range) (Figure 5d). While axial interacting pairs (γ > 50°) are more frequent, there were a few interactions with angles below 30° (displays coplanarity), possibly to maximize π stacking and packing [49]. The preferred orientations are similar to those with aromatic–aromatic interactions [50]. The geometries observed in abundance are not necessarily the ones that have the highest interaction energy between the two moieties in a pair, but can provide the maximum overall stability to the protein structure by the optimum use of all amide–π interactions.
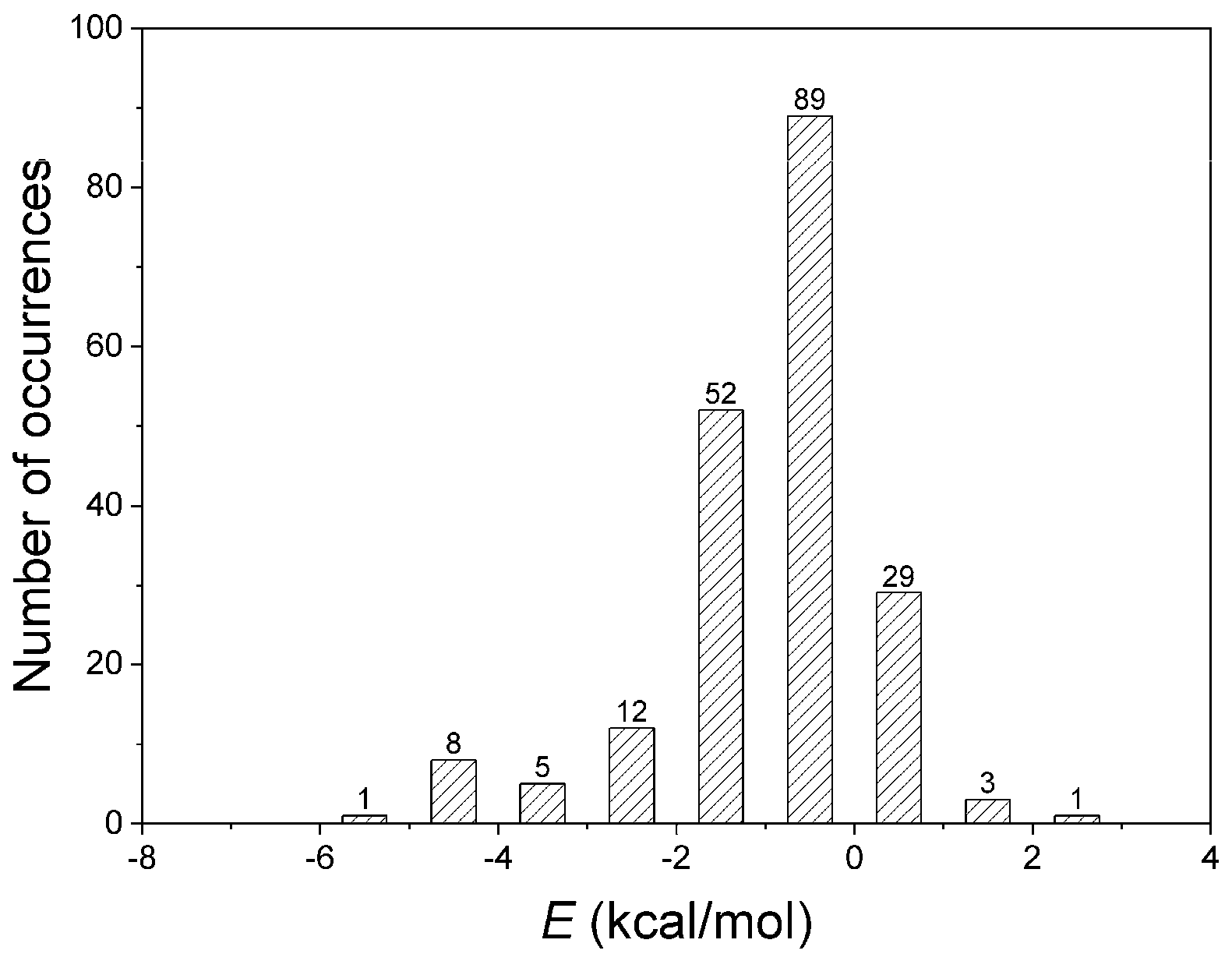
The quantification of non-covalent interactions is crucial for a rational approach to biological systems, not only for protein structure and function but also for antibody binding or drug design and further development of supramolecular chemistry [51]. Thus, the energetic contributions of residues implicated in amide–π interactions were computed using ab initio calculations at the LMP2 level. Numerous interactions are possible within a large protein structure, and sometimes, it is not easy to parse out the role of the amide–π interaction in their energetics by a simple calculation. Therefore, the interacting pair residues participating in other non-covalent interactions were not analyzed. To avoid calculating more than 2000 interactions, we carefully selected 200 structures representing nearly all interactions. The results for amide–π interacting pairs are presented in Figure 6. The energies computed for many amide–π interactions are substantially stabilizing, with 16% of the total showing positive (repulsive) interaction energies. The repulsive nature of those interactions arises from the unfavorable geometries of amide–π interactions in the crystal structures. It is usually counterbalanced by stronger interactions (salt bridge, H-bonding, or similar) [27]. Our database showed amide–π interactions showed energy less than −5 kcal/mol; most have energy in the 0 to −2 kcal/mol range. The energies of the weakly polar amide–π interactions discussed here are in the lower range of strong hydrogen bonds, as categorized by Desiraju and Steiner [52]. The energies associated with amide–π interactions may contribute to overall protein stability. It should also be considered in supramolecular chemistry and protein engineering [47].
