
Amide–π Interactions in the Structural Stability of Proteins: Role in the Oligomeric Phycocyanins


Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
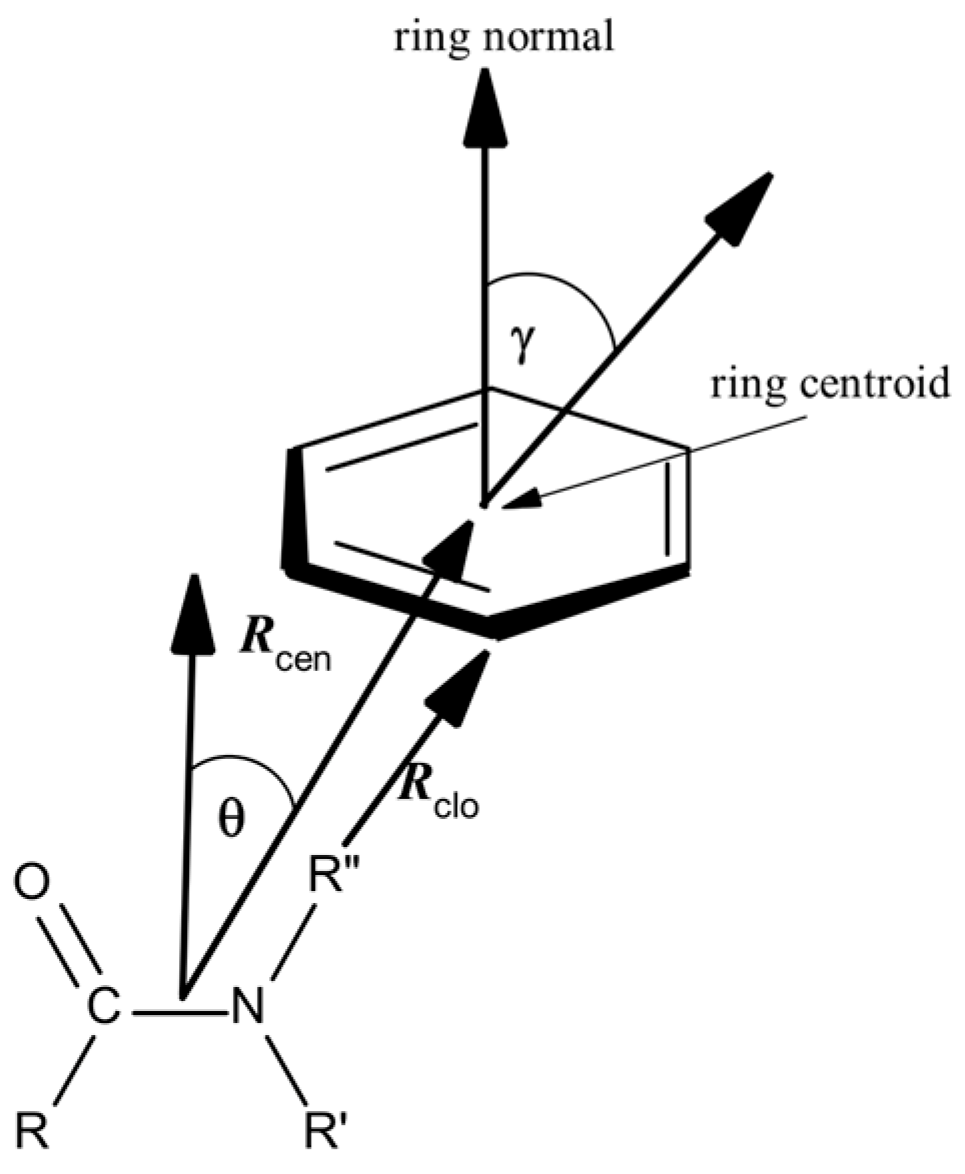
2.2. Amide–π Interaction Analysis in Phycocyanin Interfaces
2.3. Computation of Amide–π Interaction Energy in Phycocyanin Interfaces
2.4. Computation of Hot-Spot Residues
2.5. Computation of Stabilization Centers
2.6. Computation of Conservation of Amino Acid Residues
3. Results and Discussion
3.1. Distribution of Amide–π Interactions
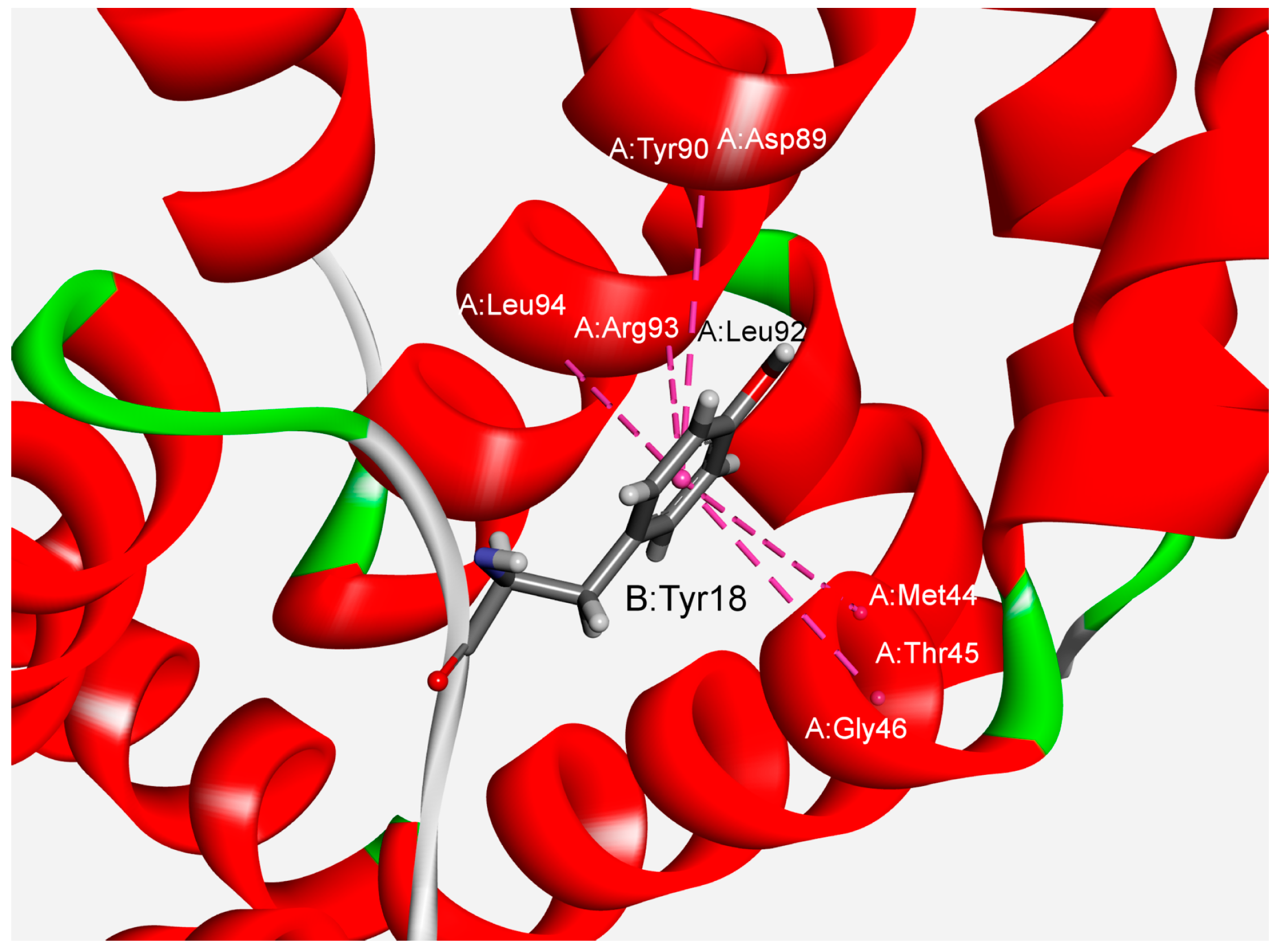
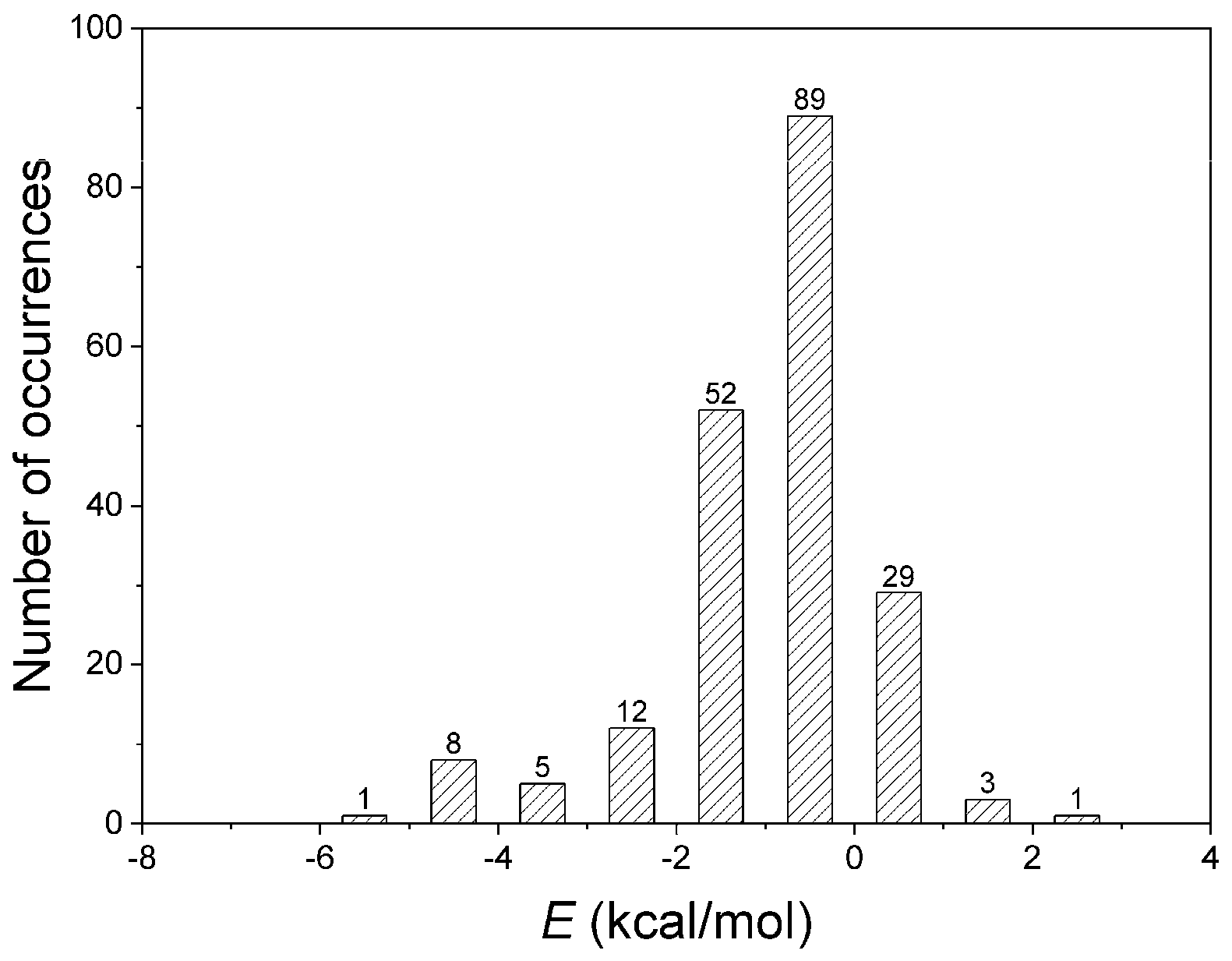
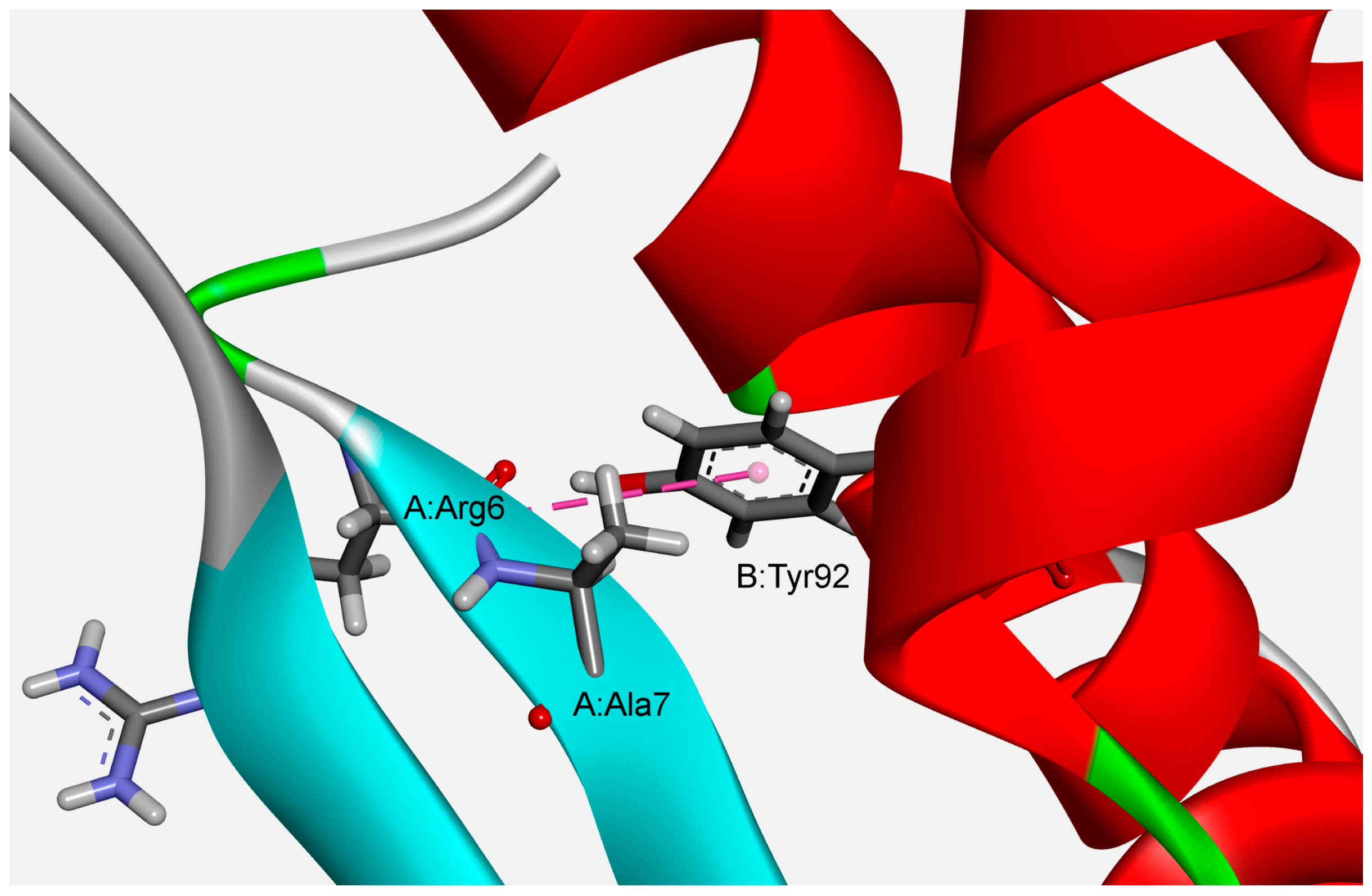
3.2. Interaction Pattern and Energetic Contribution of Amide–π Interactions
3.3. Stabilization Centers and Conservation of Amino Acid Residues
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tandeau de Marsac, N. Phycobiliproteins and phycobilisomes: The early observations. Photosynth. Res. 2003, 76, 193–205. [Google Scholar] [CrossRef]
- Kannaujiya, V.K.; Kumar, D.; Singh, V.; Sinha, R.P. Chapter 4—Advances in phycobiliproteins research: Innovations and commercialization. In Natural Bioactive Compounds; Sinha, R., Häder, D.P., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 57–81. [Google Scholar]
- Wang, X.Q.; Li, L.N.; Chang, W.R.; Zhang, J.P.; Gui, L.L.; Guo, B.J.; Liang, D.C. Structure of C-phycocyanin from Spirulina platensis at 2.2 Å resolution: A novel monoclinic crystal form for phycobiliproteins in phycobilisomes. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 784–792. [Google Scholar] [CrossRef] [PubMed]
