
Combining Subtractive Genomics with Computer-Aided Drug Discovery Techniques to Effectively Target S. sputigena in Periodontitis


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
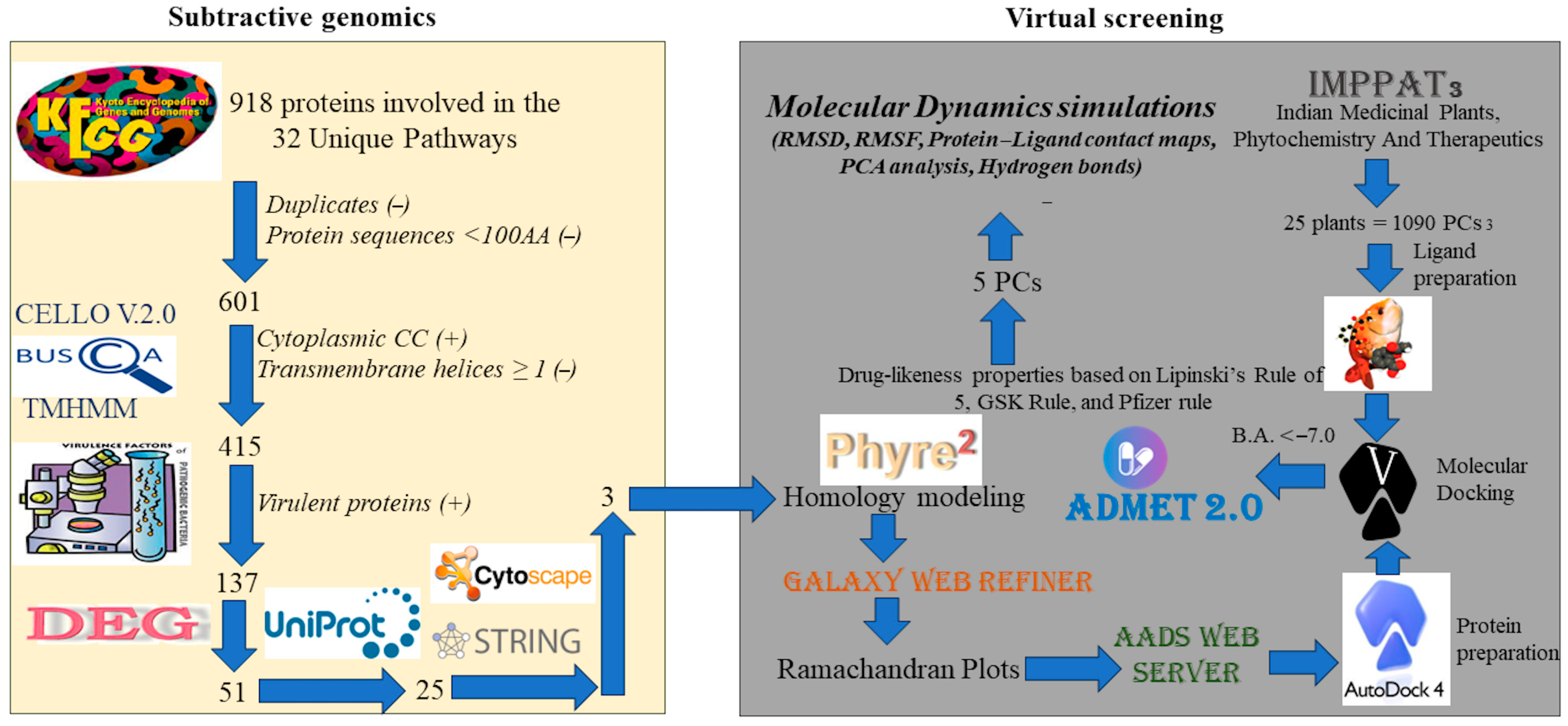
2.1. Subtractive Genomics
2.1.1. Identification of Unique Pathways
2.1.2. Identification of Protein Subcellular Localization
2.1.3. Identification of Protein Transmembrane Helices and Virulence Factors
2.1.4. Essential Gene Identification (DEG)
2.1.5. Identification of Non-Homologous Proteins
2.1.6. Protein–Protein Interaction (PPI) Network
2.2. In Silico Structure-Based Drug Discovery
2.2.1. Homology Modeling
2.2.2. Target-Based Virtual Screening
2.2.3. Molecular Dynamic Simulations
3. Results
3.1. Subtractive Genomics
3.1.1. Unique Pathways Identifications and Proteins Involved
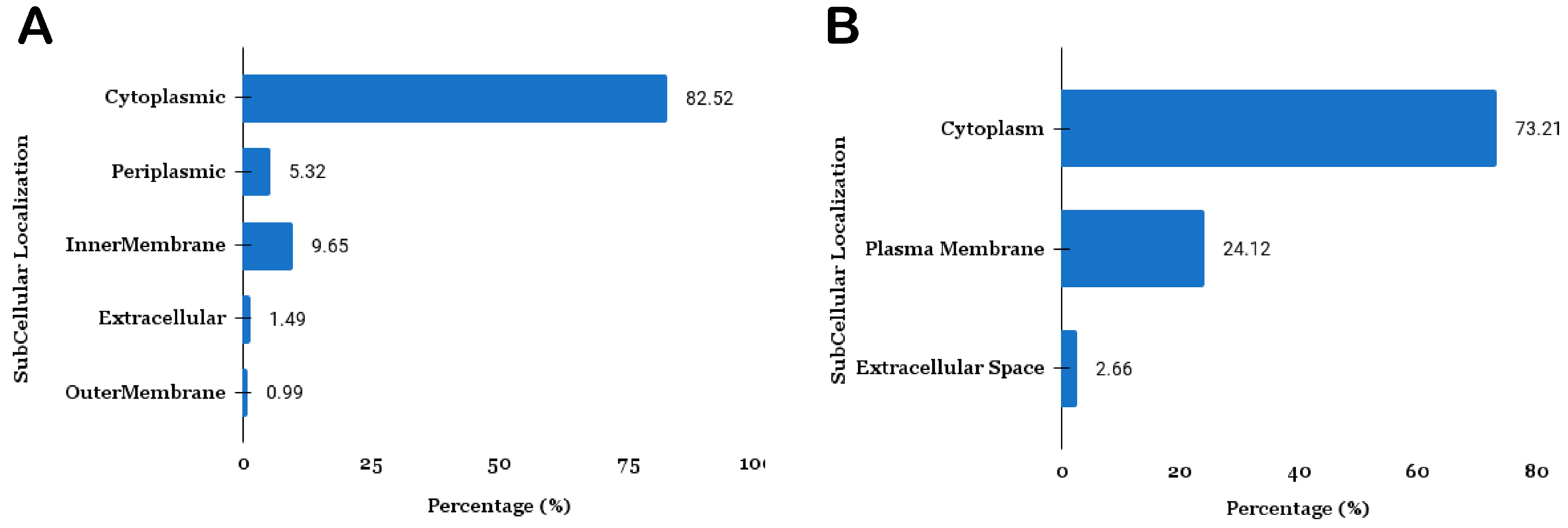
3.1.2. Subcellular Localization
3.1.3. Identification of Protein Transmembrane Helices and Virulent Factors
3.1.4. Essential Genes and Non-Homologous Protein Identification
3.1.5. Protein–Protein Interaction Analysis
3.2. Structure-Based Drug Discovery
3.2.1. Homology Modeling and Protein Optimization
3.2.2. Phytochemical Screening
3.2.3. Drug Likeness Properties
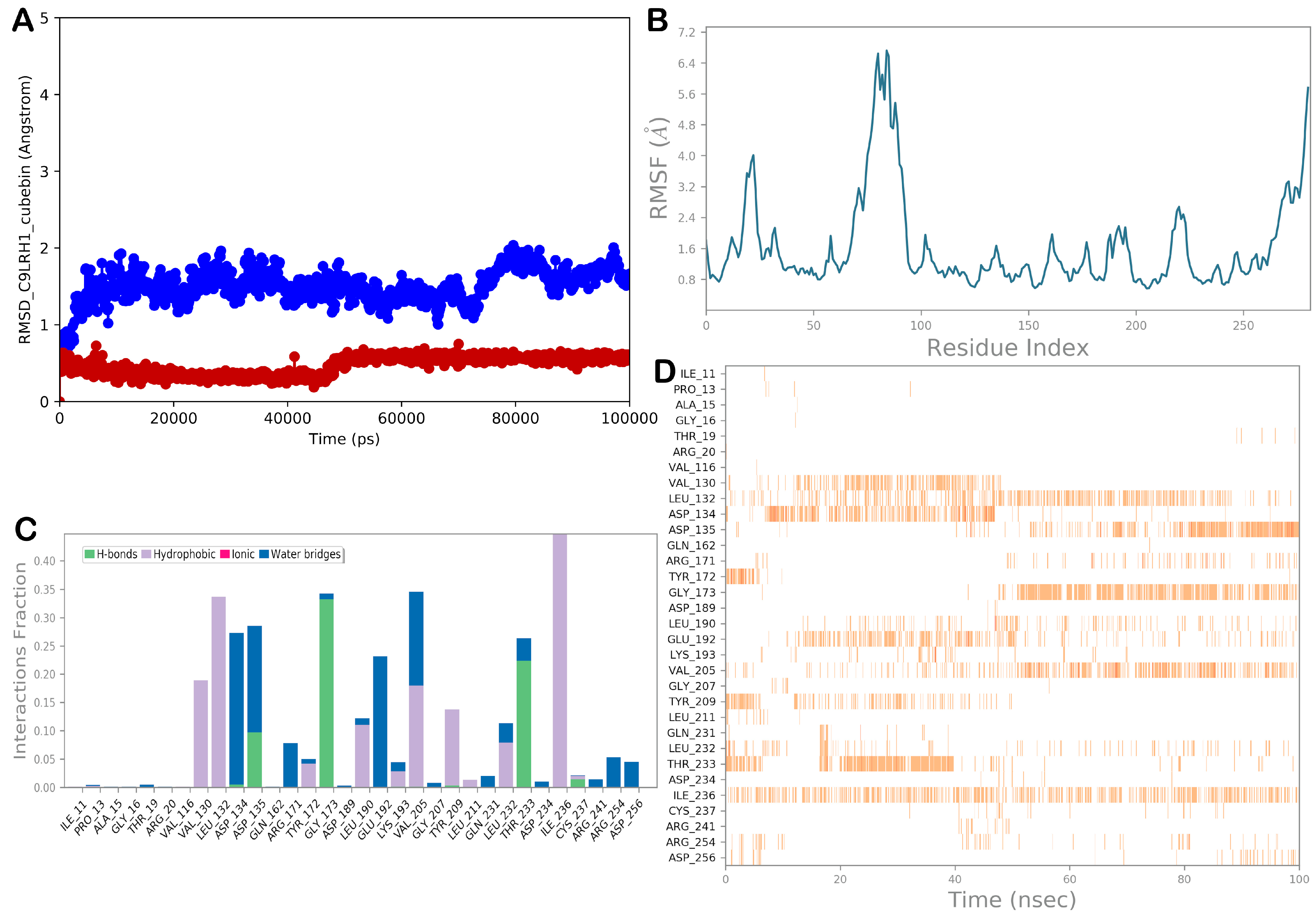
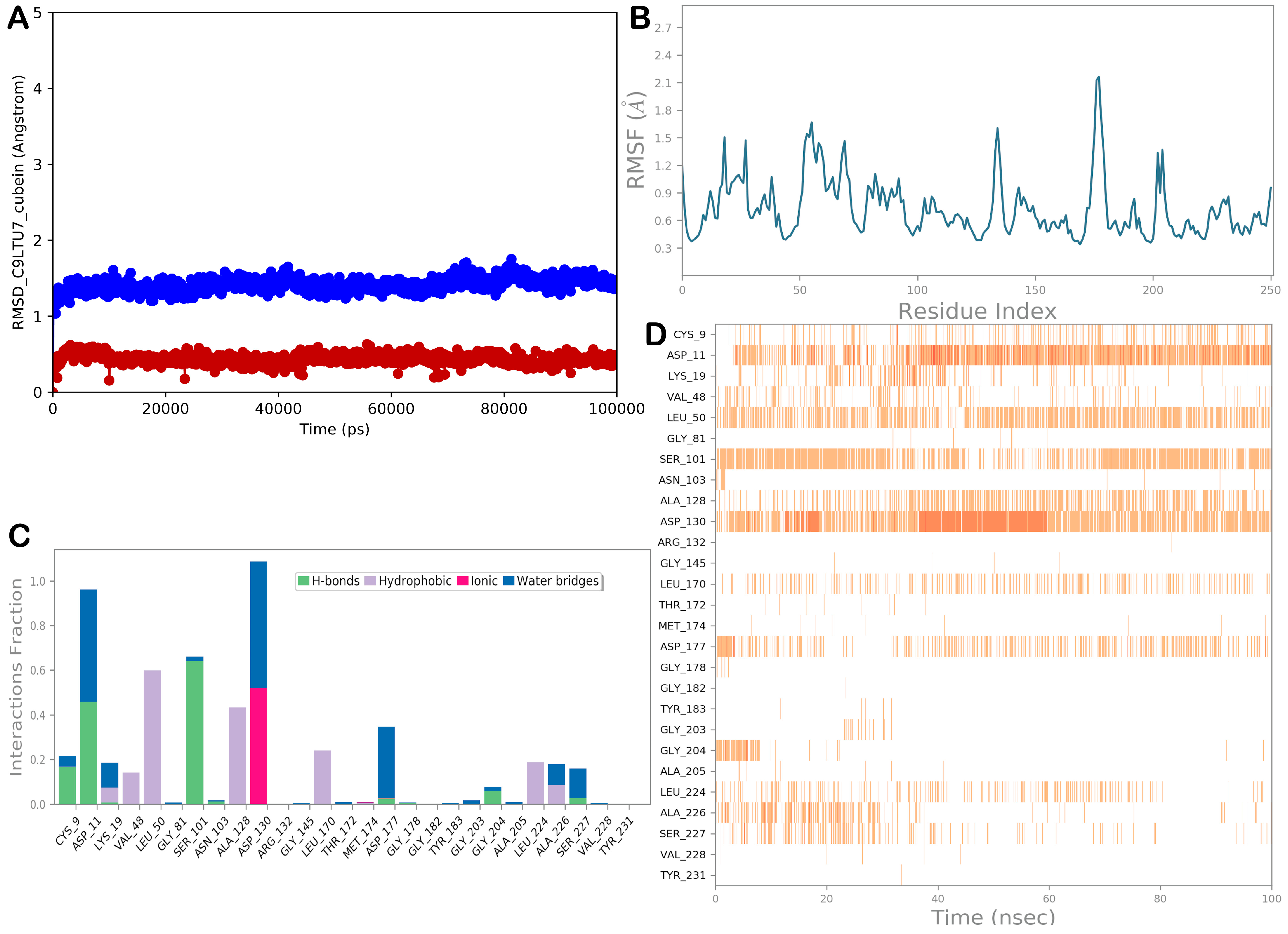
3.2.4. Molecular Dynamic Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rath, C.B.; Schirmeister, F.; Figl, R.; Seeberger, P.H.; Schaffer, C.; Kolarich, D. Flagellin Glycoproteomics of the Periodontitis Associated Pathogen Selenomonas sputigena Reveals Previously Not Described O-glycans and Rhamnose Fragment Rearrangement Occurring on the Glycopeptides. Mol. Cell. Proteom. 2018, 17, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Hiranmayi, K.V.; Sirisha, K.; Ramoji Rao, M.V.; Sudhakar, P. Novel Pathogens in Periodontal Microbiology. J. Pharm. Bioallied Sci. 2017, 9, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.F.; Karuthan, C.; Cheah, Y.K.; Ngeow, W.C.; Rosnah, Z.; Yap, S.F.; Ong, H.K.A. The oral microbiome community variations associated with normal, potentially malignant disorders and malignant lesions of the oral cavity. Malays. J. Pathol. 2017, 39, 1–15. [Google Scholar]
