Spontaneous Self-Assembly of Single-Chain Amphiphilic Polymeric Nanoparticles in Water


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
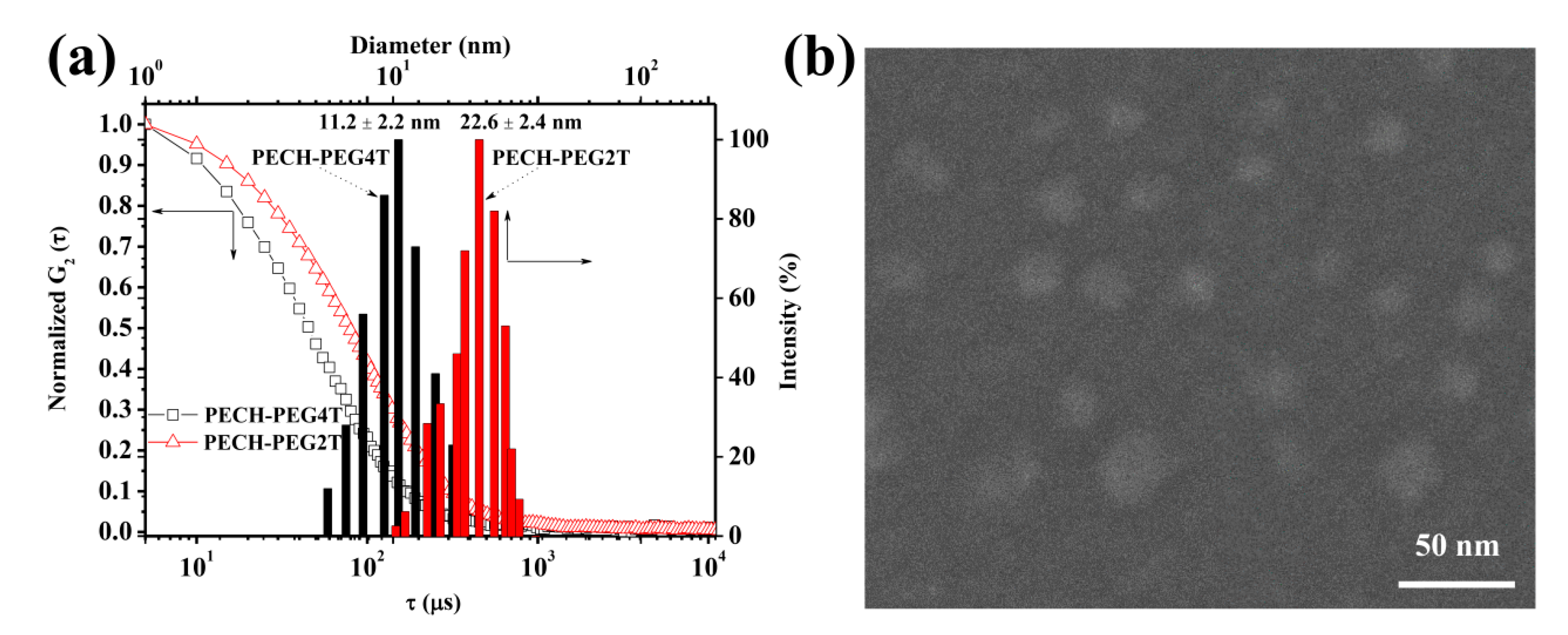
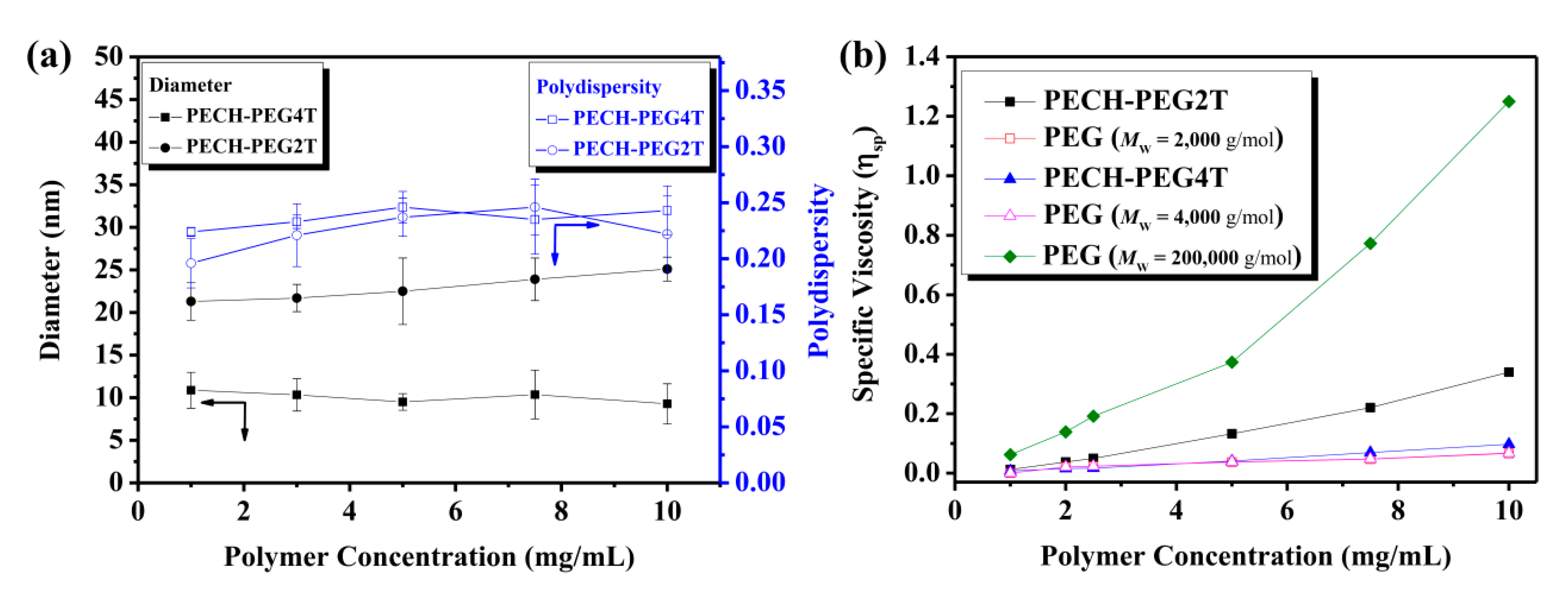
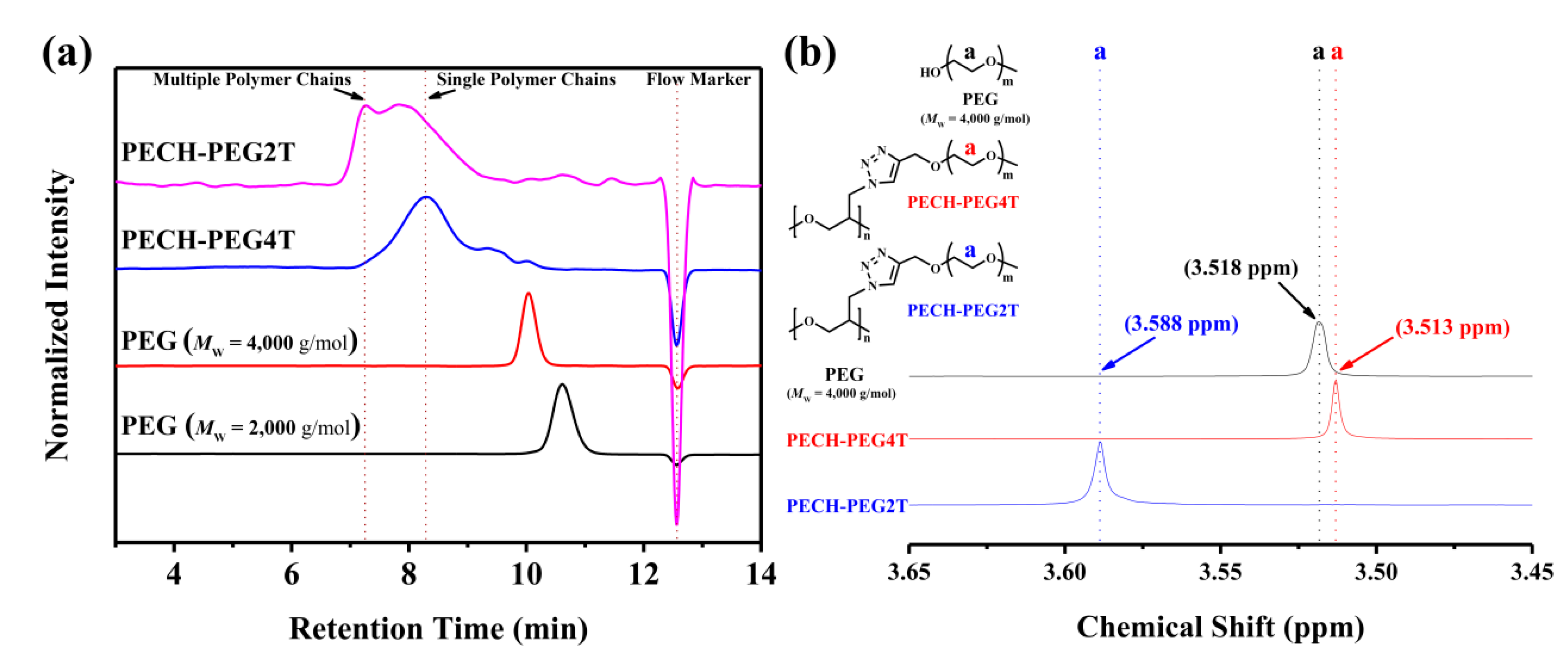
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rowan, S.J. Polymers with bio-inspired strength. Nat. Chem. 2009, 1, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sakai, F.; Su, L.; Liu, Y.; Wei, K.; Chen, G.; Jiang, M. Progressive macromolecular self-assembly: From biomimetic chemistry to bio-inspired materials. Adv. Mater. 2013, 25, 5215–5525. [Google Scholar] [CrossRef] [PubMed]
- Pieters, B.J.G.E.; van Eldijk, M.B.; Noltea, R.J.M.; Mecinović, J. Natural supramolecular protein assemblies. Chem. Soc. Rev. 2016, 45, 24–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.-X. Polymer models of protein stability, folding, and interactions. Biochemistry 2004, 43, 2141–2154. [Google Scholar] [CrossRef] [PubMed]
- Gorske, B.C.; Stringer, J.R.; Bastian, B.L.; Fowler, S.A.; Blackwell, H.E. New strategies for the design of folded peptoids revealed by a survey of noncovalent interactions in model systems. J. Am. Chem. Soc. 2009, 131, 16555–16567. [Google Scholar] [CrossRef] [Green Version]
- Knowles, T.P.J.; Oppenheim, T.W.; Buell, A.K.; Chirgadze, D.Y.; Welland, M.E. Nanostructured films from hierarchical self-assembly of amyloidogenic proteins. Nat. Nanotechnol. 2010, 5, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Altintas, O.; Barner-Kowollik, C. Single chain folding of synthetic polymers by covalent and non-covalent interactions: Current status and future perspectives. Macromol. Rapid Commun. 2012, 33, 958–971. [Google Scholar] [CrossRef]
- Huo, M.; Wang, N.; Fang, T.; Sun, M.; Wei, Y.; Yuan, J. Single-chain polymer nanoparticles: Mimic the proteins. Polymer 2015, 66, A11–A21. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, M.; Latorre-Sanchezab, A.; Pomposo, J.A. Advances in single chain technology. Chem. Soc. Rev. 2015, 44, 6122–6142. [Google Scholar] [CrossRef] [Green Version]