- de Morais, M.G.; da Fontoura Prates, D.; Moreira, J.B.; Duarte, J.H.; Costa, J.A.V. Phycocyanin from Microalgae: Properties, Extraction and Purification, with Some Recent Applications. Ind. Biotechnol. 2018, 14, 30–37. [Google Scholar] [CrossRef]
- McGregor, A.; Klartag, M.; David, L.; Adir, N. Allophycocyanin trimer stability and functionality are primarily due to polar enhanced hydrophobicity of the phycocyanobilin binding pocket. J. Mol. Biol. 2008, 384, 406–421. [Google Scholar] [CrossRef]
- Johansson, A.; Kollman, P.; Rothenberg, S.; McKelvey, J. Hydrogen bonding ability of the amide group. J. Am. Chem. Soc. 1974, 96, 3794–3800. [Google Scholar] [CrossRef]
- Eberhardt, E.S.; Raines, R.T. Amide-amide and amide-water hydrogen bonds: Implications for protein folding and stability. J. Am. Chem. Soc. 1994, 116, 2149–2150. [Google Scholar] [CrossRef] [PubMed]
- Persch, E.; Dumele, O.; Diederich, F. Molecular recognition in chemical and biological systems. Angew. Chem. Int. Ed. 2015, 54, 3290–3327. [Google Scholar] [CrossRef]
- Bootsma, A.N.; Wheeler, S.E. Stacking interactions of heterocyclic drug fragments with protein amide backbones. ChemMedChem 2018, 13, 835–841. [Google Scholar] [CrossRef]
- Imai, Y.N.; Inoue, Y.; Nakanishi, I.; Kitaura, K. Amide-pi interactions between formamide and benzene. J. Comput. Chem. 2009, 15, 2267–2276. [Google Scholar] [CrossRef]
- Ferreira de Freitas, R.; Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Med. Chem. Commun. 2017, 8, 1970–1981. [Google Scholar] [CrossRef]
- Harder, M.; Kuhn, B.; Diederich, F. Efficient stacking on protein amide fragments. ChemMedChem 2013, 8, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.M.; Waters, M.L. Effects of lysine acetylation in a beta-Hairpin peptide: Comparison of an amide–π and a cation–π interaction. J. Am. Chem. Soc. 2006, 128, 13586–13591. [Google Scholar] [CrossRef]