- Marsh, P.D.; Martin, M.V.; Lewis, M.A.O.; Williams, D. Oral Microbiology E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Praveen, M. Multi-epitope-based vaccine designing against Junín virus glycoprotein: Immunoinformatics approach. Futur. J. Pharm. Sci. 2024, 10, 29. [Google Scholar] [CrossRef]
- Lukic, D.; Karygianni, L.; Flury, M.; Attin, T.; Thurnheer, T. Endodontic-Like Oral Biofilms as Models for Multispecies Interactions in Endodontic Diseases. Microorganisms 2020, 8, 674. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, L.F.; Fermiano, D.; Feres, M.; Figueiredo, L.C.; Teles, F.R.; Mayer, M.P.; Faveri, M. Levels of Selenomonas species in generalized aggressive periodontitis. J. Periodontal Res. 2012, 47, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Kononen, E.; Gursoy, M.; Gursoy, U.K. Periodontitis: A Multifaceted Disease of Tooth-Supporting Tissues. J. Clin. Med. 2019, 8, 1135. [Google Scholar] [CrossRef] [PubMed]
- Al-Otaibi, M.; Al-Harthy, M.; Gustafsson, A.; Johansson, A.; Claesson, R.; Angmar-Mansson, B. Subgingival plaque microbiota in Saudi Arabians after use of miswak chewing stick and toothbrush. J. Clin. Periodontol. 2004, 31, 1048–1053. [Google Scholar] [CrossRef]
- Teughels, W.; Van Essche, M.; Sliepen, I.; Quirynen, M. Probiotics and oral healthcare. Periodontology 2000 2008, 48, 111–147. [Google Scholar] [CrossRef]
- Allaker, R.P.; Ian Douglas, C.W. Non-conventional therapeutics for oral infections. Virulence 2015, 6, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Abranches, J.; Zeng, L.; Kajfasz, J.K.; Palmer, S.R.; Chakraborty, B.; Wen, Z.T.; Richards, V.P.; Brady, L.J.; Lemos, J.A. Biology of Oral Streptococci. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Norskov-Lauritsen, N.; Claesson, R.; Birkeholm Jensen, A.; Aberg, C.H.; Haubek, D. Aggregatibacter Actinomycetemcomitans: Clinical Significance of a Pathobiont Subjected to Ample Changes in Classification and Nomenclature. Pathogens 2019, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D. The bacterial etiology of destructive periodontal disease: Current concepts. J. Periodontol. 1992, 63, 322–331. [Google Scholar] [CrossRef]
- Jantan, I.; Ahmad, W.; Bukhari, S.N. Plant-derived immunomodulators: An insight on their preclinical evaluation and clinical trials. Front. Plant Sci. 2015, 6, 655. [Google Scholar] [CrossRef]