- Lyon, C.K.; Prasher, A.; Hanlon, A.M.; Tuten, B.T.; Tooley, C.A.; Frank, P.G.; Berda, E.B. A brief user’s guide to single-chain nanoparticles. Polym. Chem. 2015, 6, 181–197. [Google Scholar] [CrossRef]
- Gao, Y.; Newland, B.; Zhou, D.; Matyjaszewski, K.; Wang, W. Controlled polymerization of multivinyl monomers: Formation of cyclized/knotted single-chain polymer architectures. Angew. Chem. Int. Ed. 2017, 56, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomposo, J.A. Single-Chain Polymer Nanoparticles: Synthesis, Characterization, Simulations, and Applications; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2017; pp. 1–400. [Google Scholar]
- Aiertza, M.K.; Odriozola, I.; Cabañero, G.; Grande, H.J.; Loinaz, I. Single-chain polymer nanoparticles. Cell. Mol. Life Sci. 2012, 69, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Appel, E.A.; Barrio, J.D.; Dyson, J.; Isaacs, L.; Scherman, O.A. Metastable single-chain polymer nanoparticles prepared by dynamic cross-linking with nor-seco-cucurbit[10]uril. Chem. Sci. 2012, 3, 2278–2281. [Google Scholar] [CrossRef]
- Hosono, N.; Gillissen, M.A.J.; Li, Y.; Sheiko, S.S.; Palmans, A.R.A.; Meijer, E.W. Orthogonal self-assembly in folding block copolymers. J. Am. Chem. Soc. 2013, 135, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C.; Chang, F.C.; Yen, H.C.; Lee, D.J.; Chiu, C.W.; Xin, Z. Supramolecular assembly mediates the formation of single-chain polymeric nanoparticles. ACS Macro Lett. 2015, 4, 1184–1188. [Google Scholar] [CrossRef] [Green Version]
- Murray, B.S.; Fulton, D.A. Dynamic covalent single-chain polymer nanoparticles. Macromolecules 2011, 44, 7242–7252. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, A.; Fulton, D.A.; Pomposo, J.A. pH-Responsive single-chain polymer nanoparticles utilising dynamic covalent enamine bonds. Chem. Commun. 2014, 50, 1871–1874. [Google Scholar] [CrossRef] [Green Version]
