
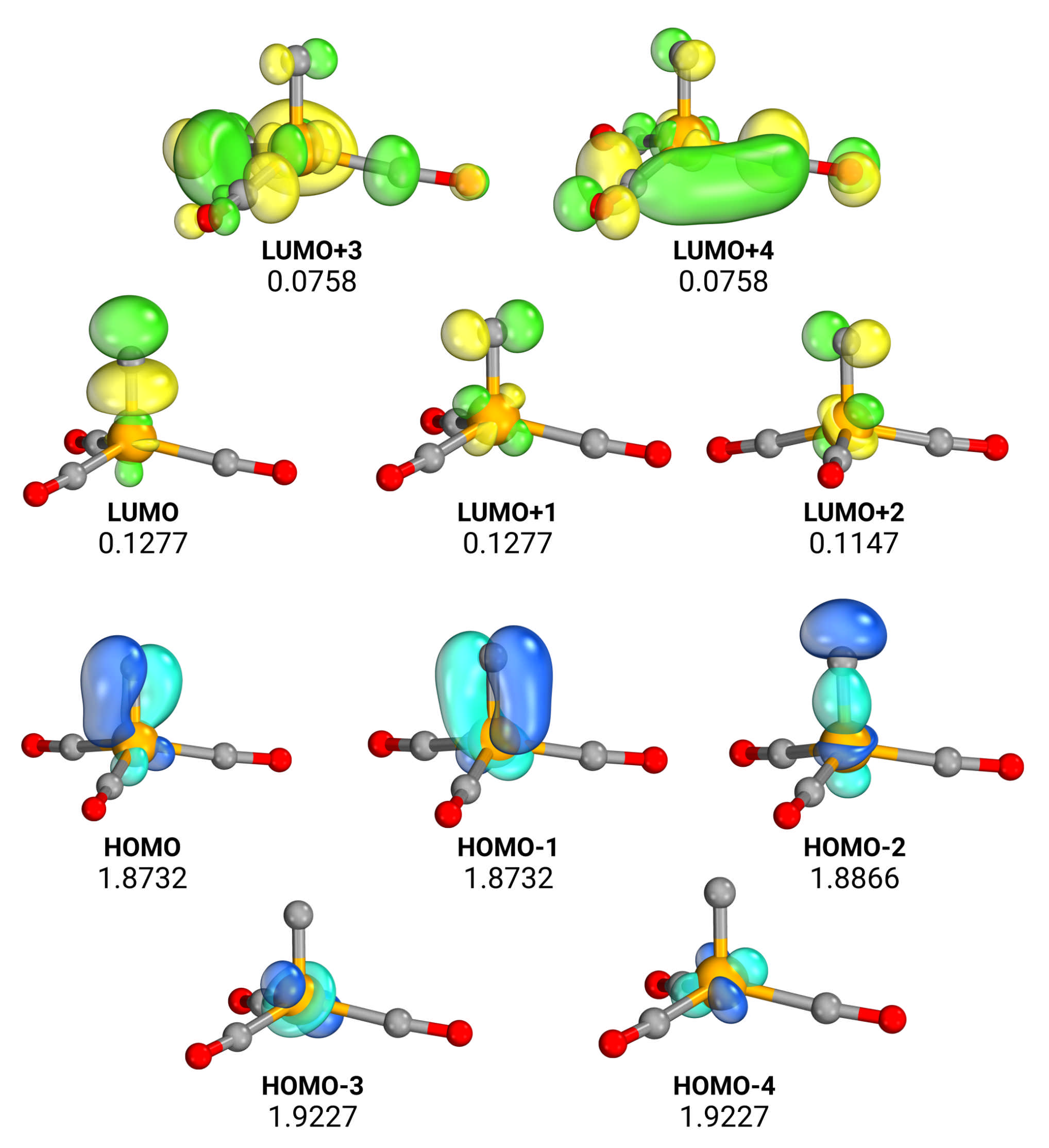
Is There a Quadruple Fe-C Bond in FeC(CO)3?


