Continuum Modelling for Interacting Coronene Molecules with a Carbon Nanotube



Abstract
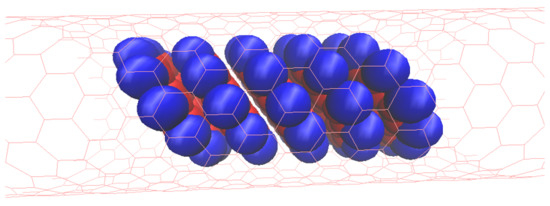
:
1. Introduction
2. Materials and Methods
2.1. Interaction in a Coronene Dimer
2.2. Interaction between a Carbon Nanotube and an Encapsulated Coronene
2.3. Interaction between a Carbon Nanotube and an Encapsulated Coronene Stack
3. Results and Discussion
3.1. Interaction in a Coronene Dimer
3.2. Interaction between a Carbon Nanotube and an Encapsulated Coronene
3.3. Interaction between a Carbon Nanotube and an Encapsulated Coronene Stack
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. Evaluation Details for the Derivation of Equation (6)
Appendix B. Evaluation Details for the Derivation of Equation (9)
References
- Rapacioli, M.; Calvo, F.; Spiegelman, F.; Joblin, C.; Wales, D. Stacked clusters of polycyclic aromatic hydrocarbon molecules. J. Phys. Chem. A 2005, 109, 2487–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, Y.M.; Lee, T.J.; Gudipati, M.S.; Allamandola, L.J.; Head-Gordon, M. Charged polycyclic aromatic hydrocarbon clusters and the galactic extended red emission. Proc. Natl. Acad. Sci. USA 2007, 104, 5274–5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Jimenez, A.J.; Sancho-Garcia, J.C. Conductance Enhancement in Nanographene- Gold Junctions by Molecular π-Stacking. J. Am. Chem. Soc. 2009, 131, 14857–14867. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Sander, M.; Janardhanan, V.; Kraft, M. A study on the coagulation of polycyclic aromatic hydrocarbon clusters to determine their collision efficiency. Combust. Flame 2010, 157, 523–534. [Google Scholar] [CrossRef]
- Richter, H.; Howard, J.B. Formation of polycyclic aromatic hydrocarbons and their growth to soot—A review of chemical reaction pathways. Prog. Energy Combust. Sci. 2000, 26, 565–608. [Google Scholar] [CrossRef]
- Guijarro, A.; Vergés, J.A.; San-Fabián, E.; Chiappe, G.; Louis, E. Herringbone Pattern and CH–π Bonding in the Crystal Architecture of Linear Polycyclic Aromatic Hydrocarbons. ChemPhysChem 2016, 17, 3548–3557. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Pisula, W.; Müllen, K. Graphenes as potential material for electronics. Chem. Rev. 2007, 107, 718–747. [Google Scholar] [CrossRef]
- Ouyang, T.; Cheng, K.; Yang, F.; Zhou, L.; Zhu, K.; Ye, K.; Wang, G.; Cao, D. From biomass with irregular structures to 1D carbon nanobelts: A stripping and cutting strategy to fabricate high performance supercapacitor materials. J. Mater. Chem. A 2017, 5, 14551–14561. [Google Scholar] [CrossRef]
- Zhang, Q.; Fang, T.; Xing, H.; Seabaugh, A.; Jena, D. Graphene nanoribbon tunnel transistors. IEEE Electron Device Lett. 2008, 29, 1344–1346. [Google Scholar] [CrossRef]
- Martinez-Blanco, J.; Mascaraque, A.; Dedkov, Y.S.; Horn, K. Ge (001) As a Template for Long-Range Assembly of π-Stacked Coronene Rows. Langmuir 2012, 28, 3840–3844. [Google Scholar] [CrossRef]
- Talyzin, A.; Luzan, S.; Leifer, K.; Akhtar, S.; Fetzer, J.; Cataldo, F.; Tsybin, Y.; Tai, C.W.; Dzwilewski, A.; Moons, E. Coronene fusion by heat treatment: Road to nanographenes. J. Phys. Chem. C 2011, 115, 13207–13214. [Google Scholar] [CrossRef]