- Wedler-Jasinski, N.; Lueckerath, T.; Mutlu, H.; Goldmann, A.S.; Walther, A.; Stenzel, M.H.; Barner-Kowollik, C. Dynamic covalent single chain nanoparticles based on hetero diels–alder chemistry. Chem. Commun. 2017, 53, 157–160. [Google Scholar] [CrossRef]
- Wong, E.H.H.; Lam, S.J.; Nam, E.; Qiao, G.G. Biocompatible single-chain polymeric nanoparticles via organo-catalyzed ring-opening polymerization. ACS Macro Lett. 2014, 3, 524–528. [Google Scholar] [CrossRef]
- Fan, W.; Tong, X.; Farnia, F.; Yu, B.; Zhao, Y. CO2-responsive polymer single-chain nanoparticles and self-assembly for gas-tunable nanoreactors. Chem. Mater. 2017, 29, 5693–5701. [Google Scholar] [CrossRef]
- Frisch, H.; Menzel, J.P.; Bloesser, F.R.; Marschner, D.E.; Mundsinger, K.; Barner-Kowollik, C. Fluorescent glyco single-chain nanoparticle-decorated nanodiamonds. J. Am. Chem. Soc. 2018, 140, 9551–9557. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Feng, X.; Xing, H.; Xu, Y.; Kim, B.K.; Baig, N.; Zhou, T.; Gewirth, A.A.; Lu, Y.; Oldfield, E.; et al. A highly efficient single-chain metal−organic nanoparticle catalyst for alkyne−azide “click” reactions in water and in cells. J. Am. Chem. Soc. 2016, 138, 11077–11080. [Google Scholar] [CrossRef] [PubMed]
- Knçfel, N.D.; Rothfuss, H.; Willenbacher, J.; Barner-Kowollik, C.; Roesky, P.W. Platinum(II)-crosslinked single-chain nanoparticles: An approach towards recyclable homogeneous catalysts. Angew. Chem. Int. Ed. 2017, 56, 4950–4954. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pujals, S.; Stals, P.J.M.; Paulöhrl, T.; Presolski, S.I.; Meijer, E.W.; Albertazzi, L.; Palmans, A.R.A. Catalytically active single-chain polymeric nanoparticles: Exploring their functions in complex biological media. J. Am. Chem. Soc. 2018, 140, 3423–3433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krögera, A.P.P.; Paulusse, J.M.J. Single-chain polymer nanoparticles in controlled drug delivery and targeted imaging. J. Control. Release 2018, 286, 326–347. [Google Scholar] [CrossRef] [PubMed]
- Kröger, A.P.P.; Hamelmann, N.M.; Juan, A.; Lindhoud, S.; Paulusse, J.M.J. Biocompatible single-chain polymer nanoparticles for drug delivery: A dual approach. ACS Appl. Mater. Interfaces 2018, 10, 30946–30951. [Google Scholar] [CrossRef] [Green Version]