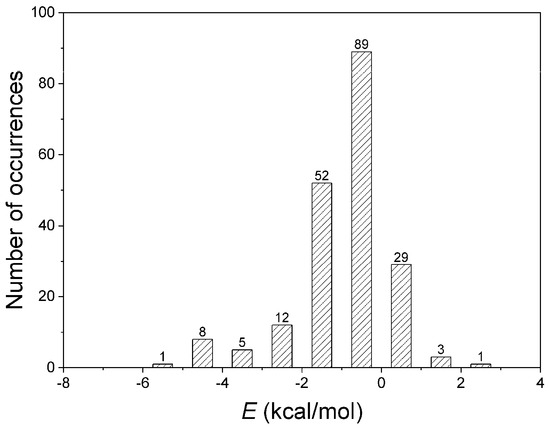
Figure 6.
Interaction energies of amide–π interactions in phycocyanin interfaces.
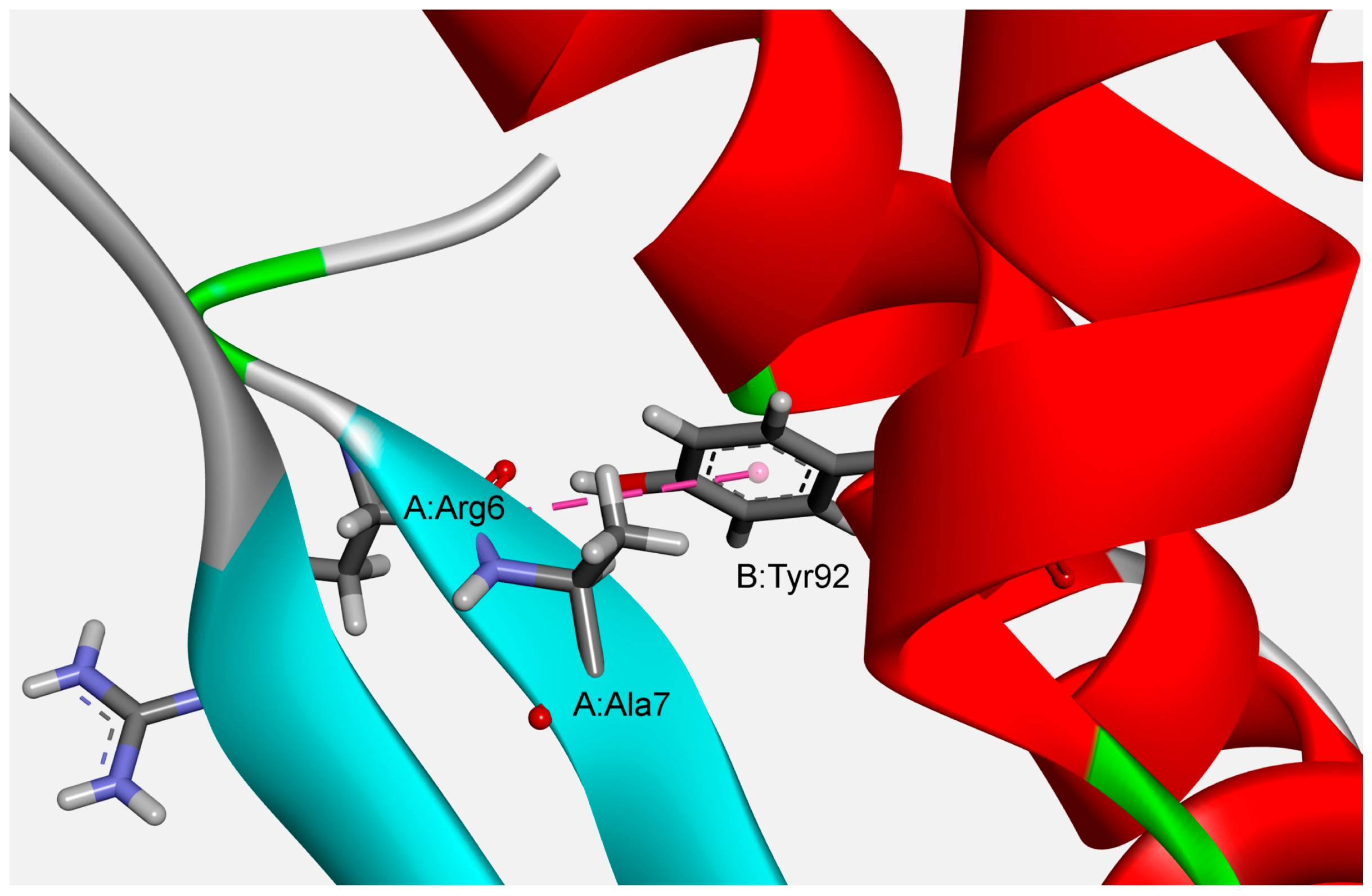
The results of our ab initio calculations of optimized structures showed that the strongest attractive amide–π interaction (−5.63 kcal/mol) exists between the A:Arg6:C,O;Ala7:N–B:Tyr92 pair in the phycocyanin PC645 from Chroomonas sp. (PDB ID code 4lms) (Figure 7). The geometrical parameters of the interaction were Rcen = 4.96 Å; Rclo = 3.34 Å; θ = 82.99°; γ = 78.25°).
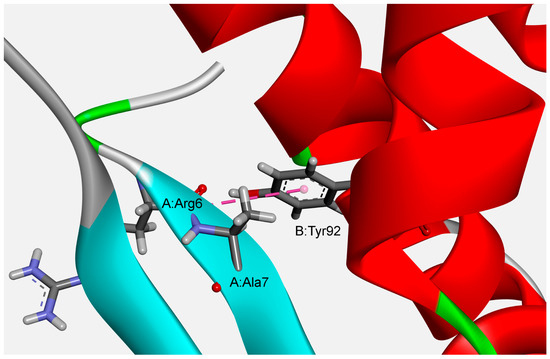
Figure 7.
Example of the structure preferred amide–π interactions of phycocyanin PC645 from Chroomonas sp. (PDB ID code 4lms).
3.3. Stabilization Centers and Conservation of Amino Acid Residues
Stabilization centers comprised of residues involved in long-range cooperative contacts are likely to play an essential role in regulating the flexibility and stability of protein structures [40]. The residues most frequently compose stabilization centers and are typically located in buried protein positions, usually having a hydrophobic or aromatic side chain. However, some polar or charged residues are found as well. The structural and sequential conservation analysis demonstrated higher stabilization center conservation over protein families [40,53]. We have calculated the stabilization centers for all predicted amide–π interactions forming residues in interfaces of phycocyanin proteins using the SCide program. It was found that 42.7% of interacting residues have one or more stabilization centers. Since many amides–π interacting residues have more than one stabilization center, these residues confer additional stability to the protein along with their participation in amide–π interactions. Interestingly, these results are similar to findings in amide–π interaction studies in superoxide dismutase enzymes [28].
The level of evolutionary conservation was frequently used to indicate the importance of the position in preserving the protein’s structure and function [54]. Functionally critical residues are identified based on the conservation score generated by the ConSurf server. Our analysis found that more than 65% of predicted amide–π interacting residues are conserved (the cut-off value used to identify the stabilizing residues was a conservation score ≥ 6). Of all the interacting residues, 14% had the highest conservation score, 9. Moreover, most additional residues incorporating the phycocyanin interfaces also show significant conservation. Since the conserved residues play a vital role in the stability and functional specificity of protein structure, results acquired from this study could help to understand the specific importance of amide–π interacting residues in the stability of oligomeric phycocyanin proteins.