- Azad, A.K.; Praveen, M.; Sulaiman, W.M.A.B.W. Assessment of Anticancer Properties of Plumbago zeylanica. In Harnessing Medicinal Plants in Cancer Prevention and Treatment; IGI Global Scientific Publishing: Hershey, PA, USA, 2024; pp. 91–121. [Google Scholar] [CrossRef]
- Kumar, R.; Mirza, M.A.; Naseef, P.P.; Kuruniyan, M.S.; Zakir, F.; Aggarwal, G. Exploring the Potential of Natural Product-Based Nanomedicine for Maintaining Oral Health. Molecules 2022, 27, 1725. [Google Scholar] [CrossRef]
- Ashraf, B.; Atiq, N.; Khan, K.; Wadood, A.; Uddin, R. Subtractive genomics profiling for potential drug targets identification against Moraxella catarrhalis. PLoS ONE 2022, 17, e0273252. [Google Scholar] [CrossRef] [PubMed]
- Uddin, R.; Siddiqui, Q.N.; Sufian, M.; Azam, S.S.; Wadood, A. Proteome-wide subtractive approach to prioritize a hypothetical protein of XDR-Mycobacterium tuberculosis as potential drug target. Genes. Genom. 2019, 41, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.A.O.; Knapp, R.B. BioTools: A Biosignal Toolbox for Composers and Performers. In Computer Music Modeling and Retrieval. Sense of Sounds. CMMR 2007; Lecture Notes in Computer Science, Volume 4969; Springer: Berlin/Heidelberg, Germany, 2008; pp. 441–452. [Google Scholar] [CrossRef]
- Javed, F.; Hayat, M. Predicting subcellular localization of multi-label proteins by incorporating the sequence features into Chou’s PseAAC. Genomics 2019, 111, 1325–1332. [Google Scholar] [CrossRef]
- Yu, C.S.; Lin, C.J.; Hwang, J.K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004, 13, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.; Dubey, S.; Munang’andu, H.M.; Karunasagar, I.; Karunasagar, I.; Evensen, O. Application of Outer Membrane Protein-Based Vaccines Against Major Bacterial Fish Pathogens in India. Front. Immunol. 2020, 11, 1362. [Google Scholar] [CrossRef]
- Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Combating tigecycline resistant Acinetobacter baumannii: A leap forward towards multi-epitope based vaccine discovery. Eur. J. Pharm. Sci. 2019, 132, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ou, H.Y.; Zhang, C.T. DEG: A database of essential genes. Nucleic Acids Res. 2004, 32, D271–D272. [Google Scholar] [CrossRef]