- Travis, C.R.; Francis, D.Y.; Williams, J.; Waters, M.L. Evaluation of acyllysine isostere interactions with the aromatic pocket of the AF9 YEATS domain. Prot. Sci. 2023, 32, e4533. [Google Scholar] [CrossRef]
- Lauber, B.S.; Hardegger, L.A.; Asraful, A.K.; Lund, B.A.; Dumele, O.; Harder, M.; Kuhn, B.; Engh, R.A.; Diederich, F. Addressing the glycine-rich loop of protein kinases by a multi-facetted interaction network: Inhibition of PKA and a PKB mimic. Chem. Eur. J. 2016, 22, 211–221. [Google Scholar] [CrossRef]
- Ehmke, V.; Winkler, E.; Banner, D.W.; Haap, W.; Schweizer, W.B.; Rottmann, M.; Kaiser, M.; Freymond, C.; Schirmeister, T.; Diederich, F. Optimization of triazine nitriles as rhodesain inhibitors: Structure-activity relationships, bioisosteric imidazopyridine nitriles, and X-ray crystal structure analysis with human cathepsin L. ChemMedChem 2013, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- DeGasparo, R.; Brodbeck-Persch, E.; Bryson, S.; Hentzen, N.B.; Kaiser, M.; Pai, E.F.; Krauth-Siegel, R.L.; Diederich, F. Biological evaluation and X-ray co-crystal structures of cyclohexylpyrrolidine ligands for trypanothione reductase, an enzyme from the redox metabolism of trypanosoma. ChemMedChem 2018, 13, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Breberina, L.M.; Zlatović, M.V.; Nikolić, M.R.; Stojanović, S.Đ. Computational analysis of non-covalent interactions in phycocyanin subunit interfaces. Mol. Inform. 2019, 38, e1800145. [Google Scholar] [CrossRef]
