1. Introduction
In recent decades, advancements in the field of biosensing have precipitated the development of point-of-care (PoC) technologies for the rapid analysis of a whole host of molecules [
1,
2]. The main driving force behind these marvels lies in the advancement of the receptor materials utilized in the transduction of a biological/chemical target into a tangible output signal [
3]. Molecularly imprinted polymers (MIPs) are a primary example of a developed synthetic receptor material, consisting of a highly crosslinked polymeric network with selective nanocavities engineered for the binding of an analyte distributed throughout [
4,
5]. These synthetic alternatives to biological recognition elements demonstrate enhanced resistance to extreme physical conditions and can operate in a wide range of scenarios, which is highlighted in the increasing number of applications that have been linked to these synthetic wonders [
6].
Traditionally, MIPs have been developed through a time-consuming trial-and-error methodology consisting of altering the chemical compositions and ratios of the monomer and crosslinker species to achieve desired results [
7,
8]. The same tedium exists for determining the performance of a MIP, having to expose it to countless molecular moieties to truly determine the selective capabilities of the polymer [
9,
10]. Due to the nature of PoC analysis, this testing process is vital, though traditional approaches have proven costly and protracted. The potential to lessen these constrictors does exist and lies within the realms of data science, more commonly referred to as computational chemistry. Many chemical processes have already benefited from the implementation of diverse algorithms, revolutionizing the fields of retrosynthesis, reaction optimization, and drug design [
11,
12,