- Sarkar, M.; Maganti, L.; Ghoshal, N.; Dutta, C. In silico quest for putative drug targets in Helicobacter pylori HPAG1: Molecular modeling of candidate enzymes from lipopolysaccharide biosynthesis pathway. J. Mol. Model. 2012, 18, 1855–1866. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein-structure coordinates. Int. Tables Crystallogr. 2012, F, 684–687. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- El-Hachem, N.; Haibe-Kains, B.; Khalil, A.; Kobeissy, F.H.; Nemer, G. AutoDock and AutoDockTools for Protein-Ligand Docking: Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1(BACE1) as a Case Study. Methods Mol. Biol. 2017, 1598, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Praveen, M.; Ullah, I.; Buendia, R.; Khan, I.A.; Sayed, M.G.; Kabir, R.; Bhat, M.A.; Yaseen, M. Exploring Potentilla nepalensis Phytoconstituents: Integrated Strategies of Network Pharmacology, Molecular Docking, Dynamic Simulations, and MMGBSA Analysis for Cancer Therapeutic Targets Discovery. Pharmaceuticals 2024, 17, 134. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Biswas, D.; Jayaram, B. AADS--an automated active site identification, docking, and scoring protocol for protein targets based on physicochemical descriptors. J. Chem. Inf. Model. 2011, 51, 2515–2527. [Google Scholar] [CrossRef]
- Tsou, C.L. Active site flexibility in enzyme catalysis. Ann. N. Y. Acad. Sci. 1998, 864, 1–8. [Google Scholar] [CrossRef]
- Praveen, M.; Morales-Bayuelo, A. Drug Designing against VP4, VP7 and NSP4 of Rotavirus Proteins –Insilico studies. Mor. J. Chem. 2023, 11, 729–741. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Brogi, S.; Sirous, H.; Calderone, V.; Chemi, G. Amyloid beta fibril disruption by oleuropein aglycone: Long-time molecular dynamics simulation to gain insight into the mechanism of action of this polyphenol from extra virgin olive oil. Food Funct. 2020, 11, 8122–8132. [Google Scholar] [CrossRef]
- Brogi, S.; Guarino, I.; Flori, L.; Sirous, H.; Calderone, V. In Silico Identification of Natural Products and World-Approved Drugs Targeting the KEAP1/NRF2 Pathway Endowed with Potential Antioxidant Profile. Computation 2023, 11, 255. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 2002, 98, 6885–6892. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- da Silva, E.R.; Brogi, S.; Lucon-Junior, J.F.; Campiani, G.; Gemma, S.; Maquiaveli, C.D.C. Dietary polyphenols rutin, taxifolin and quercetin related compounds target Leishmania amazonensis arginase. Food Funct. 