- Ruuska, H.; Pakkanen, T.A. Ab Initio Study of Interlayer Interaction of Graphite: Benzene- Coronene and Coronene Dimer Two-Layer Models. J. Phys. Chem. B 2001, 105, 9541–9547. [Google Scholar] [CrossRef]
- Paulsson, M.; Stafström, S. Theoretical study of electron transport along self-assembled graphitic nanowires. J. Phys. Condens. Matter 2000, 12, 9433. [Google Scholar] [CrossRef]
- Bag, S.; Maiti, P.K. Ultrahigh charge carrier mobility in nanotube encapsulated coronene stack. Phys. Rev. B 2017, 96, 245401. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Yang, H.; Yin, Z.; Guo, J.; Boey, F.; Zhang, H.; Zhang, Q. Preparation, characterization, and photoswitching/light-emitting behaviors of coronene nanowires. J. Mater. Chem. 2011, 21, 1423–1427. [Google Scholar] [CrossRef]
- Takazawa, K.; Inoue, J.I.; Mitsuishi, K. Self-assembled coronene nanofibers: Optical waveguide effect and magnetic alignment. Nanoscale 2014, 6, 4174–4181. [Google Scholar] [CrossRef]
- Yoshida, Y.; Isomura, K.; Kishida, H.; Kumagai, Y.; Mizuno, M.; Sakata, M.; Koretsune, T.; Nakano, Y.; Yamochi, H.; Maesato, M.; et al. Conducting π Columns of Highly Symmetric Coronene, The Smallest Fragment of Graphene. Chem. Eur. J. 2016, 22, 6023–6030. [Google Scholar] [CrossRef]
- Okazaki, T.; Iizumi, Y.; Okubo, S.; Kataura, H.; Liu, Z.; Suenaga, K.; Tahara, Y.; Yudasaka, M.; Okada, S.; Iijima, S. Coaxially Stacked Coronene Columns inside Single-Walled Carbon Nanotubes. Angew. Chem. Int. Ed. 2011, 50, 4853–4857. [Google Scholar] [CrossRef]
- Dappe, Y.J.; Martínez, J.I. Effect of van der Waals forces on the stacking of coronenes encapsulated in a single-wall carbon nanotube and many-body excitation spectrum. Carbon 2013, 54, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Chernov, A.I.; Fedotov, P.V.; Anoshkin, I.V.; Nasibulin, A.G.; Kauppinen, E.I.; Kuznetsov, V.L.; Obraztsova, E.D. Single-walled carbon nanotubes as a template for coronene stack formation. Phys. Status Solidi (B) 2014, 251, 2372–2377. [Google Scholar] [CrossRef]
- Sakane, Y.; Mouri, K.; Shintani, K. Morphology of a columnar stack of coronene molecules encapsulated in a single-walled carbon nanotube. AIP Adv. 2015, 5, 117113. [Google Scholar] [CrossRef] [Green Version]
- Verberck, B.; Okazaki, T.; Tarakina, N.V. Ordered and disordered packing of coronene molecules in carbon nanotubes. Phys. Chem. Chem. Phys. 2013, 15, 18108–18114. [Google Scholar] [CrossRef] [PubMed]
- Mouri, K.; Shintani, K. Geometrical constraint on stacking of polycyclic aromatic hydrocarbon molecules encapsulated in a single-walled carbon nanotube. Phys. Chem. Chem. Phys. 2016, 18, 31043–31053. [Google Scholar] [CrossRef] [PubMed]
- Botka, B.; Füstös, M.; Klupp, G.; Kocsis, D.; Székely, E.; Utczás, M.; Simándi, B.; Botos, Á.; Hackl, R.; Kamarás, K. Low-temperature encapsulation of coronene in carbon nanotubes. Phys. Status Solidi (B) 2012, 249, 2432–2435. [Google Scholar] [CrossRef]
- Baowan, D.; Cox, B.J.; Hilder, T.A.; Hill, J.M.; Thamwattana, N. Modelling and Mechanics of Carbon-Based Nanostructured Materials, 1st ed.; Elsevier Micro & Nano Technologies Series; William Andrew: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Tran-Duc, T.; Thamwattana, N.; Cox, B.J.; Hill, J.M. General model for molecular interactions in a benzene dimer. Math. Mech. Solids 2010, 15, 782–799. [Google Scholar] [CrossRef]