- Breberina, L.M.; Nikolić, M.R.; Stojanović, S.Đ.; Zlatović, M.V. Influence of cation–π interactions to the structural stability of phycocyanin proteins: A computational study. Comput. Biol. Chem. 2022, 100, 107752. [Google Scholar] [CrossRef]
- Breberina, L.; Nikolić, M.; Stojanović, S.Đ.; Zlatović, M. On the importance of π–π interactions in structural stability of phycocyanins. J. Serb. Chem. Soc. 2023, 88, 481–494. [Google Scholar] [CrossRef]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlić, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, D392–D401. [Google Scholar] [CrossRef]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef]
- Accelrys Software Inc. Discovery Studio Visualizer, Release 2021; Accelrys Software Inc.: San Diego, CA, USA, 2021. [Google Scholar]
- Jackson, M.R.; Beahm, R.; Duvvuru, S.; Narasimhan, C.; Wu, J.; Wang, H.N.; Philip, V.M.; Hinde, R.J.; Howell, E.E. A preference for edgewise interactions between aromatic rings and carboxylate anions: The biological relevance of anion–quadrupole interactions. J. Phys. Chem. B 2007, 111, 8242–8249. [Google Scholar] [CrossRef] [PubMed]
- Philip, V.; Harris, J.; Adams, R.; Nguyen, D.; Spiers, J.; Baudry, J.; Howell, E.E.; Hinde, R.J. A survey of aspartate-phenylalanine and glutamate-phenylalanine interactions in the protein data bank: Searching for anion-π pairs. Biochemistry 2011, 50, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Ribić, V.R.; Stojanović, S.Đ.; Zlatović, M.V. Anion–π interactions in active centers of superoxide dismutases. Int. J. Biol. Macromol. 2018, 106, 559–568. [Google Scholar] [CrossRef]
