From Chip Size to Wafer-Scale Nanoporous Gold Reliable Fabrication Using Low Currents Electrochemical Etching

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussions
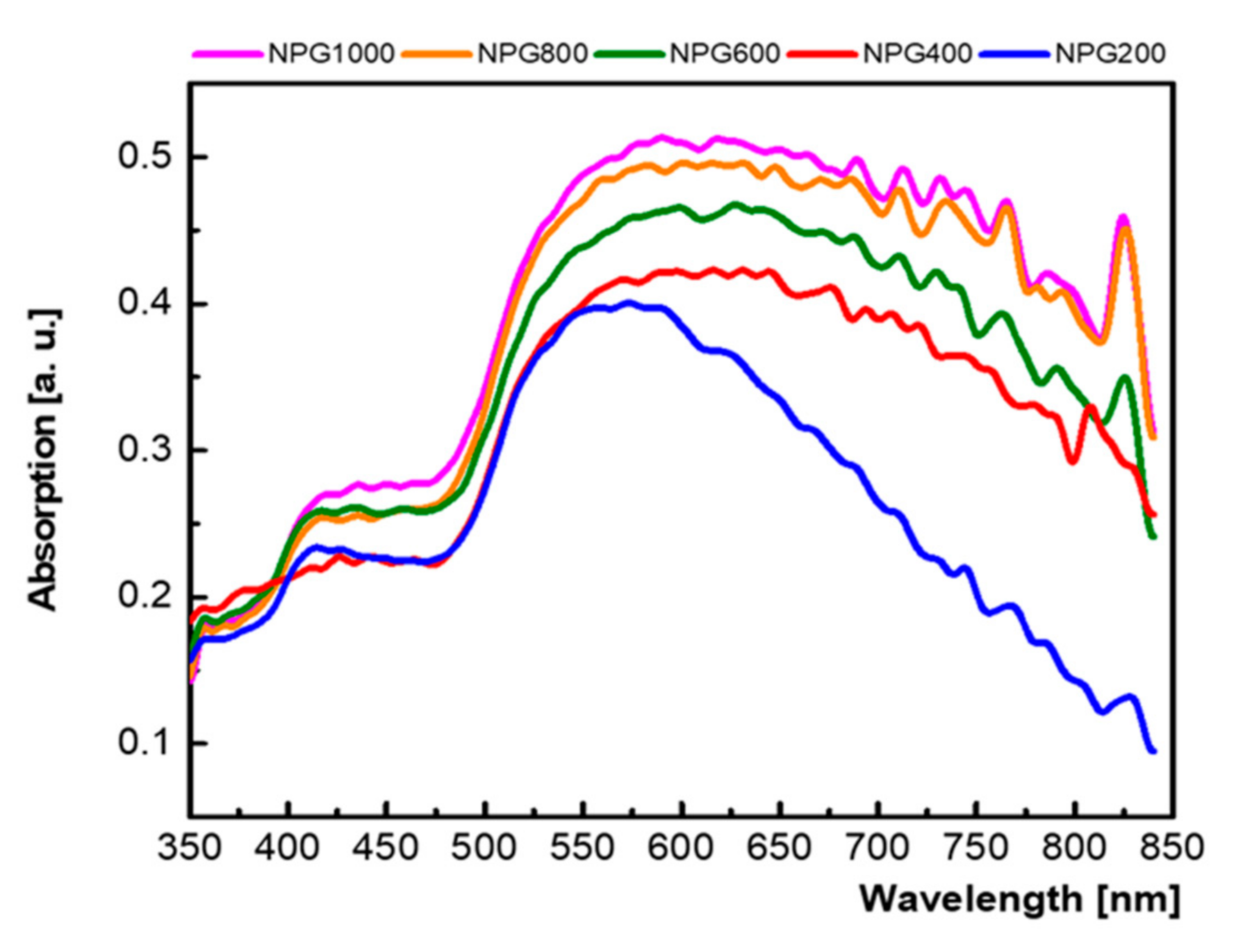
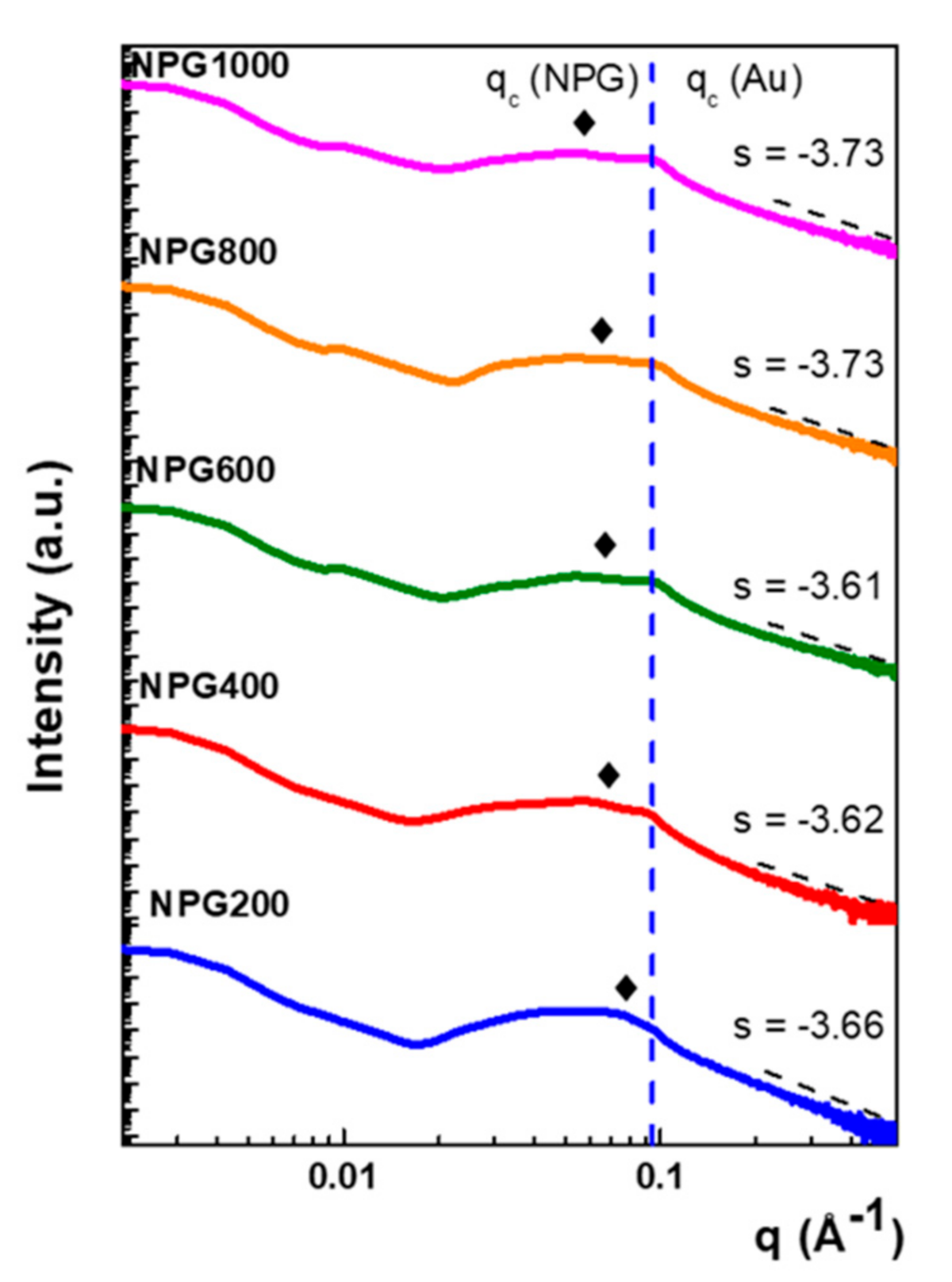
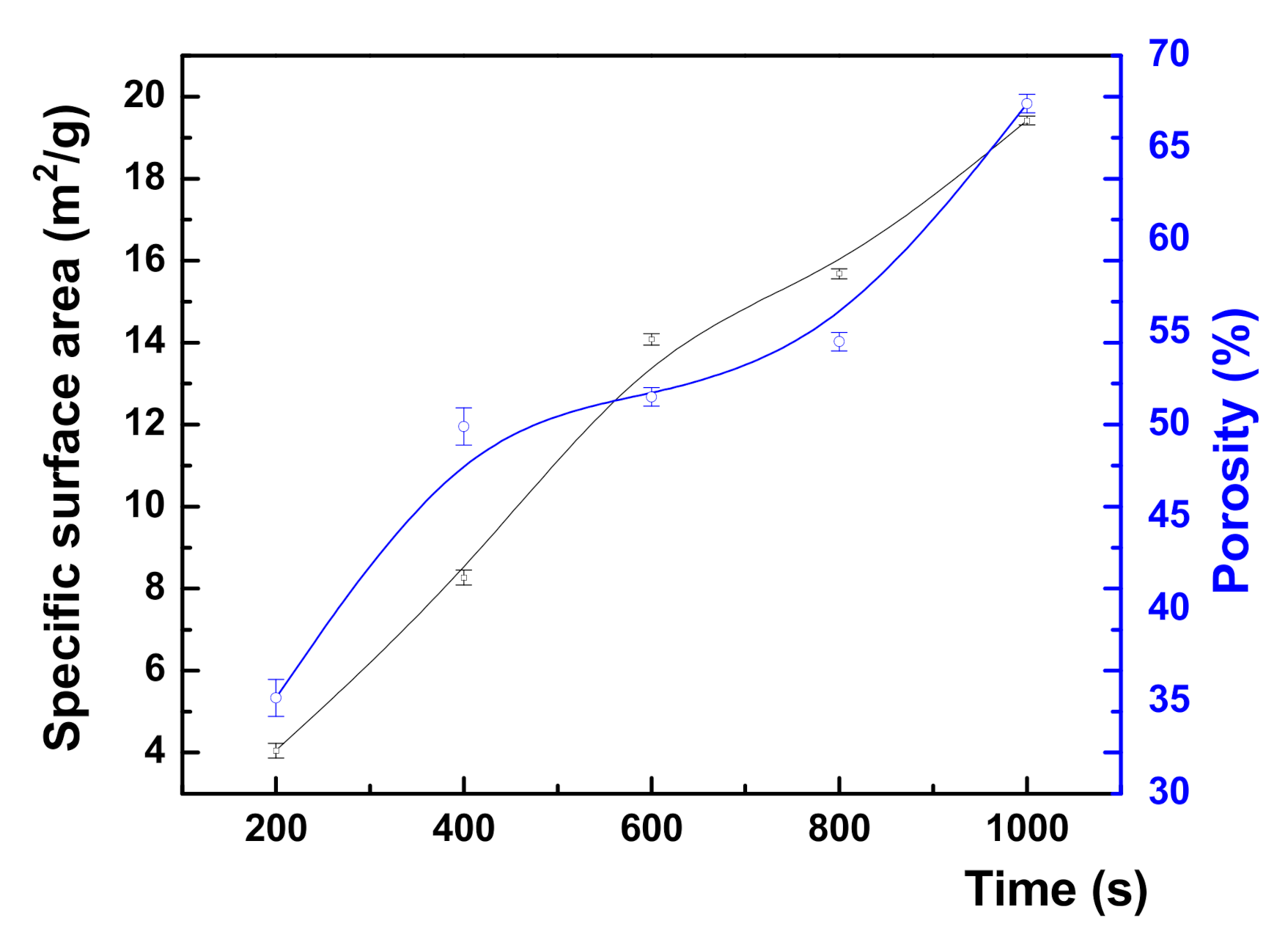
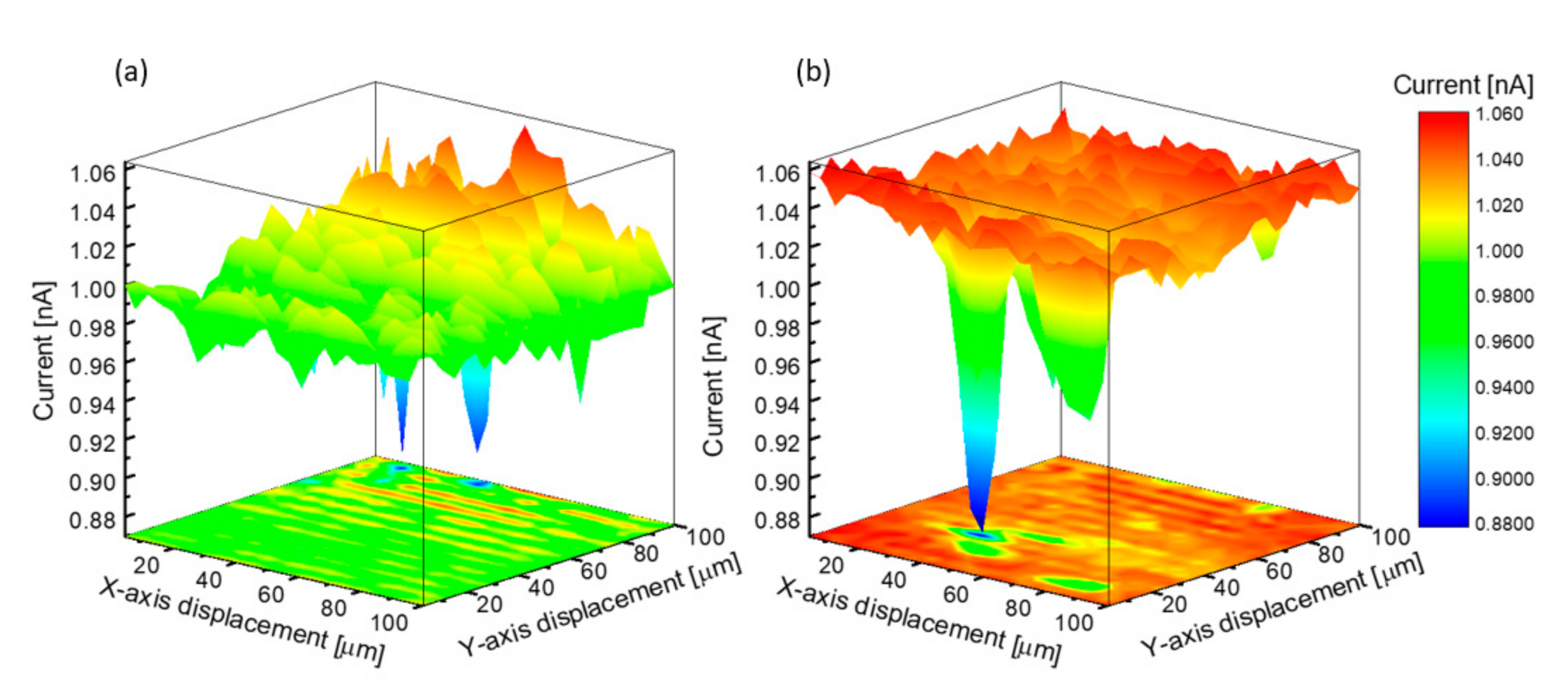
3.1. Micro-Structural and Optical Investigations
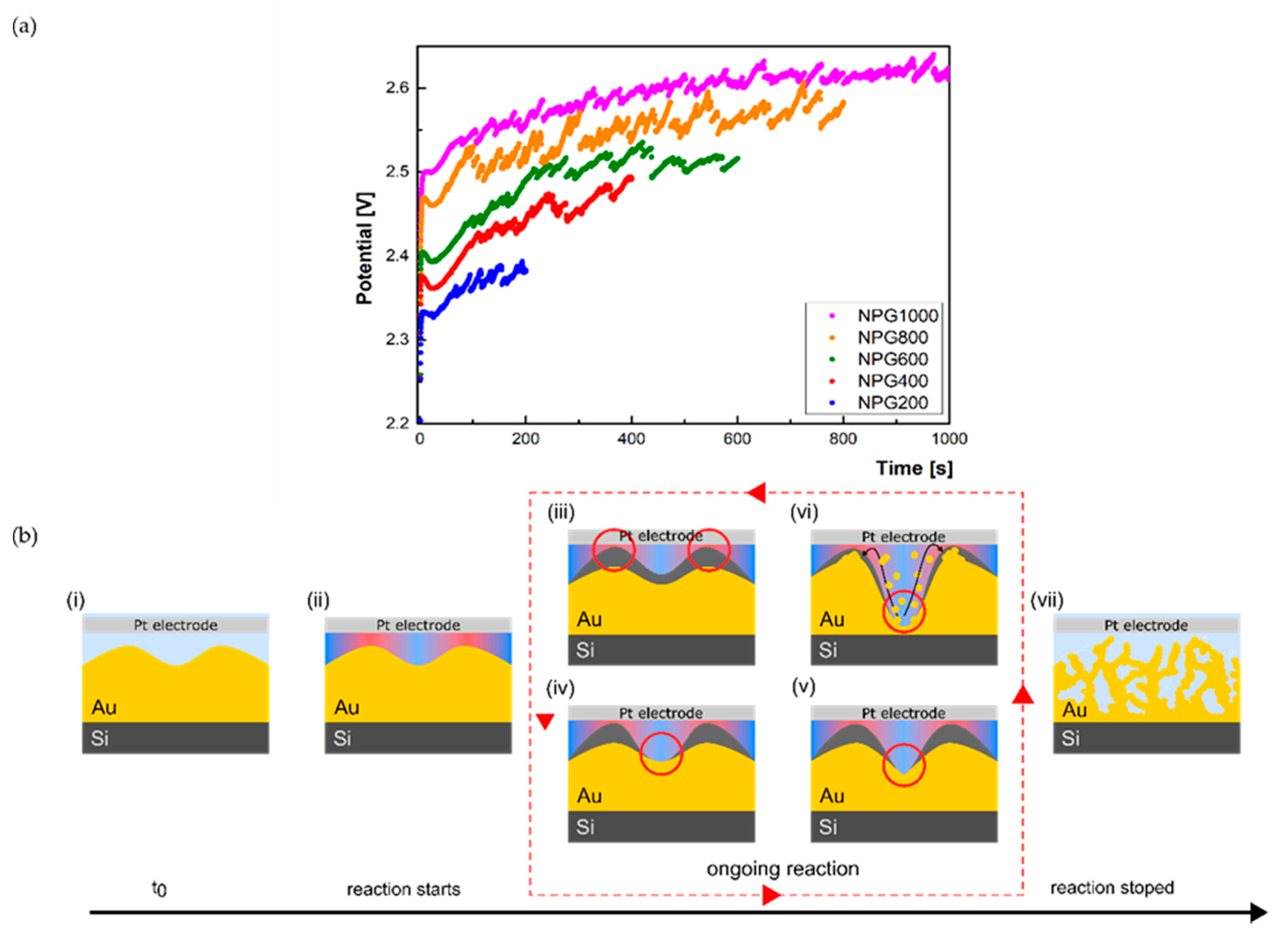
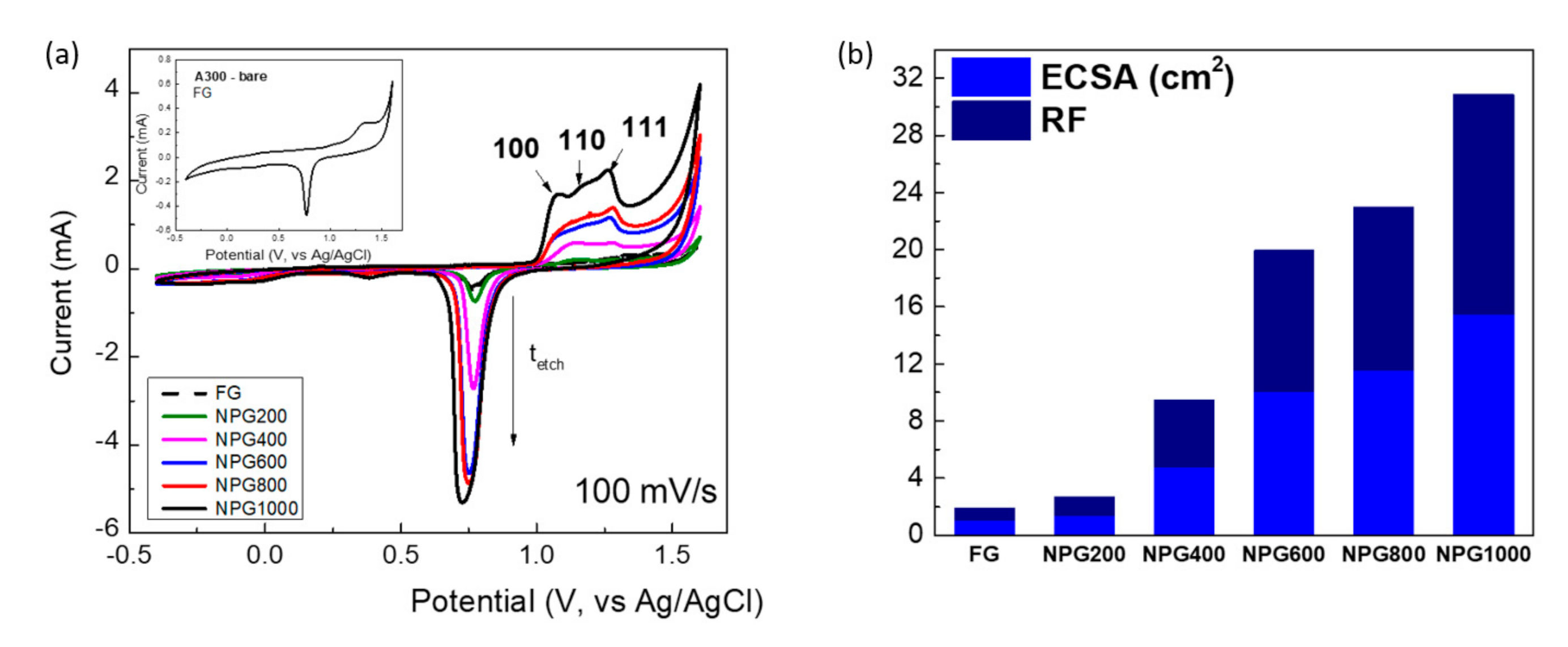
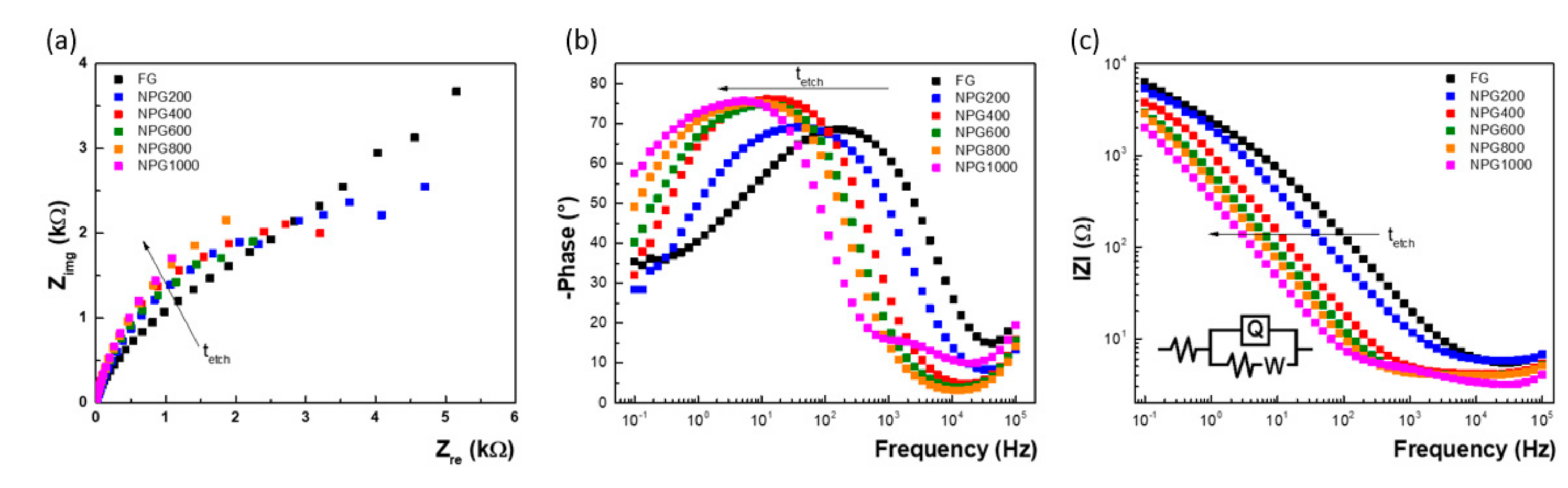
