
Grand Canonical Monte Carlo Simulation of Nitrogen Adsorption in a Silica Aerogel Model

Abstract
:1. Introduction
2. Physical and Mathematical Model
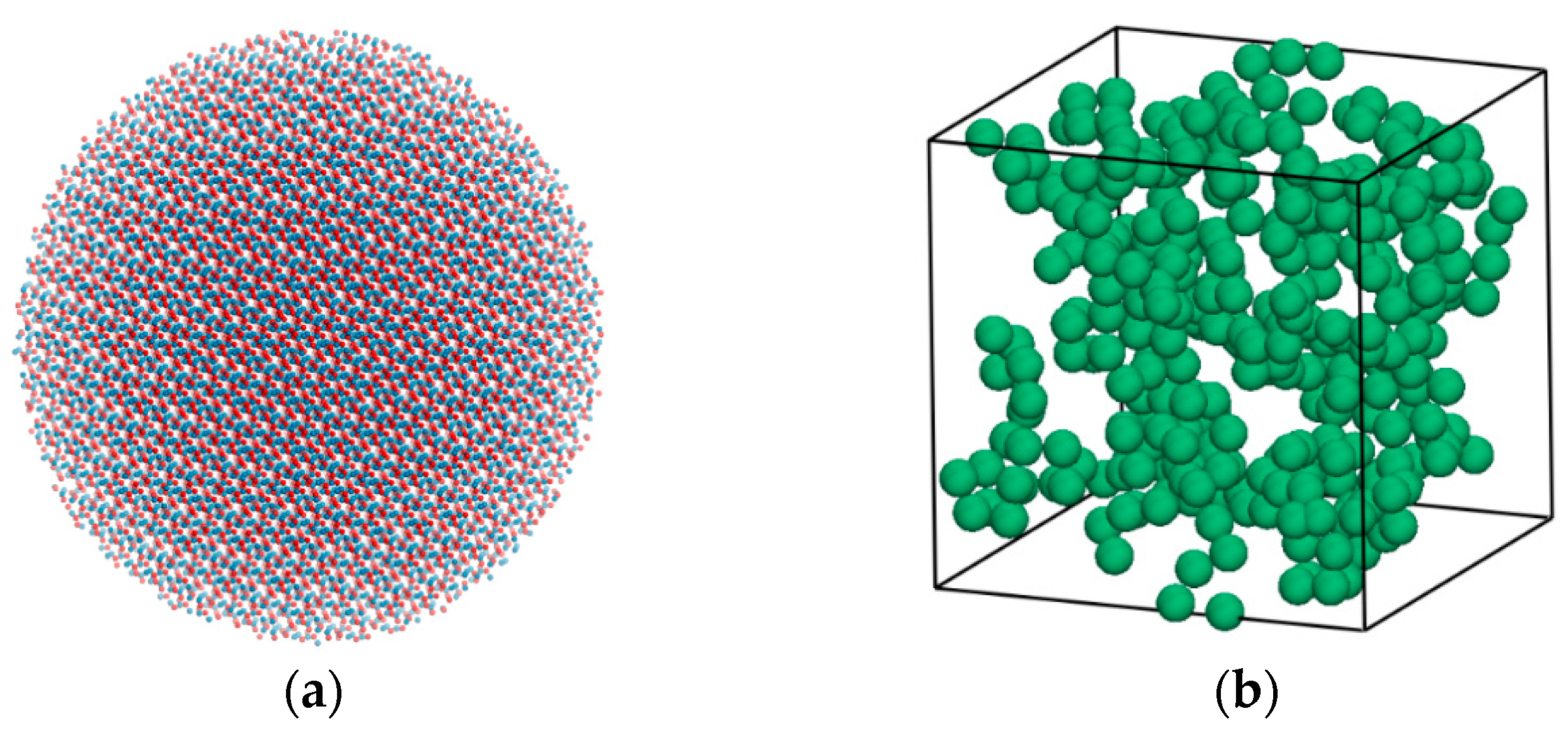
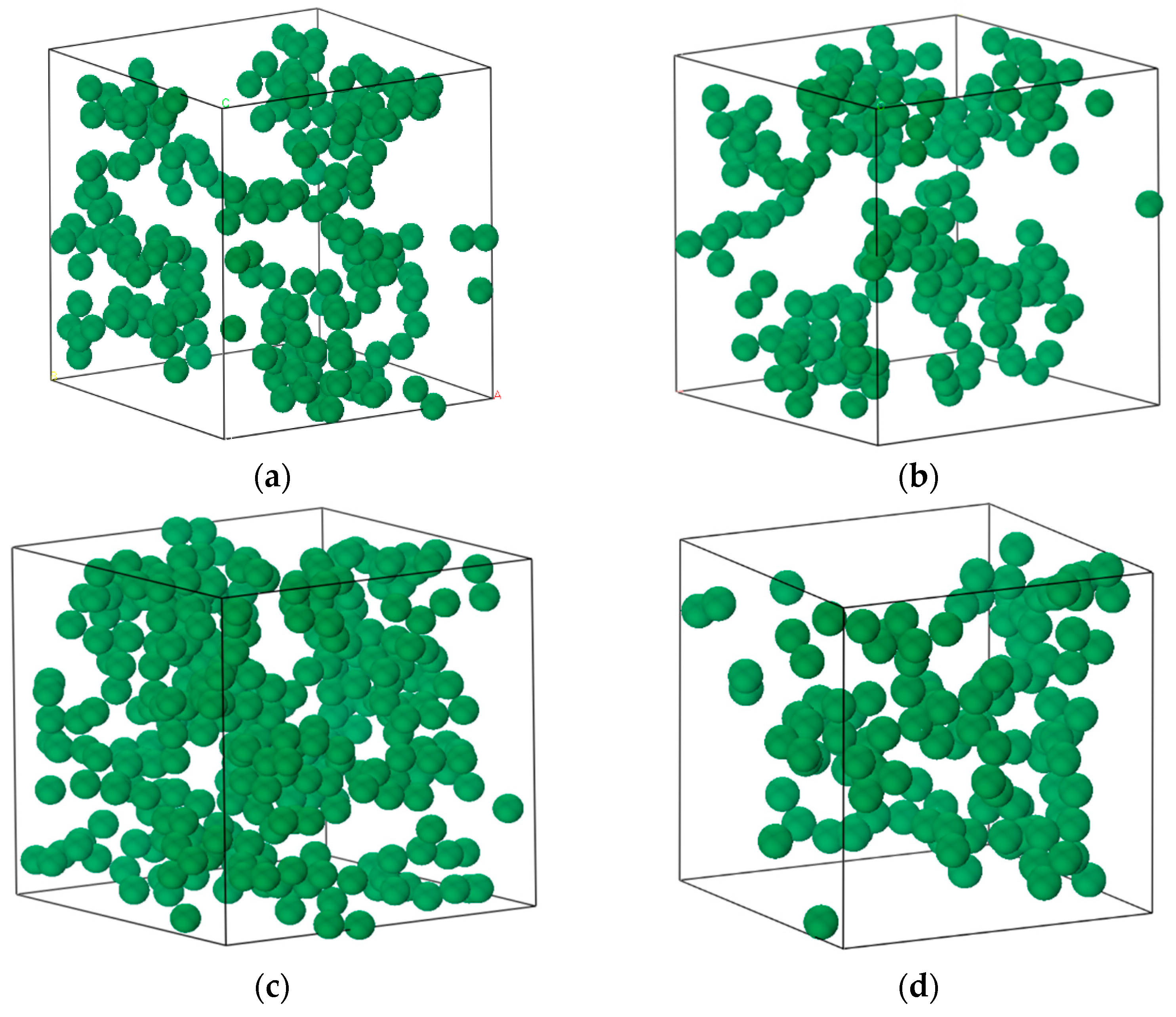
2.1. Physical Model
2.2. GCMC Method
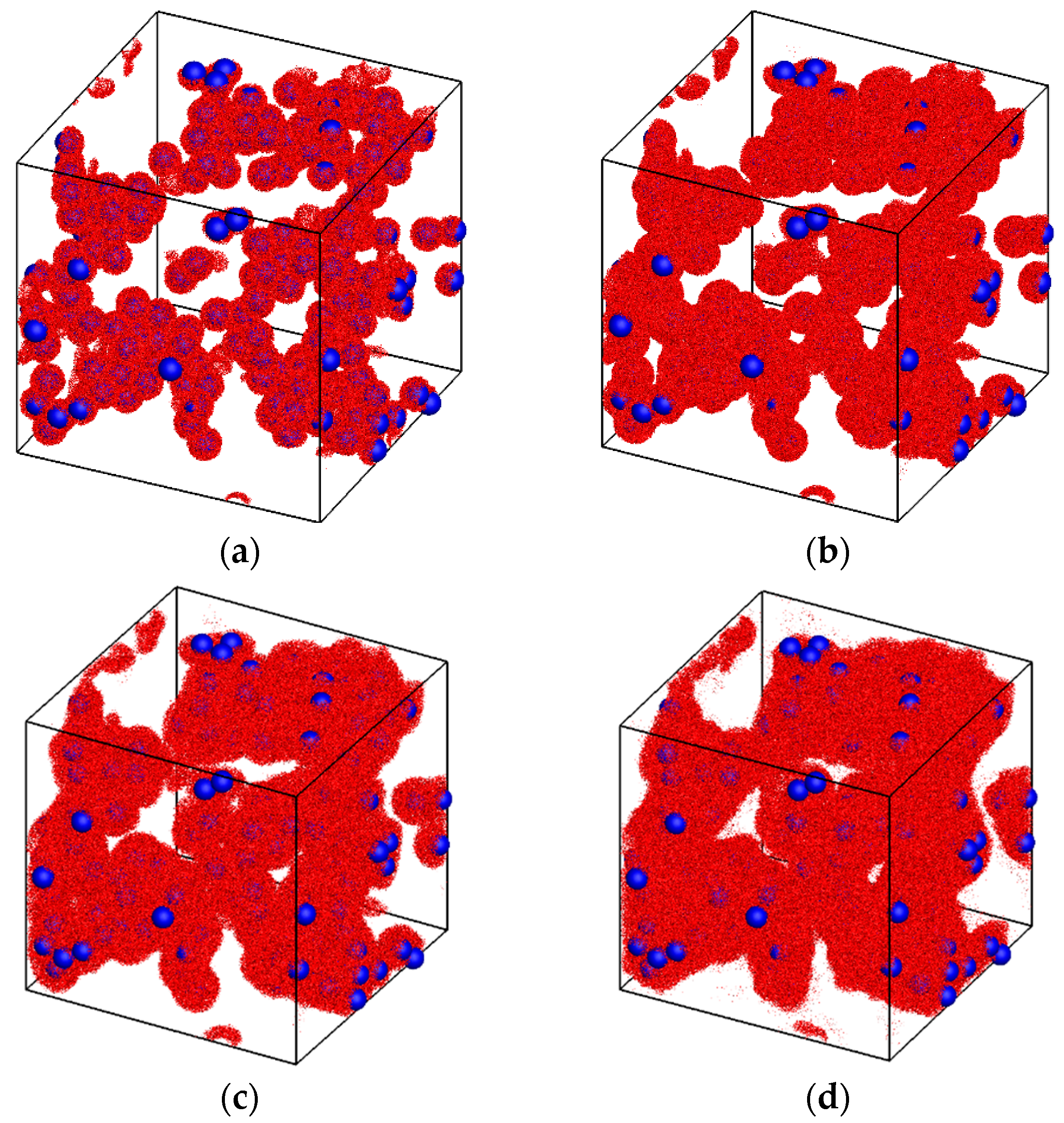
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baetens, R.; Jelle, B.P.; Gustavsen, A. Aerogel insulation for building applications: A state-of-the-art review. Energy Build. 2011, 19, 761–769. [Google Scholar] [CrossRef]
- Smirnova, I.; Mamic, J.; Arlt, W. Adsorption of drugs on silica aerogels. Langmuir 2003, 19, 8521–8525. [Google Scholar] [CrossRef]
- Standeker, S.; Novak, Z.; Knez, Z. Adsorption of toxic organic compounds from water with hydrophobic silica aerogels. J. Colloid Interface Sci. 2007, 310, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Adebajo, M.O.; Frost, R.L.; Kloprogge, J.T.; Carmody, O.; Kokot, S. Porous materials for oil spill cleanup: A review of synthesis and absorbing properties. J. Porous Mater. 2003, 10, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, Z.Y.; Zhao, X.P.; Tao, W.Q. Investigation of the effect of the gas permeation induced by pressure gradient on transient heat transfer in silica aerogel. Int. J. Heat Mass Transf. 2016, 95, 1026–1037. [Google Scholar] [CrossRef]
- Lee, J.A.; Mounce, A.M.; Oh, S.; Zimmerman, A.M.; Halperin, W.P. Enhanced self-diffusion of adsorbed methanol in silica aerogel. Phys. Rev. B 2014, 90, 174501. [Google Scholar] [CrossRef]