- Stojanović, S.Đ.; Petrović, Z.Z.; Zlatović, M.V. Amide–π interactions in active centers of superoxide dismutase. J. Serb. Chem. Soc. 2021, 86, 781–793. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Jaguar; Schrödinger, LLC: New York, NY, USA, 2018.
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.nW.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Jones, G.J.; Robertazzi, A.; Platts, J.A. Efficient and accurate theoretical methods to investigate anion-π interactions in protein model structures. J. Phys. Chem. B 2013, 117, 3315–3322. [Google Scholar] [CrossRef]
- Riley, K.E.; Platts, J.A.; Řezáč, J.; Hobza, P.; Hill, J.G. Assessment of the performance of MP2 and MP2 variants for the treatment of non-covalent interactions. J. Phys. Chem. A 2012, 116, 4159–4169. [Google Scholar] [CrossRef] [PubMed]
- Cukuroglu, E.; Gursoy, A.; Keskin, O. HotRegion: A database of predicted hot spot clusters. Nucleic Acids Res. 2012, 40, D829–D833. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Tsai, C.J.; Wolfson, H.; Nussinov, R. A new, structurally nonredundant, diverse data set of protein-protein interfaces and its implications. Protein Sci. 2004, 13, 1043–1055. [Google Scholar] [CrossRef]
- Tsai, C.J.; Lin, S.L.; Wolfson, H.J.; Nussinov, R. A dataset of protein-protein interfaces generated with a sequence-order-independent comparison technique. J. Mol. Biol. 1996, 260, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Ma, B.; Nussinov, R. Hot regions in protein-protein interactions: The organization and contribution of structurally conserved hot pot residues. J. Mol. Biol. 2005, 345, 1281–1294. [Google Scholar] [CrossRef]
- Hubbard, S.J.; Thornton, J.M. “NACCESS”, Computer Program; Department of Biochemistry and Molecular Biology, University College London: London, UK, 1993. [Google Scholar]