Abstract
:Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kalita, A.J.; Rohman, S.S.; Kashyap, C.; Ullah, S.S.; Guha, A.K. Transition metal carbon quadruple bond: Viability through single electron transmutation. Phys. Chem. Chem. Phys. 2020, 22, 24178–24180. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 1007, 4572–4585. [Google Scholar] [CrossRef]
- Balabanov, N.B.; Peterson, K.A. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc–Zn. J. Chem. Phys. 2005, 123, 064107. [Google Scholar] [CrossRef] [Green Version]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Pople, J.A.; Krishnan, R.; Schlegel, H.B.; Binkley, J.S. Derivative studies in hartree-fock and møller-plesset theories. Int. J. Quantum Chem. 1979, 16, 225–241. [Google Scholar] [CrossRef]
- Scheiner, A.C.; Scuseria, G.E.; Rice, J.E.; Lee, T.J.; Schaefer, H.F. Analytic evaluation of energy gradients for the single and double excitation coupled cluster (CCSD) wave function: Theory and application. J. Chem. Phys. 1987, 87, 5361–5373. [Google Scholar] [CrossRef]
- Scuseria, G.E. Analytic evaluation of energy gradients for the singles and doubles coupled cluster method including perturbative triple excitations: Theory and applications to FOOF and Cr2. J. Chem. Phys. 1991, 94, 442–447. [Google Scholar] [CrossRef]
- Stanton, J.F.; Gauss, J.; Cheng, L.; Harding, M.E.; Matthews, D.A.; Szalay, P.G. CFOUR, Coupled-Cluster techniques for Computational Chemistry, a quantum-chemical program package. With contributions from A.A. Auer, R.J. Bartlett, U. Benedikt, C. Berger, D.E. Bernholdt, S. Blaschke, Y. J. Bomble, S. Burger, O. Christiansen, D. Datta, F. Engel, R. Faber, J. Greiner, M. Heckert, O. Heun, M. Hilgenberg, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, T. Kirsch, K. Klein, G.M. KopperW.J. Lauderdale, F. Lipparini, T. Metzroth, L.A. Mück, D.P. O’Neill, T. Nottoli, D.R. Price, E. Prochnow, C. Puzzarini, K. Ruud, F. Schiffmann, W. Schwalbach, C. Simmons, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang, J.D. Watts and the integral packages MOLECULE (J. Almlöf and P.R. Taylor), PROPS (P.R. Taylor), ABACUS (T. Helgaker, H.J. Aa. Jensen, P. Jørgensen, and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen. Available online: http://www.cfour.de (accessed on 29 August 2021).
- Matthews, D.A.; Cheng, L.; Harding, M.E.; Lipparini, F.; Stopkowicz, S.; Jagau, T.C.; Szalay, P.G.; Gauss, J.; Stanton, J.F. Coupled-cluster techniques for computational chemistry: The CFOUR program package. J. Chem. Phys. 2020, 152, 214108. [Google Scholar] [CrossRef]
- Jensen, H.J.A.; Jorgensen, P. A direct approach to second-order MCSCF calculations using a norm extended optimization scheme. J. Chem. Phys. 1984, 80, 1204–1214. [Google Scholar] [CrossRef]
- Lipparini, F.; Gauss, J. Cost-Effective Treatment of Scalar Relativistic Effects for Multireference Systems: A CASSCF Implementation Based on the Spin-free Dirac–Coulomb Hamiltonian. J. Chem. Theory Comput. 2016, 12, 4284–4295. [Google Scholar] [CrossRef] [PubMed]
- Pulay, P.; Hamilton, T.P. UHF natural orbitals for defining and starting MC-SCF calculations. J. Chem. Phys. 1988, 88, 4926–4933. [Google Scholar] [CrossRef]
- Tóth, Z.; Pulay, P. Comparison of Methods for Active Orbital Selection in Multiconfigurational Calculations. J. Chem. Theory Comput. 2020, 16, 7328–7341. [Google Scholar] [CrossRef] [PubMed]
- Knizia, G. Intrinsic atomic orbitals: An unbiased bridge between quantum theory and chemical concepts. J. Chem. Theory Comput. 2013, 9, 4834–4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knizia, G.; Klein, J.E. Electron flow in reaction mechanisms—Revealed from first principles. Angew. Chem. Int. Ed. 2015, 54, 5518–5522. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Frenking, G. The Chemical Bond in C2. Chem. Eur. J. 2016, 22, 4100–4108. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Rzepa, H.S.; Hoffmann, R. One Molecule, Two Atoms, Three Views, Four Bonds? Angew. Chem. Int. Ed. 2013, 52, 3020–3033. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Method | ||
|---|---|---|
| M062X | 1.482 | 1208 |
| M062X * | 1.481 | 1211 |
| B3LYP | 1.526 | 1113 |
| MN15 | 1.501 | 1165 |
| PBE0 | 1.514 | 1146 |
| B97X-D | 1.510 | 1154 |
| Method | ||
|---|---|---|
| HF | 1.489 | 1041 |
| MP2 | 1.654 | 591 |
| CCSD | 1.539 | 1039 |
| CCSD(T) | 1.559 | 926 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nottoli, T.; Lipparini, F. Is There a Quadruple Fe-C Bond in FeC(CO)3? Computation 2021, 9, 95. https://doi.org/10.3390/computation9090095
Nottoli T, Lipparini F. Is There a Quadruple Fe-C Bond in FeC(CO)3? Computation. 2021; 9(9):95. https://doi.org/10.3390/computation9090095
Chicago/Turabian StyleNottoli, Tommaso, and Filippo Lipparini. 2021. "Is There a Quadruple Fe-C Bond in FeC(CO)3?" Computation 9, no. 9: 95. https://doi.org/10.3390/computation9090095
APA StyleNottoli, T., & Lipparini, F. (2021). Is There a Quadruple Fe-C Bond in FeC(CO)3? Computation, 9(9), 95. https://doi.org/10.3390/computation9090095