- Sun, C.; Boutilier, M.S.; Au, H.; Poesio, P.; Bai, B.; Karnik, R.; Hadjiconstantinou, N.G. Mechanisms of molecular permeation through nanoporous graphene membranes. Langmuir 2014, 30, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Deng, W.Q.; Goddard, W.A. Improved designs of metal-organic frameworks for hydrogen storage. Angew. Chem. 2007, 119, 6405–6408. [Google Scholar] [CrossRef]
- Düren, T.; Bae, Y.S.; Snurr, R.Q. Using molecular simulation to characterise metal-organic frameworks for adsorption applications. Chem. Soc. Rev. 2009, 38, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Mpourmpakis, G.; Froudakis, G.E.; Lithoxoos, G.P.; Samios, J. SiC nanotubes: A novel material for hydrogen storage. Nano. Lett. 2006, 6, 1581–1583. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Grajciar, L.; Nachtigall, P.; Düren, T. Accurate prediction of methane adsorption in a metal-organic framework with unsaturated metal sites by direct implementation of an ab initio derived potential energy surface in GCMC simulation. J. Phys. Chem. C 2011, 115, 23074–23080. [Google Scholar] [CrossRef]
- Ravikovitch, P.I.; Vishnyakov, A.; Russo, R.; Neimark, A.V. Unified approach to pore size characterization of microporous carbonaceous materials from N2, Ar, and CO2 adsorption isotherms. Langmuir 2000, 16, 2311–2320. [Google Scholar] [CrossRef]