- Dosztányi, Z.; Fiser, A.; Simon, I. Stabilization centers in proteins: Identification, characterization and predictions. J. Mol. Biol. 1997, 272, 597–612. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Magyar, C.; Tusnady, G.; Simon, I. SCide: Identification of stabilization centers in proteins. Bioinformatics 2003, 19, 899–900. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef]
- Tóth, G.; Watts, C.R.; Murphy, R.F.; Lovas, S. Significance of aromatic-backbone amide interactions in protein structure. Proteins 2001, 43, 373–381. [Google Scholar] [CrossRef]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot spots—A review of the protein-protein interface determinant amino-acid residues. Proteins 2007, 68, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Mahadevi, A.S.; Sastry, G.N. Cooperativity in non-covalent interactions. Chem. Rev. 2016, 116, 2775–2825. [Google Scholar] [CrossRef]
- Mitchell, J.B.; Laskowski, R.A.; Thornton, J.M. Non-randomness in side-chain packing: The distribution of interplanar angles. Proteins 1997, 29, 370–380. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Gagné, M.; Rappé, A.K. π-stacking interactions: Alive and well in proteins. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef]
- Dimitrijević, B.P.; Borozan, S.Z.; Stojanović, S. π–π and cation–π interactions in protein–porphyrin complex crystal structures. RSC Adv. 2012, 2, 12963–12972. [Google Scholar] [CrossRef]