13]. Computational methods have also been applied to molecularly imprinted polymers, though they focus on the compositional nature rather than the binding of various analytes [
14,
15,
16,
17].
One computational tool that can aid the development of MIPs is machine learning, and in particular the use of neural networks (NN). This kind of machine learning methodology is most commonly applied in the fields of chemo-informatics, enabling the prediction of the physical and chemical properties of molecules, or even how a compound is likely to react [
18,
19,
20]. As more datasets on molecules have become available, the use of NN has become increasingly more popular, with this approach offering more reliable chemical predictions [
21,
22]. This success can be derived from the different NN architectures possible, enabling chemical information to be layered and processed in numerous ways that allow new trends and relationships to be perceived [
23]. The dataset and desired outcomes of analysis dictate which model is selected, though the selection may not have an obvious choice and requires investigation.
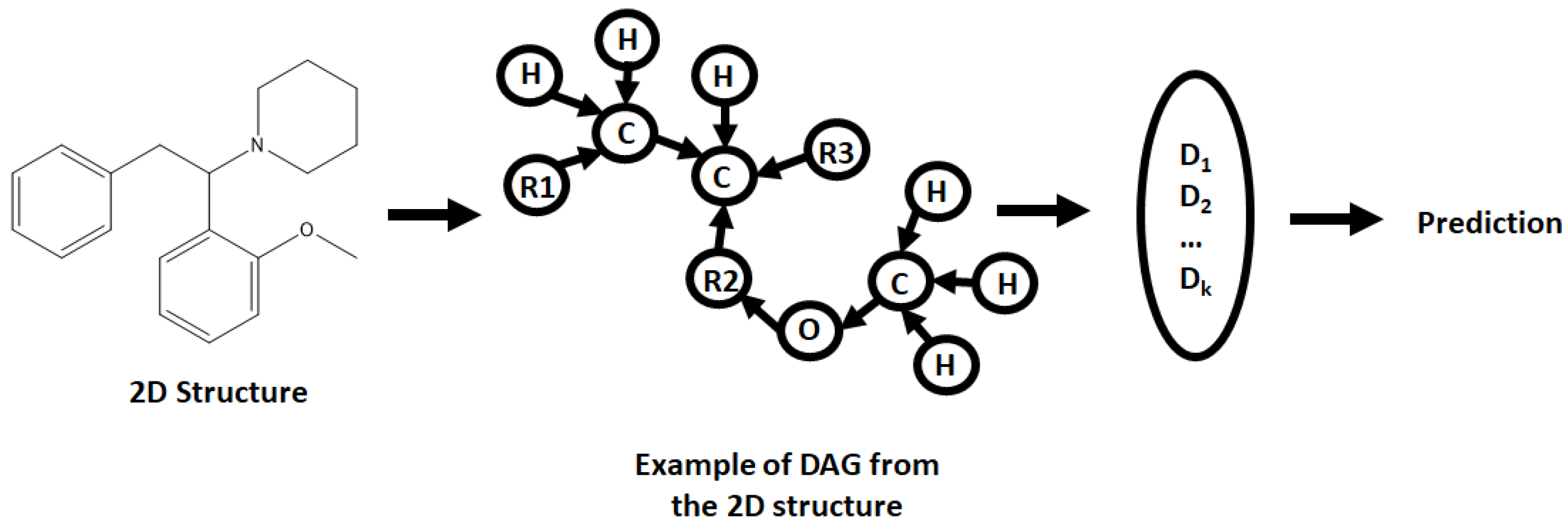
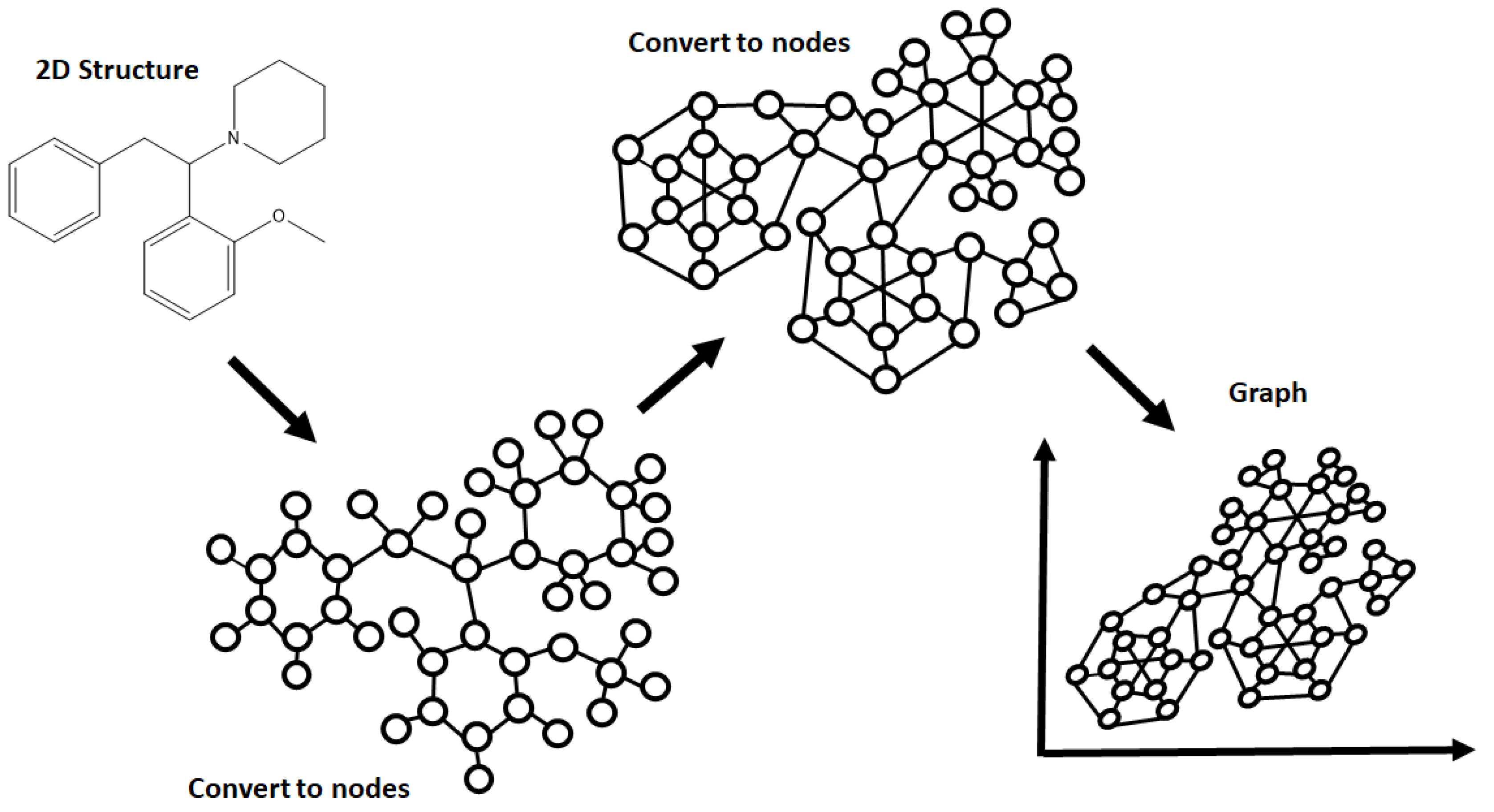
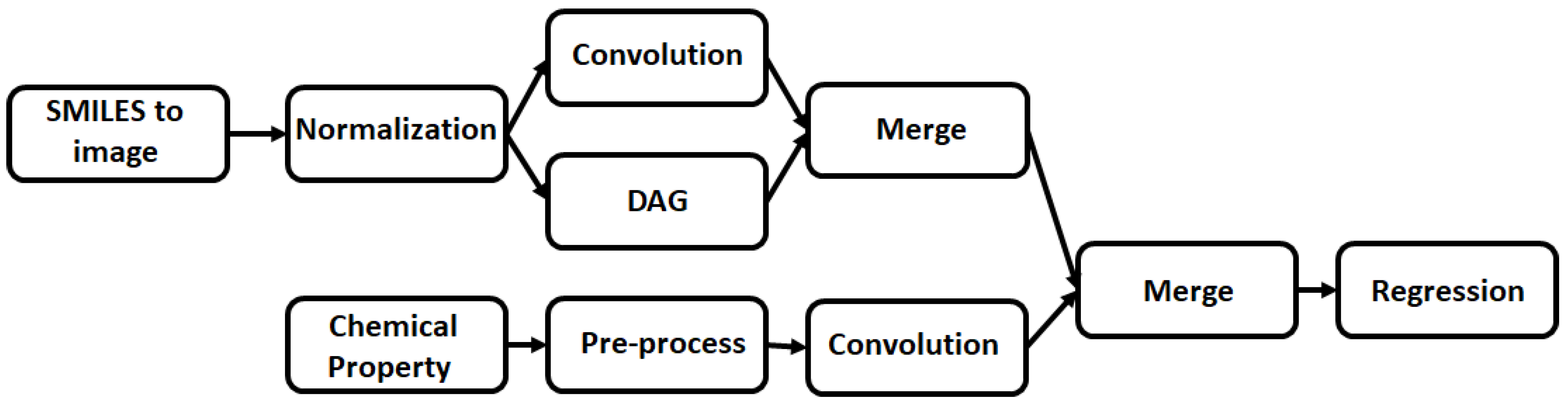
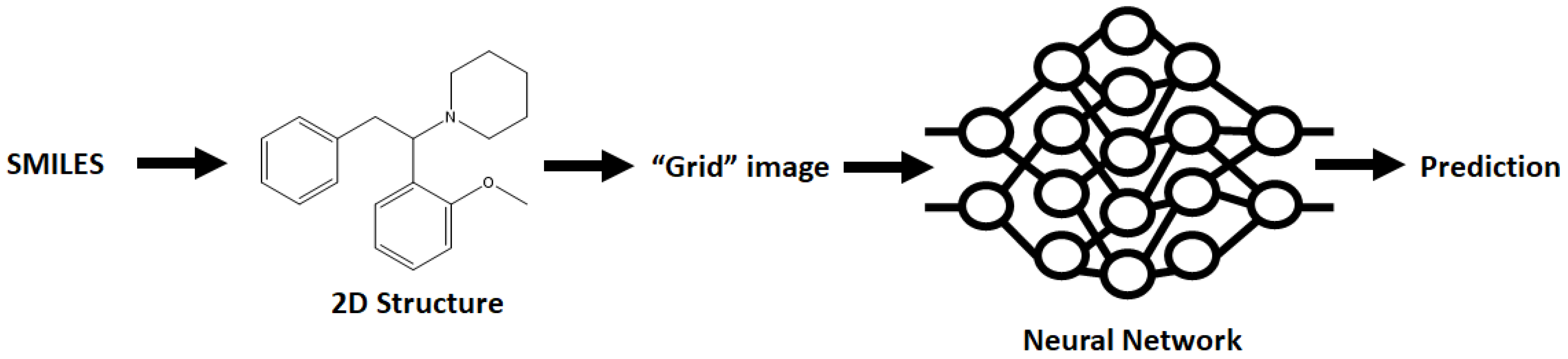
A wide range of NN architectures has been documented, including Directed Acyclic Graphs (DAG), Spatial Graph convolution, Multitask Regressors, and image classification models, to name just a few [
24,
25,
26,
27]. Each has a unique manner in which it processes data, though all require molecular structures and other variables to be preprocessed prior to analysis. This preprocessing is known as featurizing, a process that can also affect the way the model selected interprets the data [
28,
29]. Molecular structures are featurized by converting them into Simplified Molecular Input Line Entry Specification (SMILES), transforming a molecular structure into a line of text that the models can interpret [