2019, 10, 3172–3180. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, B.; Sirous, H.; Brogi, S.; Calderone, V. Structure-Based High-Throughput Virtual Screening and Molecular Dynamics Simulation for the Discovery of Novel SARS-CoV-2 NSP3 Mac1 Domain Inhibitors. Viruses 2023, 15, 2291. [Google Scholar] [CrossRef]
- Yazdani, B.; Sirous, H.; Enguita, F.J.; Brogi, S.; Wing, P.A.C.; Fassihi, A. Discovery of novel direct small-molecule inhibitors targeting HIF-2alpha using structure-based virtual screening, molecular dynamics simulation, and MM-GBSA calculations. Mol. Divers. 2024, 28, 1203–1224. [Google Scholar] [CrossRef]
- Wehmeier, U.F.; Piepersberg, W. Enzymology of aminoglycoside biosynthesis-deduction from gene clusters. Methods Enzymol. 2009, 459, 459–491. [Google Scholar] [CrossRef] [PubMed]
- Samuel, G.; Reeves, P. Biosynthesis of O-antigens: Genes and pathways involved in nucleotide sugar precursor synthesis and O-antigen assembly. Carbohydr. Res. 2003, 338, 2503–2519. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn, F.; Maertens, J.; Beauprez, J.; Soetaert, W.; De Mey, M. Biotechnological advances in UDP-sugar based glycosylation of small molecules. Biotechnol. Adv. 2015, 33, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Ahangar, M.S.; Vyas, R.; Nasir, N.; Biswal, B.K. Structures of native, substrate-bound and inhibited forms of Mycobacterium tuberculosis imidazoleglycerol-phosphate dehydratase. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2461–2467. [Google Scholar] [CrossRef]
- Lonergan, Z.R.; Palmer, L.D.; Skaar, E.P. Histidine Utilization Is a Critical Determinant of Acinetobacter Pathogenesis. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [PubMed]
- Alkatheri, A.H.; Yap, P.S.; Abushelaibi, A.; Lai, K.S.; Cheng, W.H.; Erin Lim, S.H. Microbial Genomics: Innovative Targets and Mechanisms. Antibiotics 2023, 12, 190. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.L.; Coimbra, H.S.; Pereira, A.C.; Almeida, V.A.; Lima, T.C.; Costa, E.S.; Vinholis, A.H.; Royo, V.A.; Silva, R.; Filho, A.A.; et al. Evaluation of piper cubeba extract, (-)-cubebin and its semi-synthetic derivatives against oral pathogens. Phytother. Res. 2007, 21, 420–422. [Google Scholar] [CrossRef]