- Maury, P.A.; Reinhoudt, D.N.; Huskens, J. Assembly of nanoparticles on patterned surfaces by non-covalent interactions. Curr. Opin. Colloid Interface Sci. 2008, 13, 74–80. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Magyar, C.; Gromiha, M.M.; Pujadas, G.; Tusnady, G.E.; Simon, I. SRide: A server for identifying stabilizing residues in proteins. Nucleic Acids Res. 2005, 33, W303–W305. [Google Scholar] [CrossRef]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Amide | π | ||||
|---|---|---|---|---|---|---|
| Number a | % b | Number a | % b | Nhot-spot c | %hot-spot d | |
| Backbone | 2086 | 100 | 0 | 0 | 0 | 0 |
| Side-chain | ||||||
| His | 0 | 0 | 12 | 0.58 | 0 | 0 |
| Phe | 0 | 0 | 885 | 42.42 | 744 | 45.7 |
| Trp | 0 | 0 | 9 | 0.43 | 0 | 0 |
| Tyr | 0 | 0 | 1180 | 56.57 | 884 | 54.3 |
| Total | 2086 | 100 | 2086 | 100 | 1628 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breberina, L.M.; Zlatović, M.V.; Stojanović, S.Đ.; Nikolić, M.R. Amide–π Interactions in the Structural Stability of Proteins: Role in the Oligomeric Phycocyanins. Computation 2024, 12, 172. https://doi.org/10.3390/computation12090172
Breberina LM, Zlatović MV, Stojanović SĐ, Nikolić MR. Amide–π Interactions in the Structural Stability of Proteins: Role in the Oligomeric Phycocyanins. Computation. 2024; 12(9):172. https://doi.org/10.3390/computation12090172
Chicago/Turabian StyleBreberina, Luka M., Mario V. Zlatović, Srđan Đ. Stojanović, and Milan R. Nikolić. 2024. "Amide–π Interactions in the Structural Stability of Proteins: Role in the Oligomeric Phycocyanins" Computation 12, no. 9: 172. https://doi.org/10.3390/computation12090172
APA StyleBreberina, L. M., Zlatović, M. V., Stojanović, S. Đ., & Nikolić, M. R. (2024). Amide–π Interactions in the Structural Stability of Proteins: Role in the Oligomeric Phycocyanins. Computation, 12(9), 172. https://doi.org/10.3390/computation12090172