- Ravikovitch, P.I.; Wei, D.; Chueh, W.T.; Haller, G.L.; Neimark, A.V. Evaluation of pore structure parameters of MCM-41 catalyst supports and catalysts by means of nitrogen and argon adsorption. J. Phys. Chem. B 1997, 101, 3671–3679. [Google Scholar] [CrossRef]
- Ohba, T.; Kaneko, K. Internal surface area evaluation of carbon nanotube with GCMC simulation-assisted N2 adsorption. J. Phys. Chem. B 2002, 106, 7171–7176. [Google Scholar] [CrossRef]
- MacElroy, J.M.D.; Raghavan, K. Adsorption and diffusion of a Lennard-Jones vapor in microporous silica. J. Chem. Phys. 1990, 93, 2068–2079. [Google Scholar] [CrossRef]
- Ulker, Z.; Erucar, I.; Keskin, S.; Erkey, C. Novel nanostructured composites of silica aerogels with a metal organic framework. Microporous Mesop. Mater. 2013, 170, 52–358. [Google Scholar] [CrossRef]
- Le, T.; Striolo, A.; Cole, D.R. Propane simulated in silica pores: Adsorption isotherms, molecular structure, and mobility. Chem. Eng. Sci. 2015, 121, 292–299. [Google Scholar] [CrossRef]
- Gavalda, S.; Gubbins, K.E.; Hanzawa, Y.; Kaneko, K.; Thomson, K.T. Nitrogen adsorption in carbon aerogels: A molecular simulation study. Langmuir 2002, 18, 2141–2151. [Google Scholar] [CrossRef]
- Zhang, G.; Dass, A.; Rawashdeh, A.M.M.; Thomas, J.; Counsil, J.A.; Sotiriou-Leventis, C.; Fabrizio, E.F.; Ilhan, F.; Vassilaras, P.; Scheiman, D.A.; et al. Isocyanate-crosslinked silica aerogel monoliths: Preparation and characterization. J. Non-Cryst. Solids 2004, 350, 152–164. [Google Scholar] [CrossRef]