- Rajalekshmi, D.S.; Kabeer, F.A.; Madhusoodhanan, A.R.; Bahulayan, A.K.; Prathapan, R.; Prakasan, N.; Varughese, S.; Nair, M.S. Anticancer activity studies of cubebin isolated from Piper cubeba and its synthetic derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Daliri, E.B.; Kim, N.; Kim, J.R.; Yoo, D.; Oh, D.H. Microbial Etiology and Prevention of Dental Caries: Exploiting Natural Products to Inhibit Cariogenic Biofilms. Pathogens 2020, 9, 569. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.I.; Ha, J.Y.; Min, K.R.; Nakagawa, H.; Tsurufuji, S.; Chang, I.M.; Kim, Y. Inhibitory effects of Oriental herbal medicines on IL-8 induction in lipopolysaccharide-activated rat macrophages. Planta Med. 1995, 61, 26–30. [Google Scholar] [CrossRef]
- Ware, I.; Franke, K.; Dube, M.; Ali El Enshasy, H.; Wessjohann, L.A. Characterization and Bioactive Potential of Secondary Metabolites Isolated from Piper sarmentosum Roxb. Int. J. Mol. Sci. 2023, 24, 1328. [Google Scholar] [CrossRef] [PubMed]
- Zoric, N.; Kosalec, I.; Tomic, S.; Bobnjaric, I.; Jug, M.; Vlainic, T.; Vlainic, J. Membrane of Candida albicans as a target of berberine. BMC Complement. Altern. Med. 2017, 17, 268. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt ID | Description | Localization 1,2 | TM Helices 3 | Virulent 4 | Essential 5 | Non-Homologous 6 |
|---|---|---|---|---|---|---|
| C9LY48 | Histidinol-phosphate aminotransferase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LUR0 | dTDP-4-dehydrorhamnose 3,5-epimerase | Cytoplasmic | 0 | Yes | Yes | Yes |
| F4EVW5 | KpsF/GutQ family protein | Cytoplasmic | 0 | Yes | Yes | Yes |
| F4EYF8 | ADP-L-glycero-D-manno-heptose-6-epimerase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LRN8 | 3-deoxy-manno-octulosonate cytidylyltransferase | Cytoplasmic | 0 | Yes | Yes | Yes |
| F4EW90 | 2-dehydro-3-deoxyphosphooctonate aldolase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LVT5 | Nucleotide sugar dehydrogenase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LXZ9 | DegT/DnrJ/EryC1/StrS aminotransferase | Cytoplasmic | 0 | Yes | Yes | Yes |
| F4EYU7 | Glucose-1-phosphate cytidylyltransferase | Cytoplasmic | 0 | Yes | Yes | Yes |