4. Conclusions
We have systematically analyzed the influence of amide–π interactions on the stability of commercially attractive phycocyanins and allophycocyanins. The characteristic features of residues involved in amide–π interactions have been assessed regarding the distribution of amide–π interactions, interaction geometry, energetic contribution, hot-spot regions, stabilizing centers, and conservation score of interacting residues. A significant number of amide–π interactions in all phycocyanins was found. Amide–π interaction forming residues are highly conserved, indicating these interactions’ vital role in the complex structure of phycocyanins. All of the interactions occur in the backbone of proteins. The multiple interaction patterns found in the present study indicate that around 76% of the total interacting residues participate in multiple amide–π interactions.
The results show that approximately 78% of the total interacting residues in the dataset are involved in forming hot-spot regions. When investigating the preferences of residues to form amide–π interactions, it was found that Phe and Tyr have a higher occurrence than His and Trp. Our analysis of interaction distance between the interacting pairs suggests that most distances were most favorable in the distance range of 5–7 Å. The centroid–centroid distance distribution for amide–π interactions was bimodal with a prominent minimum between 6.0 and 6.5 Å. Considering angle distribution, effective amide–π interactions can be realized above a wider area above the π ring, showing well-defined represented states, parallel-displaced and T-shaped.
Analysis of amide–π interaction energy informed that favorable energy interactions were less than −5 kcal/mol, while most have energy from 0 to −2 kcal/mol. These residues might provide additional stability to these proteins and their energetic contribution due to amide–π interactions. Stabilization centers are proposed to contribute to the stability of proteins; a significant percentage of amide–π interacting residues are also located in stabilization centers. To conclude, our observations with phycocyanins in the present study identify amide–π interactions and structural motifs that contribute to stabilizing increasingly used phycocyanins, relevant to understanding structure–function relationships. They are also helpful to the efforts made to design and engineer complex protein–protein aggregates.
Author Contributions
Conceptualization, S.Đ.S. and M.R.N.; methodology, S.Đ.S.; software, M.V.Z. and S.Đ.S.; validation, M.V.Z. and S.Đ.S.; formal analysis, L.M.B., M.V.Z., S.Đ.S. and M.R.N.; investigation, L.M.B. and S.Đ.S.; resources, M.V.Z. and S.Đ.S.; data curation, L.M.B. and S.Đ.S.; writing—original draft preparation, L.M.B.; writing—review and editing, M.V.Z., S.Đ.S. and M.R.N.; visualization, L.M.B. and S.Đ.S.; supervision, M.R.N.; funding acquisition, M.R.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science, Technological Development and Innovation of the Republic of Serbia (Contract Nos: 451-03-66/2024-03/200168 and 451-03-66/2024-03/200026).
Data Availability Statement
All the data generated in the current research is presented in the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tandeau de Marsac, N. Phycobiliproteins and phycobilisomes: The early observations. Photosynth. Res. 2003, 76, 193–205. [Google Scholar] [CrossRef]
- Kannaujiya, V.K.; Kumar, D.; Singh, V.; Sinha, R.P. Chapter 4—Advances in phycobiliproteins research: Innovations and commercialization. In Natural Bioactive Compounds; Sinha, R., Häder, D.P., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 57–81. [Google Scholar]
- Wang, X.Q.; Li, L.N.; Chang, W.R.; Zhang, J.P.; Gui, L.L.; Guo, B.J.; Liang, D.C. Structure of C-phycocyanin from Spirulina platensis at 2.2 Å resolution: A novel monoclinic crystal form for phycobiliproteins in phycobilisomes. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 784–792. [Google Scholar] [CrossRef] [PubMed]
- de Morais, M.G.; da Fontoura Prates, D.; Moreira, J.B.; Duarte, J.H.; Costa, J.A.V. Phycocyanin from Microalgae: Properties, Extraction and Purification, with Some Recent Applications. Ind. Biotechnol. 2018, 14, 30–37. [Google Scholar] [CrossRef]
- McGregor, A.; Klartag, M.; David, L.; Adir, N. Allophycocyanin trimer stability and functionality are primarily due to polar enhanced hydrophobicity of the phycocyanobilin binding pocket. J. Mol. Biol. 2008, 384, 406–421. [Google Scholar] [CrossRef]
- Johansson, A.; Kollman, P.; Rothenberg, S.; McKelvey, J. Hydrogen bonding ability of the amide group. J. Am. Chem. Soc. 1974, 96, 3794–3800. [Google Scholar] [CrossRef]