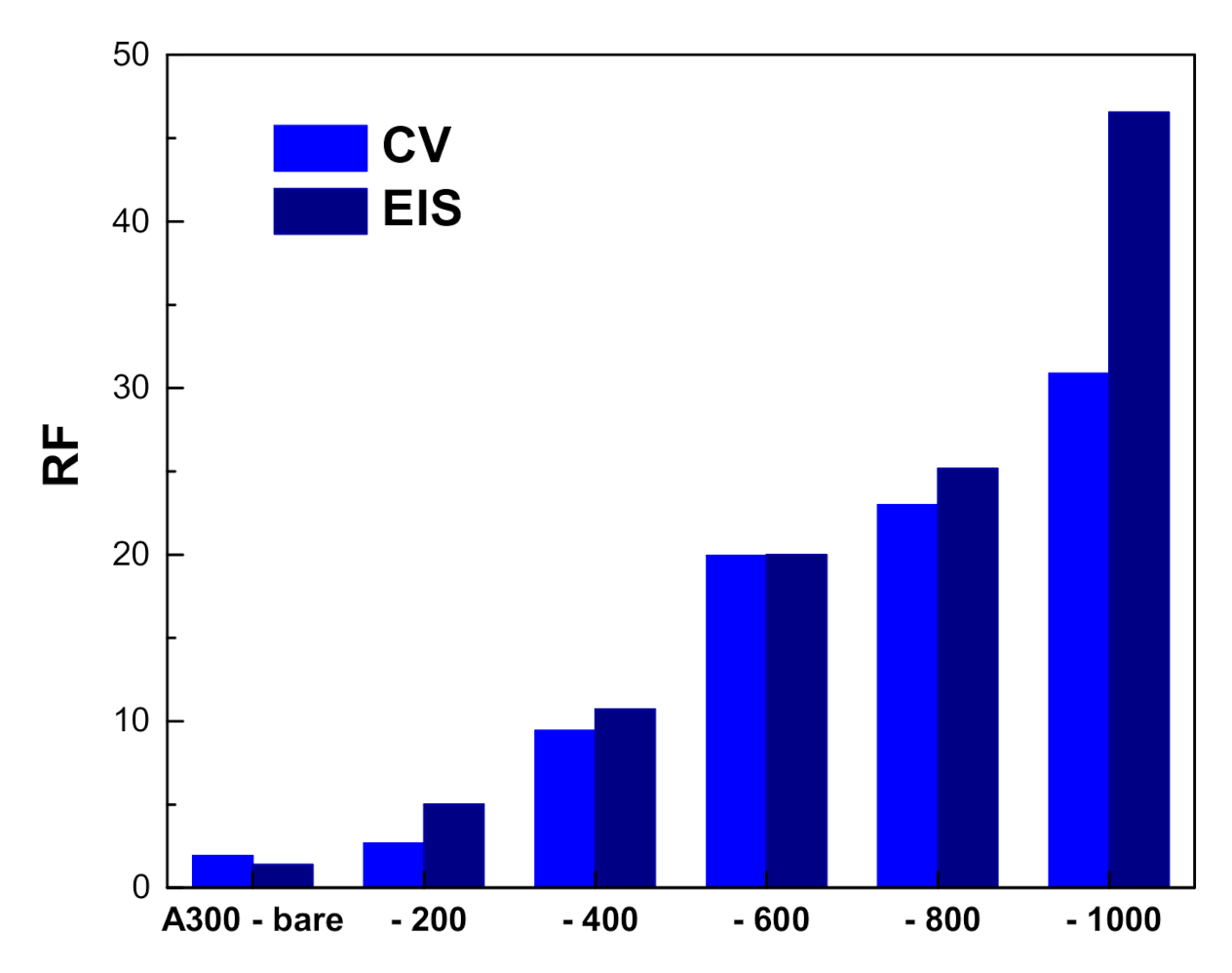
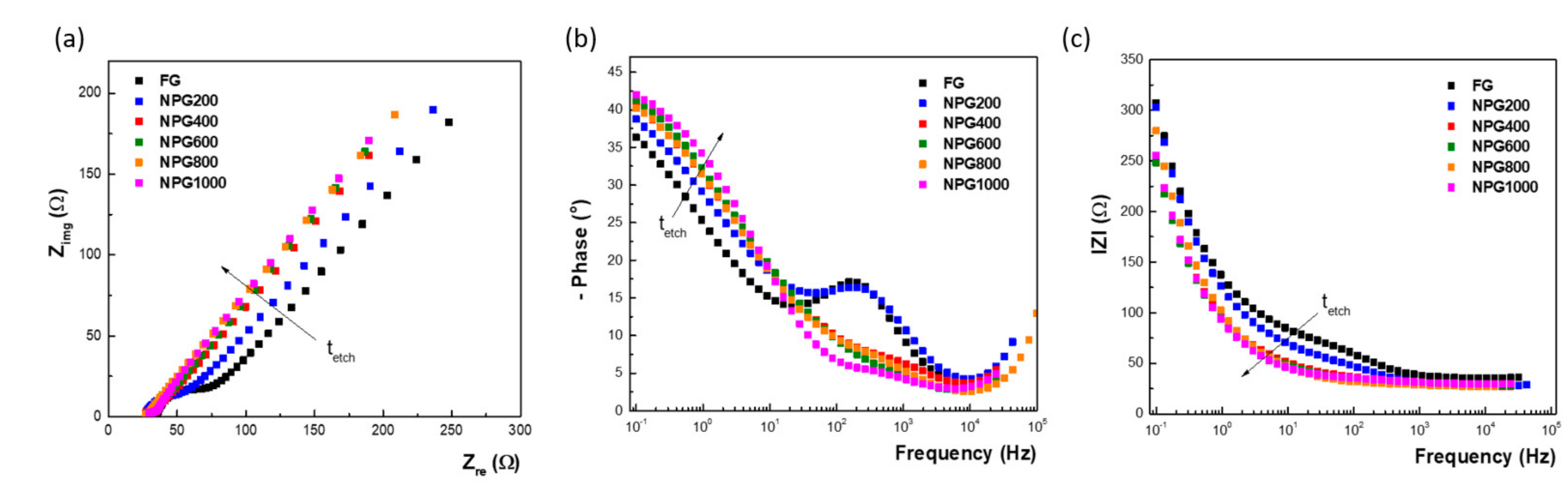
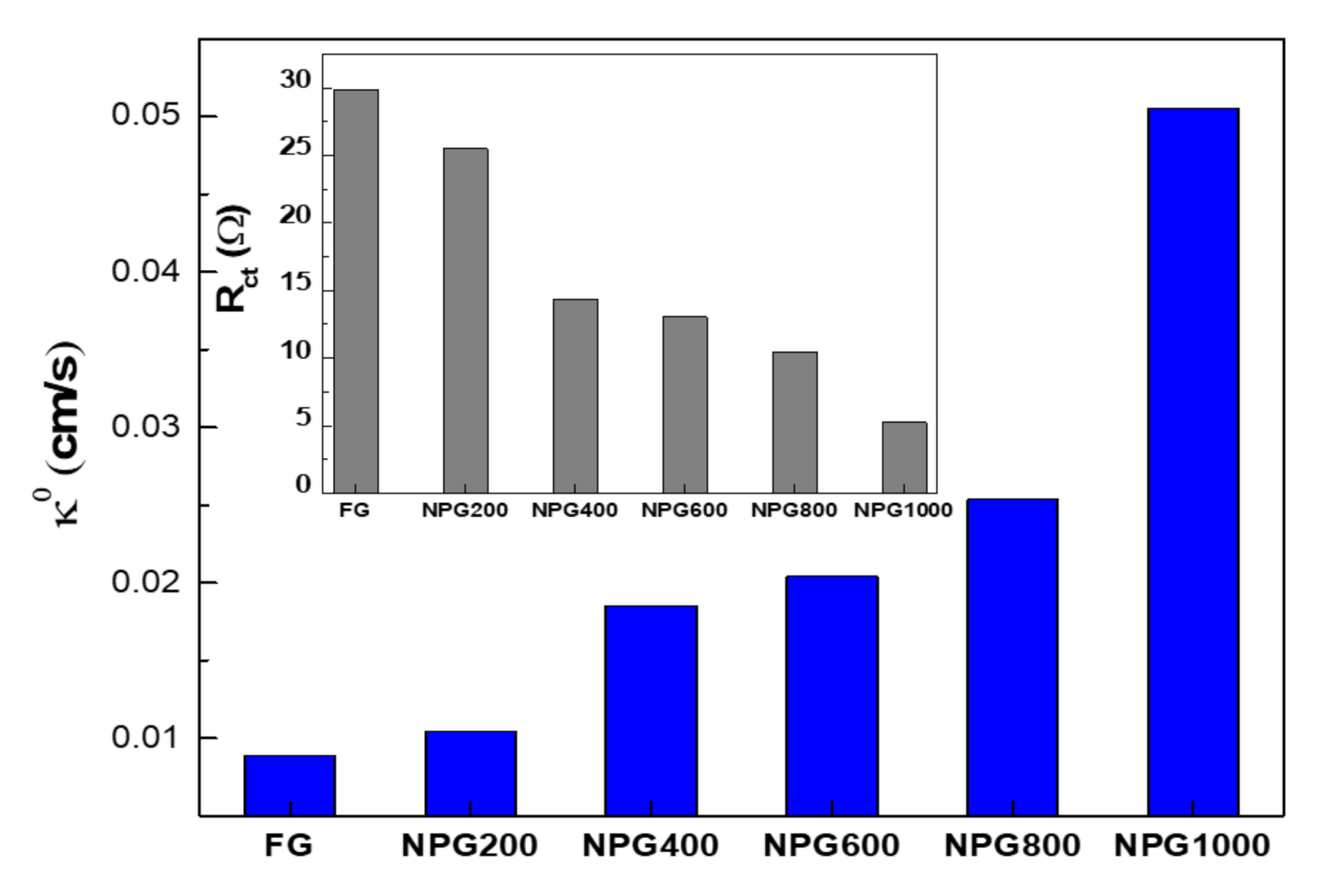
3.2. Electrochemical Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Forty, A.J. Corrosion micromorphology of noble metal alloys and depletion gilding. Nature 1979, 282, 597–598. [Google Scholar] [CrossRef]
- Pickering, H.W.; Swann, P.R. Electron Metallography of Chemical Attack Upon Some Alloys Susceptible to Stress Corrosion Cracking. Corrosion 1963, 19, 373t–389t. [Google Scholar] [CrossRef]