- Zeng, S.Q.; Hunt, A.; Greif, R. Geometric structure and thermal conductivity of porous medium silica aerogel. J. Heat Transf. 1995, 117, 1055–1058. [Google Scholar] [CrossRef]
- Park, I.-A.; MacElroy, J.M.D. Simulation of a hard-sphere fluid in bicontinuous random media. Mol. Simul. 1989, 2, 105–145. [Google Scholar] [CrossRef]
- Schaefer, D.W.; Keefer, K.D. Structure of random porous materials: Silica aerogel. Phys. Rev. Lett. 1986, 56, 2199–2202. [Google Scholar] [CrossRef] [PubMed]
- Paul, M. Formation of fractal clusters and networks by irreversible diffusion-limited aggregation. Phys. Rev. Lett. 1983, 51, 1119–1122. [Google Scholar]
- Good, B. Structure and thermal conductivity of silica aerogels from computer simulations. MRS Proc. 2005, 885. [Google Scholar] [CrossRef]
- Hou, T.J.; Zhu, L.L.; Li, Y.Y.; Xu, X.J. The localization and adsorption of benzene and propylene in ITQ-1 zeolite: Grand canonical Monte Carlo simulations. J. Mol. Struct. Theochem 2001, 535, 9–23. [Google Scholar] [CrossRef]
- Makrodimitris, K.; Papadopoulos, G.K.; Theodorou, D.N. Prediction of permeation properties of CO2 and N2 through silicalite via molecular simulations. J. Phys. Chem. B 2001, 105, 777–788. [Google Scholar] [CrossRef]
- Kaminsky, R.D.; Monson, P.A. The influence of adsorbent microstructure upon adsorption equilibria: Investigations of a model system. J. Phys. Chem. 1991, 95, 2936–2948. [Google Scholar] [CrossRef]