30]. The manner in which the rest of the data (including the SMILES) is featurized depends on the model selected, adding to the complexity of model selection. All in all, the best method of selecting a model is a trial and error approach, attempting different models and selecting the best performing as a basis to be built upon.
Due to the complex nature of NN, knowledge in the field is critical for success. As the field of neural networking has expanded, an increasing number of research groups in many different areas has adopted these algorithms for data analysis, making the demand for experts in NN higher than ever before [
31]. This increase in demand has led to the development of deep learning frameworks such as DeepChem that offer a host of deep learning architectures that can be modified and used with little experience [
32,
33,
34]. DeepChem is a python-based framework that specifically targets chemistry, offering deep learning algorithms tailored towards the analysis of molecular datasets, thus making it a perfect platform for building and testing molecular NN models for the analysis and prediction of MIP binding behavior.
The presented research sets focus on using this framework to evaluate which model and featurizer simulates best the binding of specific substrates to a MIP when compared to real world data. To this end, four different featurizers were utilized in the modelling of this data: the RDKitDescriptors, weave featurizer, ConvMolFeaturizer, and the SmilesToImage featurizers. The featurized datasets were then coupled to three different NN models: the Directed Acyclic Graph (DAG) model, Graph Convolution model, and multitask regressor. These models were trained and tested with a combination of data from previous publications and data collected specifically for the purpose of this publication.