| F4EVY9 | Bifunctional protein GlmU | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LRH0 | Phosphoglucomutase/phosphomannomutase alpha/beta/alpha domain I | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LRH1 | UTP-glucose-1-phosphate uridylyltransferase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LS71 | UDP-N-acetylglucosamine 2-epimerase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LXV5 | Aspartate 1-decarboxylase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LTU8 | 1-(5-phosphoribosyl)-5-[(5-phosphoribosylamino) methylideneamino]imidazole-4-carboxamide isomerase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LTU7 | Imidazole glycerol phosphate synthase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LT30 | RelA/SpoT domain protein | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LRN0 | 3-hydroxyacyl-[acyl-carrier-protein] dehydratase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LSI7 | Anthranilate synthase glutamine amidotransferase | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LSQ0 | Response regulator receiver domain protein | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LYT4 | CheB methylesterase | Cytoplasmic | 0 | Yes | Yes | Yes |
| F4EXR6 | Methyl-accepting chemotaxis sensory transducer | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LTV8 | Signal recognition particle protein | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LW81 | Signal recognition particle receptor FtsY | Cytoplasmic | 0 | Yes | Yes | Yes |
| C9LTA9 | RNA polymerase sigma factor SigA | Cytoplasmic | 0 | Yes | Yes | Yes |
| UniProt ID | Coverage (%) | Identity (%) | Residues in Favored Regions (%, Ramachandran Plot) | |
|---|---|---|---|---|
| Before Optimization | After Optimization | |||
| C9LUR0 | 94 | 56 | 89.8 | 95.3 |
| C9LRH1 | 92 | 63 | 84.6 | 93.1 |
| C9LTU7 | 99 | 63 | 89.3 | 95.8 |
| Plant Name | Plant Part | Phytochemical Name | C9LUR0 | C9LRH1 | C9LTU7 |
|---|---|---|---|---|---|
| Azadirachta indica | Bark | (4bS,8aR)-2,4b,8,8-tetramethyl-7,10-dioxo-5,6,8a,9-tetrahydrophenanthrene-3-carboxylic acid (IMPHY005303) | −7.2 | −8.6 | −8.3 |
| Commiphora wightii | plant exudate | Pluviatilol (IMPHY006624) | −7.0 | −8.3 | −8.0 |
| 5-[3-(1,3-Benzodioxol-5-yl)-1,3,3a,4,6,6a-hexahydrofuro [3,4-c]furan-6-yl]-1,3-benzodioxole (IMPHY014895) | −7.6 | −8.7 | −9.0 | ||
| Jatropha gossypiifolia | stem | Gadain (IMPHY004244) | −7.1 | −8.6 | −8.2 |
| Mimusops elengi | bark | Cubebin (IMPHY001912) | −7.1 | −9.0 | −9.1 |