- El Rassy, H.; Buisson, P.; Bouali, B.; Perrard, A.; Pierre, A.C. Surface characterization of silica aerogels with different proportions of hydrophobic groups, dried by the CO2 supercritical method. Langmuir 2003, 19, 358–363. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Value | ||||
|---|---|---|---|---|---|
| Experimental Study [13] | M1 | M2 | M3 | M4 | |
| Porosity (%) | 94 | 94.00 | 94.00 | 89.70 | 93.46 |
| Specific Surface (m2/g) | 837 | 930 | 930 | 895 | 547 |
| Sphere Radius (nm) | / | 4.05 | 4.05 | 4.15 | 5.00 |
| Number of Particles | / | 216 | 216 | 343 | 125 |
| Atom | ||
|---|---|---|
| N2 | 0.375 | 95.20 |
| N2 (dimer) | 0.330 | 36.20 |
| O | 0.330 | 82.98 |
| Si | 0.420 | 23.61 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, W.-L.; Chen, Z.-J.; Li, Z.Y.; Tao, W.-Q. Grand Canonical Monte Carlo Simulation of Nitrogen Adsorption in a Silica Aerogel Model. Computation 2016, 4, 18. https://doi.org/10.3390/computation4020018
Xie W-L, Chen Z-J, Li ZY, Tao W-Q. Grand Canonical Monte Carlo Simulation of Nitrogen Adsorption in a Silica Aerogel Model. Computation. 2016; 4(2):18. https://doi.org/10.3390/computation4020018
Chicago/Turabian StyleXie, Wen-Li, Zheng-Ji Chen, Zeng Yao Li, and Wen-Quan Tao. 2016. "Grand Canonical Monte Carlo Simulation of Nitrogen Adsorption in a Silica Aerogel Model" Computation 4, no. 2: 18. https://doi.org/10.3390/computation4020018
APA StyleXie, W.-L., Chen, Z.-J., Li, Z. Y., & Tao, W.-Q. (2016). Grand Canonical Monte Carlo Simulation of Nitrogen Adsorption in a Silica Aerogel Model. Computation, 4(2), 18. https://doi.org/10.3390/computation4020018