- Eberhardt, E.S.; Raines, R.T. Amide-amide and amide-water hydrogen bonds: Implications for protein folding and stability. J. Am. Chem. Soc. 1994, 116, 2149–2150. [Google Scholar] [CrossRef] [PubMed]
- Persch, E.; Dumele, O.; Diederich, F. Molecular recognition in chemical and biological systems. Angew. Chem. Int. Ed. 2015, 54, 3290–3327. [Google Scholar] [CrossRef]
- Bootsma, A.N.; Wheeler, S.E. Stacking interactions of heterocyclic drug fragments with protein amide backbones. ChemMedChem 2018, 13, 835–841. [Google Scholar] [CrossRef]
- Imai, Y.N.; Inoue, Y.; Nakanishi, I.; Kitaura, K. Amide-pi interactions between formamide and benzene. J. Comput. Chem. 2009, 15, 2267–2276. [Google Scholar] [CrossRef]
- Ferreira de Freitas, R.; Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Med. Chem. Commun. 2017, 8, 1970–1981. [Google Scholar] [CrossRef]
- Harder, M.; Kuhn, B.; Diederich, F. Efficient stacking on protein amide fragments. ChemMedChem 2013, 8, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.M.; Waters, M.L. Effects of lysine acetylation in a beta-Hairpin peptide: Comparison of an amide–π and a cation–π interaction. J. Am. Chem. Soc. 2006, 128, 13586–13591. [Google Scholar] [CrossRef]
- Travis, C.R.; Francis, D.Y.; Williams, J.; Waters, M.L. Evaluation of acyllysine isostere interactions with the aromatic pocket of the AF9 YEATS domain. Prot. Sci. 2023, 32, e4533. [Google Scholar] [CrossRef]
- Lauber, B.S.; Hardegger, L.A.; Asraful, A.K.; Lund, B.A.; Dumele, O.; Harder, M.; Kuhn, B.; Engh, R.A.; Diederich, F. Addressing the glycine-rich loop of protein kinases by a multi-facetted interaction network: Inhibition of PKA and a PKB mimic. Chem. Eur. J. 2016, 22, 211–221. [Google Scholar] [CrossRef]
- Ehmke, V.; Winkler, E.; Banner, D.W.; Haap, W.; Schweizer, W.B.; Rottmann, M.; Kaiser, M.; Freymond, C.; Schirmeister, T.; Diederich, F. Optimization of triazine nitriles as rhodesain inhibitors: Structure-activity relationships, bioisosteric imidazopyridine nitriles, and X-ray crystal structure analysis with human cathepsin L. ChemMedChem 2013, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- DeGasparo, R.; Brodbeck-Persch, E.; Bryson, S.; Hentzen, N.B.; Kaiser, M.; Pai, E.F.; Krauth-Siegel, R.L.; Diederich, F. Biological evaluation and X-ray co-crystal structures of cyclohexylpyrrolidine ligands for trypanothione reductase, an enzyme from the redox metabolism of trypanosoma. ChemMedChem 2018, 13, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Breberina, L.M.; Zlatović, M.V.; Nikolić, M.R.; Stojanović, S.Đ. Computational analysis of non-covalent interactions in phycocyanin subunit interfaces. Mol. Inform. 2019, 38, e1800145. [Google Scholar] [CrossRef]
- Breberina, L.M.; Nikolić, M.R.; Stojanović, S.Đ.; Zlatović, M.V. Influence of cation–π interactions to the structural stability of phycocyanin proteins: A computational study. Comput. Biol. Chem. 2022, 100, 107752. [Google Scholar] [CrossRef]
- Breberina, L.; Nikolić, M.; Stojanović, S.Đ.; Zlatović, M. On the importance of π–π interactions in structural stability of phycocyanins. J. Serb. Chem. Soc. 2023, 88, 481–494. [Google Scholar] [CrossRef]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlić, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, D392–D401. [Google Scholar] [CrossRef]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef]
- Accelrys Software Inc. Discovery Studio Visualizer, Release 2021; Accelrys Software Inc.: San Diego, CA, USA, 2021. [Google Scholar]
- Jackson, M.R.; Beahm, R.; Duvvuru, S.; Narasimhan, C.; Wu, J.; Wang, H.N.; Philip, V.M.; Hinde, R.J.; Howell, E.E. A preference for edgewise interactions between aromatic rings and carboxylate anions: The biological relevance of anion–quadrupole interactions. J. Phys. Chem. B 2007, 111, 8242–8249. [Google Scholar] [CrossRef] [PubMed]
- Philip, V.; Harris, J.; Adams, R.; Nguyen, D.; Spiers, J.; Baudry, J.; Howell, E.E.; Hinde, R.J. A survey of aspartate-phenylalanine and glutamate-phenylalanine interactions in the protein data bank: Searching for anion-π pairs. Biochemistry 2011, 50, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Ribić, V.R.; Stojanović, S.Đ.; Zlatović, M.V. Anion–π interactions in active centers of superoxide dismutases. Int. J. Biol. Macromol. 2018, 106, 559–568. [Google Scholar] [CrossRef]
- Stojanović, S.Đ.; Petrović, Z.Z.; Zlatović, M.V. Amide–π interactions in active centers of superoxide dismutase. J. Serb. Chem. Soc. 2021, 86, 781–793. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Jaguar; Schrödinger, LLC: New York, NY, USA, 2018.