- Tran-Duc, T.; Thamwattana, N.; Hill, J.M. Orientation of a benzene molecule inside a carbon nanotube. J. Math. Chem. 2011, 49, 1115–1127. [Google Scholar] [CrossRef]
- Heinemann, T.; Palczynski, K.; Dzubiella, J.; Klapp, S.H. Coarse-grained electrostatic interactions of coronene: Towards the crystalline phase. J. Chem. Phys. 2015, 143, 174110. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Rojas, J.; Calvo, F.; Wales, D. Coarse-graining the structure of polycyclic aromatic hydrocarbons clusters. Phys. Chem. Chem. Phys. 2016, 18, 13736–13740. [Google Scholar] [CrossRef]
- Toxvaerd, S.; Dyre, J.C. Communication: Shifted forces in molecular dynamics. J. Chem. Phys. 2011, 134, 081102. [Google Scholar] [CrossRef] [Green Version]
- Anoshkin, I.V.; Talyzin, A.V.; Nasibulin, A.G.; Krasheninnikov, A.V.; Jiang, H.; Nieminen, R.M.; Kauppinen, E.I. Coronene Encapsulation in Single-Walled Carbon Nanotubes: Stacked Columns, Peapods, and Nanoribbons. ChemPhysChem 2014, 15, 1660–1665. [Google Scholar] [CrossRef]
- Kigure, S.; Iizumi, Y.; Okazaki, T.; Okada, S. Energetics and electronic structures of carbon nanotubes encapsulating polycyclic aromatic hydrocarbon molecules. J. Phys. Soc. Jpn. 2014, 83, 124709. [Google Scholar] [CrossRef]
- Gradshteyn, I.S.; Ryzhik, I.M. Table of Integrals, Series, and Products; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First carbon ring radius | Å |
| Second carbon ring radius | Å |
| Third carbon ring radius | Å |
| Hydrogen ring radius | Å |
| First carbon ring atomic line density | Å−1 |
| Second carbon ring atomic line density | Å−1 |
| Third carbon ring atomic line density | Å−1 |
| Hydrogen ring atomic line density | Å−1 |
| CNT atomic surface density | Å−2 |
| CNT radius | Å |
| CNT radius | Å |
| CNT radius | Å |
| CNT radius | Å |
| CNT radius | Å |
| C-C attractive constant | Å6 |
| C-C repulsive constant | Å12 |
| C-H attractive constant | Å6 |
| C-H repulsive constant | Å12 |
| Angle (radians) | Displacement (Å) | Energy () |
|---|---|---|
| 0 | ||
| Nanotube (n,m) | Angle (radians) | Energy () |
|---|---|---|
| 0 |
| Angle (radians) | Coronene Spacing (Å) | Energy () |
|---|---|---|
| Angle (radians) | Coronene Spacing (Å) | Energy () |
|---|---|---|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens, K.; Tran-Duc, T.; Thamwattana, N.; Hill, J.M. Continuum Modelling for Interacting Coronene Molecules with a Carbon Nanotube. Nanomaterials 2020, 10, 152. https://doi.org/10.3390/nano10010152
Stevens K, Tran-Duc T, Thamwattana N, Hill JM. Continuum Modelling for Interacting Coronene Molecules with a Carbon Nanotube. Nanomaterials. 2020; 10(1):152. https://doi.org/10.3390/nano10010152
Chicago/Turabian StyleStevens, Kyle, Thien Tran-Duc, Ngamta Thamwattana, and James M. Hill. 2020. "Continuum Modelling for Interacting Coronene Molecules with a Carbon Nanotube" Nanomaterials 10, no. 1: 152. https://doi.org/10.3390/nano10010152
APA StyleStevens, K., Tran-Duc, T., Thamwattana, N., & Hill, J. M. (2020). Continuum Modelling for Interacting Coronene Molecules with a Carbon Nanotube. Nanomaterials, 10(1), 152. https://doi.org/10.3390/nano10010152