| Phytochemical Name | MW | HBA | HBD | LogP | TPSA | QPPCaco | QPPMDCK |
|---|---|---|---|---|---|---|---|
| (4bS,8aR)-2,4b,8,8-tetramethyl-7,10-dioxo-5,6,8a,9-tetrahydrophenanthrene-3-carboxylic acid (IMPHY005303) | 314.15 | 4 | 1 | 3.33 | 71.44 | 49 | 24 |
| Pluviatilol (IMPHY006624) | 356.13 | 6 | 1 | 3.21 | 66.38 | 3204 | 1741 |
| 5-[3-(1,3-Benzodioxol-5-yl)-1,3,3a,4,6,6a-hexahydrofuro [3,4-c]furan-6-yl]-1,3-benzodioxole (IMPHY014895) | 354.11 | 6 | 0 | 3.06 | 55.38 | 9906 | 5899 |
| Gadain (IMPHY004244) | 352.09 | 6 | 0 | 3.99 | 63.22 | 3756 | 2068 |
| Cubebin (IMPHY001912) | 356.13 | 6 | 1 | 3.60 | 66.38 | 4681 | 2623 |
| Complex | ΔGvdw a (kcal/mol) | ΔGcoul b (kcal/mol) | ΔGHbond c (kcal/mol) | ΔGLipo d (kcal/mol) | ΔGPack e (kcal/mol) | ΔGSolGB f (kcal/mol) | ΔGbind g (kcal/mol) |
|---|---|---|---|---|---|---|---|
| C9LRH1/IMPHY001912 | −50.10 | −37.66 | −1.91 | −28.16 | −1.56 | 26.04 | −68.89 |
| C9LTU7/IMPHY001912 | −49.12 | −26.19 | −2.89 | −22.34 | −2.17 | 29.73 | −64.73 |
| C9LUR0/IMPHY001912 | −22.87 | −10.38 | −1.03 | −11.27 | −1.02 | 25.88 | −27.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Praveen, M.; Sree, C.G.; Brogi, S.; Calderone, V.; Dalei, K.P.K.P. Combining Subtractive Genomics with Computer-Aided Drug Discovery Techniques to Effectively Target S. sputigena in Periodontitis. Computation 2025, 13, 34. https://doi.org/10.3390/computation13020034
Praveen M, Sree CG, Brogi S, Calderone V, Dalei KPKP. Combining Subtractive Genomics with Computer-Aided Drug Discovery Techniques to Effectively Target S. sputigena in Periodontitis. Computation. 2025; 13(2):34. https://doi.org/10.3390/computation13020034
Chicago/Turabian StylePraveen, Mallari, Chendruru Geya Sree, Simone Brogi, Vincenzo Calderone, and Kamakshya Prasad Kanchan Prava Dalei. 2025. "Combining Subtractive Genomics with Computer-Aided Drug Discovery Techniques to Effectively Target S. sputigena in Periodontitis" Computation 13, no. 2: 34. https://doi.org/10.3390/computation13020034
APA StylePraveen, M., Sree, C. G., Brogi, S., Calderone, V., & Dalei, K. P. K. P. (2025). Combining Subtractive Genomics with Computer-Aided Drug Discovery Techniques to Effectively Target S. sputigena in Periodontitis. Computation, 13(2), 34. https://doi.org/10.3390/computation13020034