- Gracia, R.; Marradi, M.; Salerno, G.; Pérez-Nicado, R.; Vicente, A.P.-S.; Dupin, D.; Rodriguez, J.; Loinaz, I.; Chiodo, F.; Nativi, C. Biocompatible single-chain polymer nanoparticles loaded with an antigen mimetic as potential anticancer vaccine. ACS Macro Lett. 2018, 7, 196–200. [Google Scholar] [CrossRef]
- Perez-Baena, I.; Loinaz, I.; Padro, D.; García, I.; Grandea, H.J.; Odriozola, I. Single-chain polyacrylic nanoparticles with multiple Gd(III) centres as potential MRI contrast agents. J. Mater. Chem. 2010, 20, 6916–6922. [Google Scholar] [CrossRef]
- Benito, A.B.; Aiertza, M.K.; Marradi, M.; Gil-Iceta, L.; Zahavi, T.S.; Szczupak, B.; Jiménez-González, M.; Reese, T.; Scanziani, E.; Passoni, L.; et al. Functional single-chain polymer nanoparticles: Targeting and imaging pancreatic tumors in vivo. Biomacromolecules 2016, 17, 3213–3221. [Google Scholar] [CrossRef]
- Özenler, S.; Yucel, M.; Tünc, Ö.; Kaya, H.; Özçelik, S.; Yildiz, U.H. Single chain cationic polymer dot as a fluorescent probe for cell imaging and selective determination of hepatocellular carcinoma cells. Anal. Chem. 2019, 91, 10357–10360. [Google Scholar] [CrossRef] [Green Version]
- Appel, E.A.; Dyson, J.; Delbarrio, J.; Walsh, Z.; Scherman, O.A. Formation of single-chain polymer nanoparticles in water through host–guest interactions. Angew. Chem. Int. Ed. 2012, 51, 4185–4189. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, D.E.; Mahon, C.S.; Fulton, D.A. Thermoresponsive dynamic covalent single-chain polymer nanoparticles reversibly transform into a hydrogel. Angew. Chem. Int. Ed. 2013, 52, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Sugita, T.; Fukae, K.; Sawamoto, M. Synthesis and single-chain folding of amphiphilic random copolymers in water. Macromolecules 2014, 47, 589–600. [Google Scholar] [CrossRef]
- Hirai, Y.; Terashima, T.; Takenaka, M.; Sawamoto, M. Precision self-assembly of amphiphilic random copolymers into uniform and self-sorting nanocompartments in water. Macromolecules 2016, 49, 5084–5091. [Google Scholar] [CrossRef]