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.nW.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Jones, G.J.; Robertazzi, A.; Platts, J.A. Efficient and accurate theoretical methods to investigate anion-π interactions in protein model structures. J. Phys. Chem. B 2013, 117, 3315–3322. [Google Scholar] [CrossRef]
- Riley, K.E.; Platts, J.A.; Řezáč, J.; Hobza, P.; Hill, J.G. Assessment of the performance of MP2 and MP2 variants for the treatment of non-covalent interactions. J. Phys. Chem. A 2012, 116, 4159–4169. [Google Scholar] [CrossRef] [PubMed]
- Cukuroglu, E.; Gursoy, A.; Keskin, O. HotRegion: A database of predicted hot spot clusters. Nucleic Acids Res. 2012, 40, D829–D833. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Tsai, C.J.; Wolfson, H.; Nussinov, R. A new, structurally nonredundant, diverse data set of protein-protein interfaces and its implications. Protein Sci. 2004, 13, 1043–1055. [Google Scholar] [CrossRef]
- Tsai, C.J.; Lin, S.L.; Wolfson, H.J.; Nussinov, R. A dataset of protein-protein interfaces generated with a sequence-order-independent comparison technique. J. Mol. Biol. 1996, 260, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Ma, B.; Nussinov, R. Hot regions in protein-protein interactions: The organization and contribution of structurally conserved hot pot residues. J. Mol. Biol. 2005, 345, 1281–1294. [Google Scholar] [CrossRef]
- Hubbard, S.J.; Thornton, J.M. “NACCESS”, Computer Program; Department of Biochemistry and Molecular Biology, University College London: London, UK, 1993. [Google Scholar]
- Dosztányi, Z.; Fiser, A.; Simon, I. Stabilization centers in proteins: Identification, characterization and predictions. J. Mol. Biol. 1997, 272, 597–612. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Magyar, C.; Tusnady, G.; Simon, I. SCide: Identification of stabilization centers in proteins. Bioinformatics 2003, 19, 899–900. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Tóth, G.; Watts, C.R.; Murphy, R.F.; Lovas, S. Significance of aromatic-backbone amide interactions in protein structure. Proteins 2001, 43, 373–381. [Google Scholar] [CrossRef]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot spots—A review of the protein-protein interface determinant amino-acid residues. Proteins 2007, 68, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Mahadevi, A.S.; Sastry, G.N. Cooperativity in non-covalent interactions. Chem. Rev. 2016, 116, 2775–2825. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Laskowski, R.A.; Thornton, J.M. Non-randomness in side-chain packing: The distribution of interplanar angles. Proteins 1997, 29, 370–380. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Gagné, M.; Rappé, A.K. π-stacking interactions: Alive and well in proteins. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef]
- Dimitrijević, B.P.; Borozan, S.Z.; Stojanović, S. π–π and cation–π interactions in protein–porphyrin complex crystal structures. RSC Adv. 2012, 2, 12963–12972. [Google Scholar] [CrossRef]
- Maury, P.A.; Reinhoudt, D.N.; Huskens, J. Assembly of nanoparticles on patterned surfaces by non-covalent interactions. Curr. Opin. Colloid Interface Sci. 2008, 13, 74–80. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Magyar, C.; Gromiha, M.M.; Pujadas, G.; Tusnady, G.E.; Simon, I. SRide: A server for identifying stabilizing residues in proteins. Nucleic Acids Res. 2005, 33, W303–W305. [Google Scholar] [CrossRef]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).