2. Dataset Selection
Over the years, MIPs have been applied to the detection of many different small molecules, demonstrating how they can be used as synthetic receptors in many different fields [
35]. A lot of data therefore exists in regard to different compositions of MIPs with a corresponding template. The data however lack depth, with specific MIP binding data generally only extending towards the template molecule and a few analogues/interferants [
36]. Finding datasets that provide a sufficient amount of molecular species binding to a MIP is therefore a tricky task.
An area where extensive binding data is of high importance is the field of drug screening. This area has seen a surge of activity in recent years with the “umbrella” of illicit substances being expanded to encompass new chemical architectures beyond classical drugs of abuse [
37]. This extended prohibition now covers compounds coined New Psychoactive Substances (NPS), being compounds that are synthetically modified variants of classical compounds that contain a psychoactive pharmacophore with an overall different chemical architecture [
38]. A prime example of such a compound is 2-methoxphenidine (2-MXP). Being akin to the classical compounds ketamine and phencyclidine (PCP), 2-MXP acts on the NMDA receptors and has dissociative effects on the user [
39]. Other positional isomers of the compound exist, and current preliminary test methodologies can easily mistake the compound for a whole variety of compounds, including caffeine, which is not controlled [
40]. It is therefore key to have binding data that covers a large array of molecules for sensors designed for these kinds of compounds, as false positives/negatives have become a greater issue as chemical architectures stray away from the norm.
With this in mind, the data selected for the training and testing of the generated neural network models are derived from the NPS field. With the research groups’ prior experience in this area, there is already MIP based data covering a range of chemical structures that can be used for training and test the models. The data were compiled from multiple NPS-based papers [
41,
42] and unpublished results to produced datasets for the training of the models. This training set was randomly selected from the data collected from the sources, introducing the models to a variety of structures in a bid to increase the models’ recognition of a diverse range of molecules. Each molecule introduced has a corresponding SMILES, initial concentration of substrate (C
i), amount of substrate bound to the MIP at equilibrium (S
b), and amount of substrate remaining in solution at equilibrium (C
f). The same was true for the testing dataset, though the number of molecules for this was dramatically reduced, as the bulk of the molecular data was used for the training of the models.