- Heiler, C.; Offenloch, J.T.; Blasco, E.; Barner-Kowollik, C. Photochemically induced folding of single chain polymer nanoparticles in water. ACS Macro Lett. 2017, 6, 56–61. [Google Scholar] [CrossRef]
- Stals, P.J.M.; Gillissen, M.A.J.; Paffen, T.F.E.; de Greef, T.F.A.; Lindner, P.; Meijer, E.W.; Palmans, A.R.A.; Voets, I.K. Folding polymers with pendant hydrogen bonding motifs in water: The effect of polymer length and concentration on the shape and size of single-chain polymeric nanoparticles. Macromolecules 2014, 47, 2947–2954. [Google Scholar] [CrossRef]
- Ter Huurne, G.M.; de Windt, L.N.J.; Liu, Y.; Meijer, E.W.; Voets, I.K.; Palmans, A.R.A. Improving the folding of supramolecular copolymers by controlling the assembly pathway complexity. Macromolecules 2017, 50, 8562–8569. [Google Scholar] [CrossRef]
- Liao, Z.S.; Huang, S.Y.; Huang, J.J.; Chen, J.K.; Lee, A.W.; Lai, J.Y.; Lee, D.J.; Cheng, C.C. Self-assembled pH-responsive polymeric micelles for highly efficient, non-cytotoxic delivery of doxorubicin chemotherapy to inhibit macrophage activation: In vitro investigation. Biomacromolecules 2018, 19, 2772–2781. [Google Scholar] [CrossRef]
- Brochu, S.; Ampleman, G. Synthesis and characterization of glycidyl azide polymers using isotactic and chiral poly(epichlorohydrin)s. Macromolecules 1996, 29, 5539–5545. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.-Y.; Cheng, C.-C. Spontaneous Self-Assembly of Single-Chain Amphiphilic Polymeric Nanoparticles in Water. Nanomaterials 2020, 10, 2006. https://doi.org/10.3390/nano10102006
Huang S-Y, Cheng C-C. Spontaneous Self-Assembly of Single-Chain Amphiphilic Polymeric Nanoparticles in Water. Nanomaterials. 2020; 10(10):2006. https://doi.org/10.3390/nano10102006
Chicago/Turabian StyleHuang, Shan-You, and Chih-Chia Cheng. 2020. "Spontaneous Self-Assembly of Single-Chain Amphiphilic Polymeric Nanoparticles in Water" Nanomaterials 10, no. 10: 2006. https://doi.org/10.3390/nano10102006