The following research therefore outlines the methods used to train and test models based on specific algorithms, evaluating these models using statistical techniques (R2, Pearson R2, and MAE score), alongside comparing the predicted values to those of the experimental data. Overall, the research endeavored to identify which of the algorithms are the most promising towards modelling MIP binding affinities based on limited datasets.
6. Conclusions
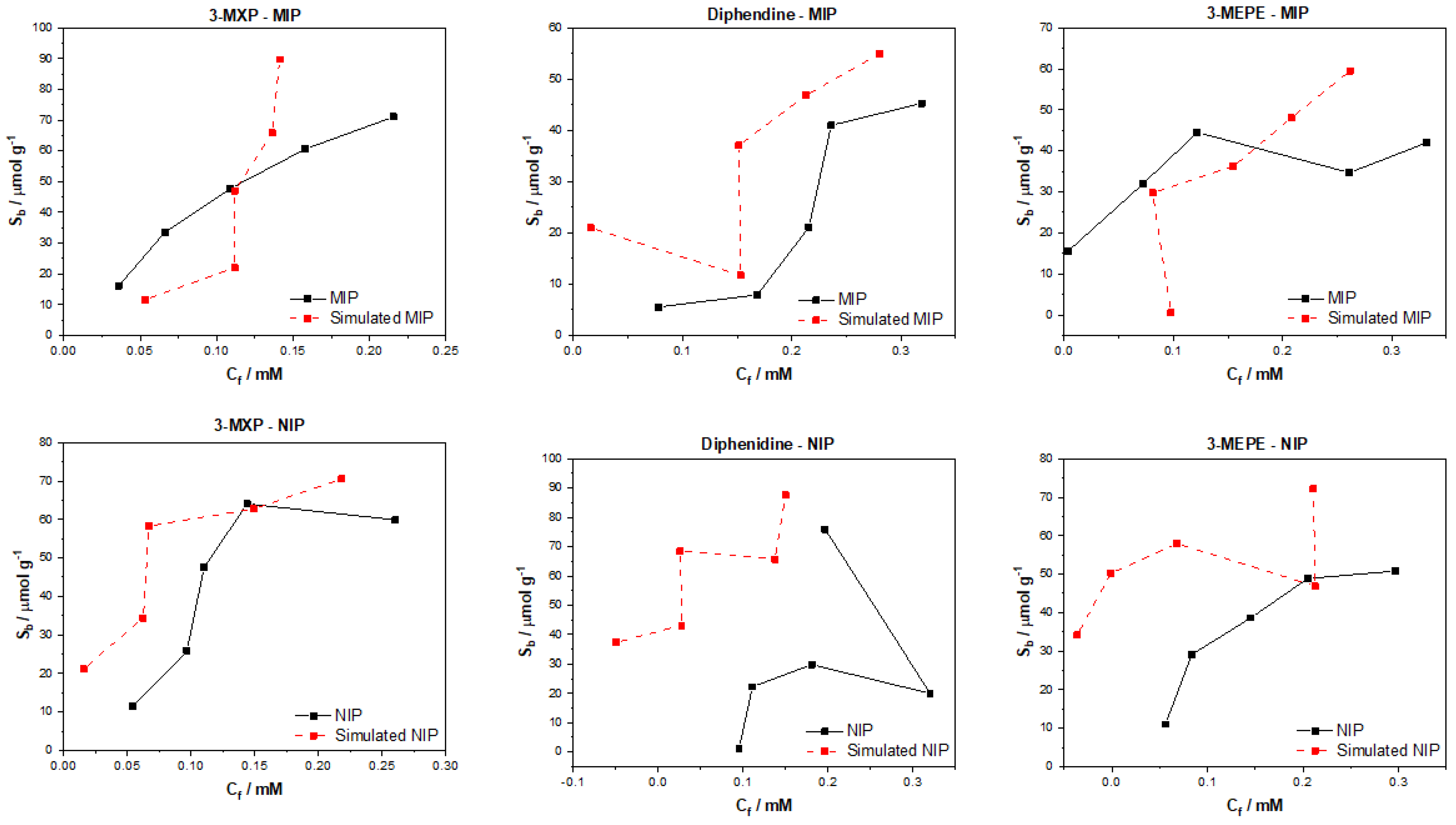
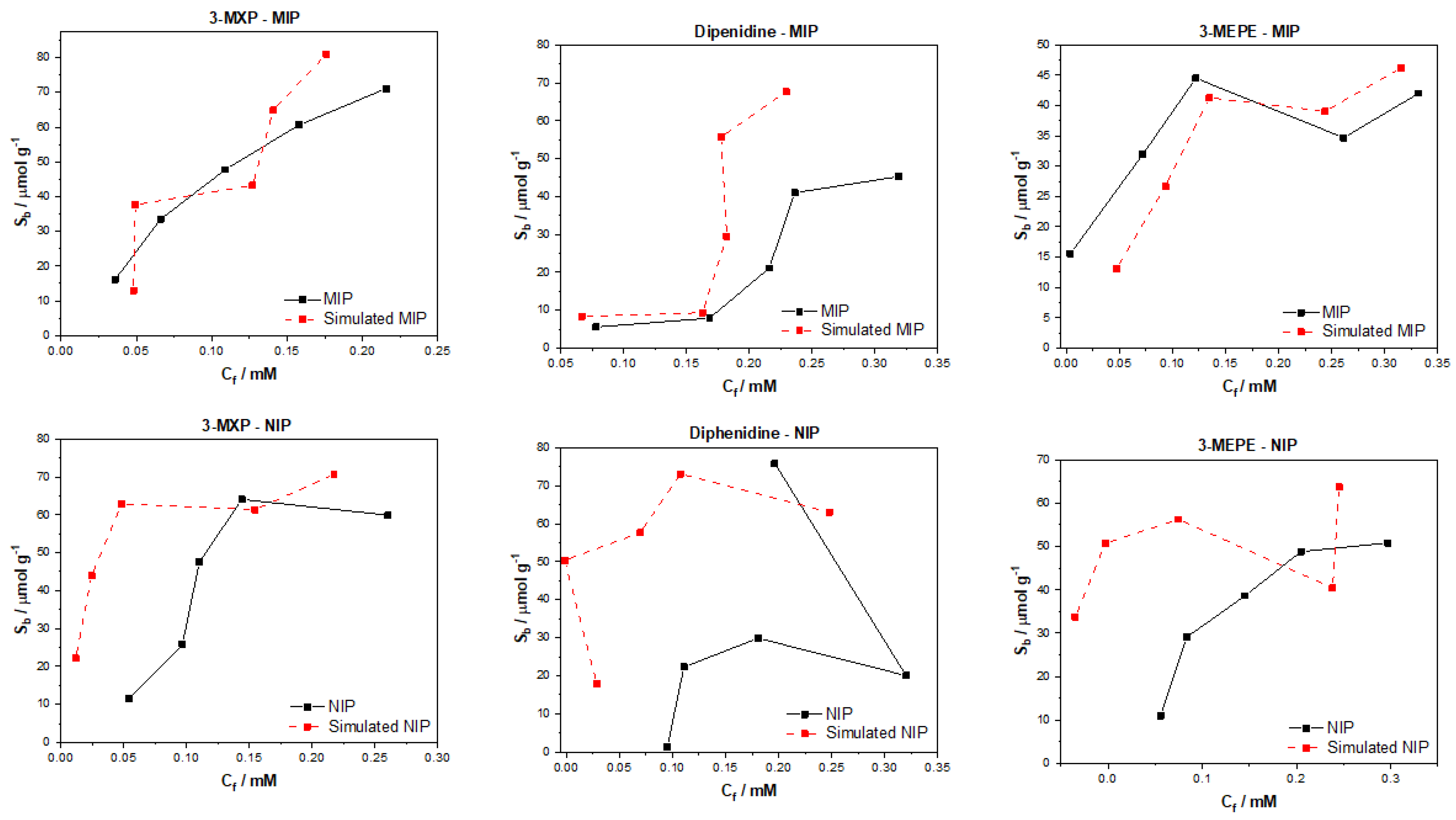
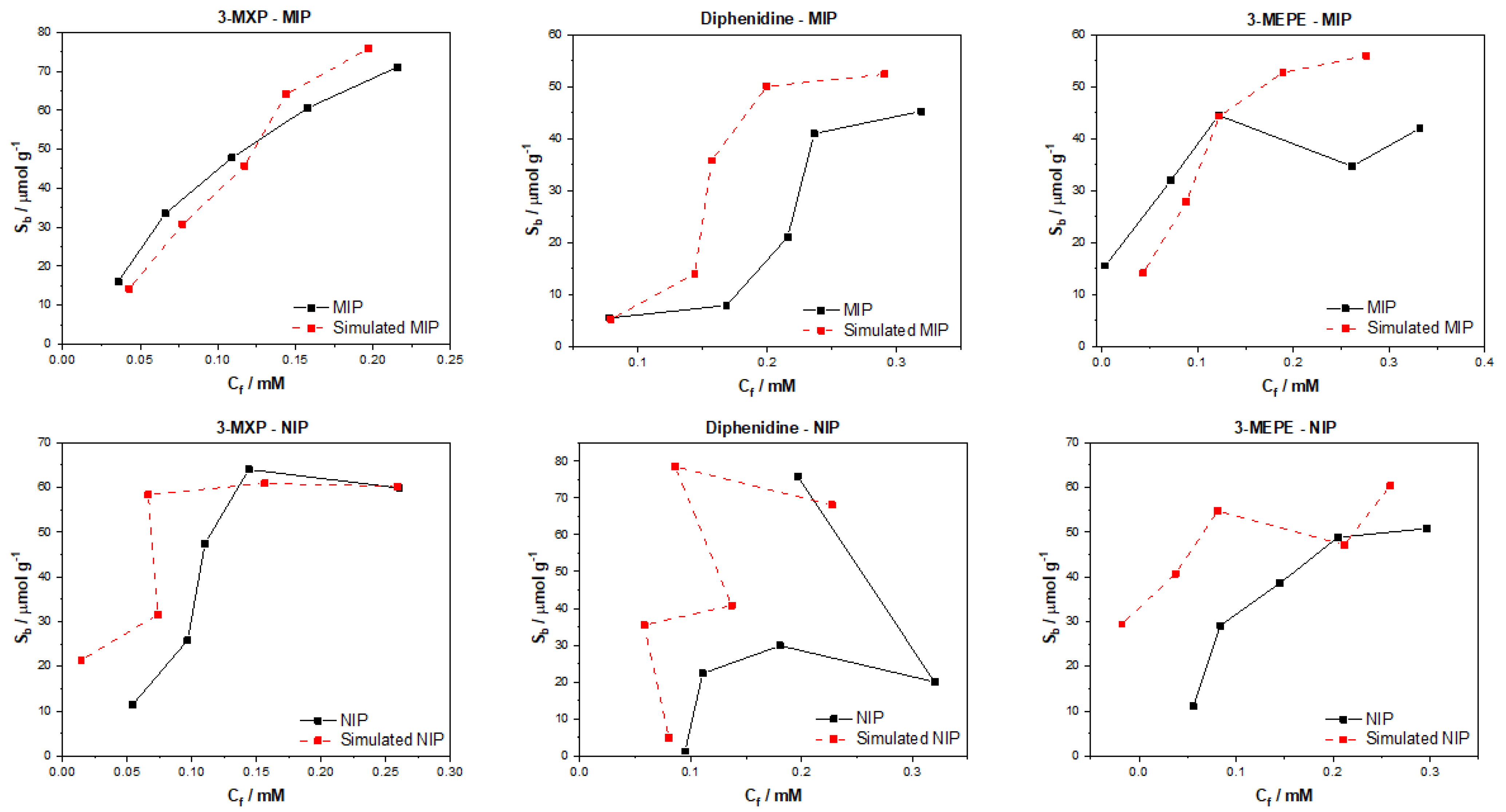
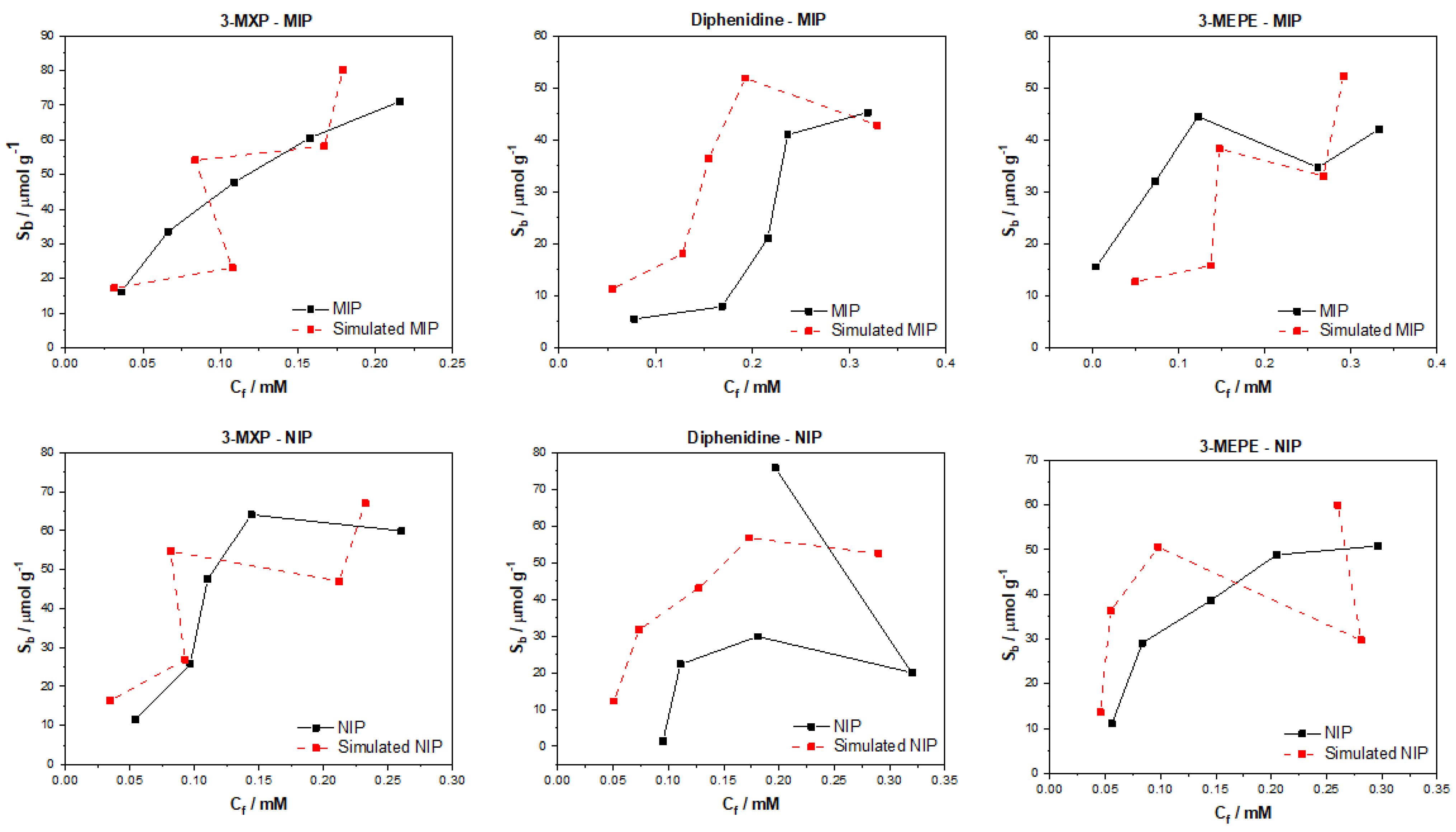
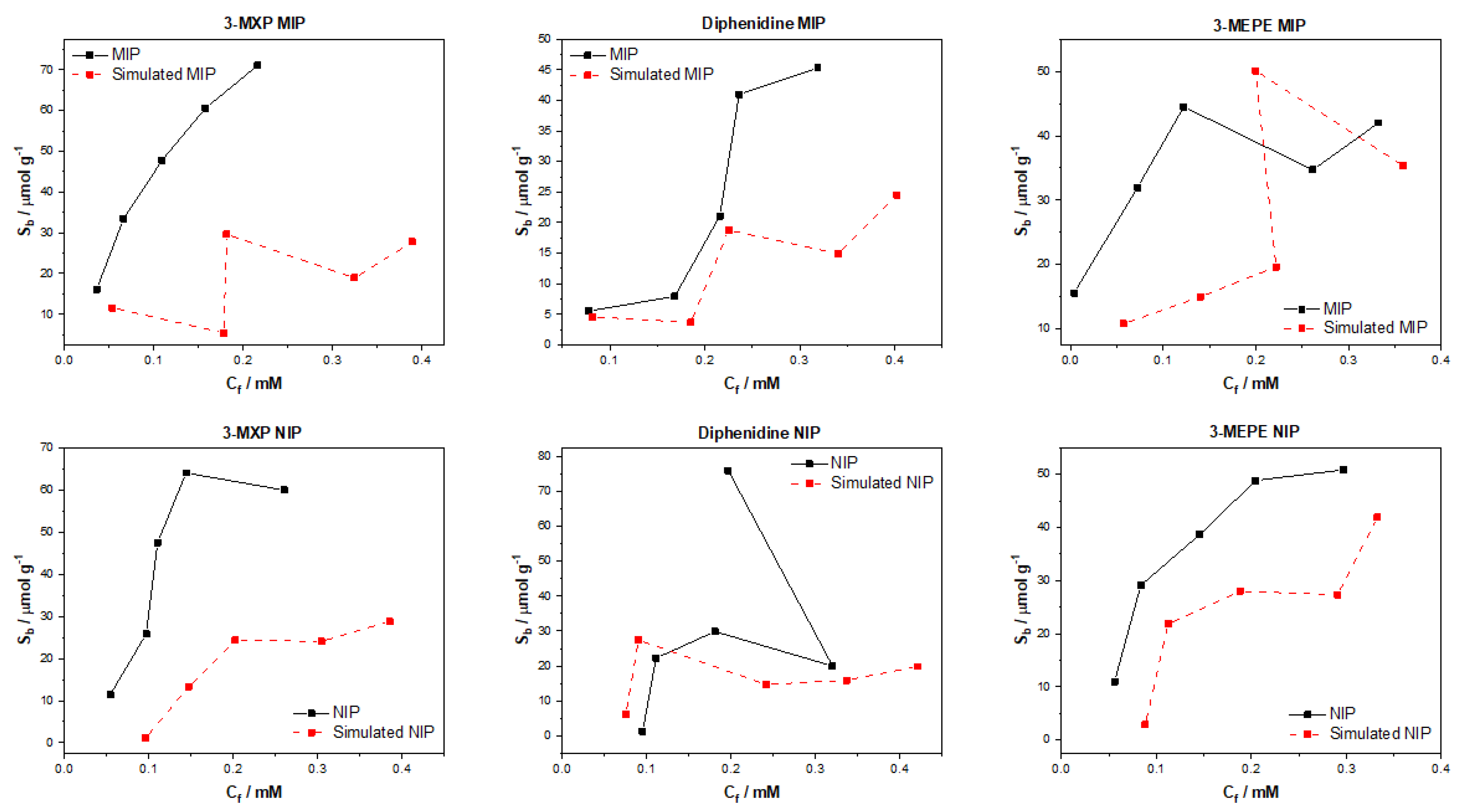
After testing the multiple algorithms for their potential in predicting MIP/NIP binding affinities, it can be concluded that there is great potential in these approaches for the simulation of MIP data. The most promising of these algorithms include the multitask regression and graph convolution models, with the simulated MIP data proving to be the most accurate representations of the experimental data discussed. The DAG, ChemCeption model and graph convolution with weave featurizer proved to have a harder time predicting the binding affinities of molecules, though DAG with modification could have the potential to predict the desired values better. The graph convolution and multitask regressor approaches could predict the trends in the experimental data, producing predicted values that were a fair representation of the testing data. The statistical analysis of all trained and tested models reveals that the models perform well when training but are weak when being tested. As previously mentioned, this is due to the limited dataset and the poor generalization of the models towards different molecules/data. Overall, though the models struggled with over or under predicting the Sb and Cf values, suggesting that additional chemical properties or a combination of algorithms might lead to a more rounded model for the prediction of these values and expansion of the datasets toward other molecules would be fruitful.
The data analyzed and simulated using these approaches were derived from MIP/NIPs synthesized by monolithic bulk polymerization. It should be mentioned that this approach produces relatively heterogeneous MIP/NIP samples with particle sizes and binding cavities randomly distributed throughout the material. The heterogeneous nature is followed into the generated binding isotherms with some of the experimental isotherms proving to have poor trends, and thus making them harder to learn via computational methods. In the future, a more homogenous MIP synthesis could be selected that would therefore lend itself to more homogenous MIP data that could prove easier for machine learning algorithms to learn and predict. Other factors such as MIP composition (monomers, crosslinkers, and porogens used) and MIP porosity could be factored into the models, building upon reliability and the precision of the approach.
The main take away from the research is that these algorithms can be applied to real world MIP/NIP binding data and can predict affinities that resemble experimental values. This opens up the possibility of utilizing this approach in real-world scenarios, with the manuscript highlighting that it is possible to computationally predict the binding of molecules to MIPs. This is the first instance of this to the authors’ knowledge and highlights the potential of machine learning algorithms in this field. The machine learning experiments presented have been conducted by scientists with limit knowledge in the field of machine learning, and have arguably produced results that warrant further investigation into the use of these algorithms in a deep context. If this knowledge was applied by users who have more experience with these models, then we believe that an algorithm and/or approach could easily be identified that could help predict the binding of molecules to specific MIP/NIP compositional chemistries. Thus, this basic research has the potential to open the door for rapid computational simulations of molecular binding affinities and could be used in the rapid analysis of MIPs, reducing material costs, analysis time, and chemical wastage.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}