- Fang, C.; Bandaru, N.M.; Ellis, A.V.; Voelcker, N.H. Electrochemical fabrication of nanoporous gold. J. Mater. Chem. 2012, 22, 2952–2957. [Google Scholar] [CrossRef]
- Wittstock, A.; Biener, J.; Bäumer, M. Nanoporous gold: A new material for catalytic and sensor applications. Phys. Chem. Chem. Phys. 2010, 12, 12919–12930. [Google Scholar] [CrossRef]
- Xiao-ting, Z.H.U.; Lu-jia, Z.; Hong, T.A.O.; Jun-wei, D.I. Synthesis of Nanoporous Gold Electrode and Its Application in Electrochemical Sensor. Chin. J. Anal. Chem. 2013, 41, 693–697. [Google Scholar] [CrossRef]
- Collinson, M.M. Nanoporous Gold Electrodes and Their Applications in Analytical Chemistry. Int. Sch. Res. Not. 2013, 2013, 1–21. [Google Scholar] [CrossRef]
- Lee, K.U.; Byun, J.Y.; Shin, H.J.; Kim, S.H. A high-performance supercapacitor based on polyaniline-nanoporous gold. J. Alloys Compd. 2019, 779, 74–80. [Google Scholar] [CrossRef]
- Ferris, A.; Bourrier, D.; Garbarino, S.; Guay, D.; Pech, D. 3D Interdigitated Microsupercapacitors with Record Areal Cell Capacitance. Small 2019, 15, 1901224. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Kim, S.W.; Jung, K.; Kim, J.B.; Jang, J.H. Ideal nanoporous gold based supercapacitors with theoretical capacitance and high energy/power density. Nano Energy 2016, 24, 17–24. [Google Scholar] [CrossRef]
- Garoli, D.; Calandrini, E.; Giovannini, G.; Hubarevich, A.; Caligiuri, V.; De Angelis, F. Nanoporous gold metamaterials for high sensitivity plasmonic sensing. Nanoscale Horizons 2019, 4, 1153–1157. [Google Scholar] [CrossRef]
- Garoli, D.; Calandrini, E.; Bozzola, A.; Toma, A.; Cattarin, S.; Ortolani, M.; De Angelis, F.; Fisica, D.; Universita, S.; Moro, P.A. Fractal-Like Plasmonic Metamaterial with a Tailorable Plasma Frequency in the near-Infrared. ACS Photonics 2018, 5, 3408–3414. [Google Scholar] [CrossRef]
- Falsig, H.; Hvolbæk, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. Int. Ed. 2008, 47, 4835–4839. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Su, J.; Xu, X.; Liu, P.; Zhao, H.; Tian, F.; Ding, Y. Low temperature CO oxidation over unsupported nanoporous gold. J. Am. Chem. Soc. 2007, 129, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Zielasek, V.; Jürgens, B.; Schulz, C.; Biener, J.; Biener, M.M.; Hamza, A.V.; Bäumer, M. Gold catalysts: Nanoporous gold foams. Angew. Chem. Int. Ed. 2006, 45, 8241–8244. [Google Scholar] [CrossRef]
- Qadir, K.; Quynh, B.T.P.; Lee, H.; Moon, S.Y.; Kim, S.H.; Park, J.Y. Tailoring metal–oxide interfaces of inverse catalysts of TiO 2 /nanoporous-Au under hydrogen oxidation. Chem. Commun. 2015, 51, 9620–9623. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.C.; Village, N.T.; Hill, W.; Rg, R. Gold-Catalysed Oxidation of and David T Thompson. Gold Bull. 2000, 33, 41–50. [Google Scholar] [CrossRef]
- Kosinova, A.; Wang, D.; Baradács, E.; Parditka, B.; Kups, T.; Klinger, L.; Erdélyi, Z.; Schaaf, P.; Rabkin, E. Tuning the nanoscale morphology and optical properties of porous gold nanoparticles by surface passivation and annealing. Acta Mater. 2017, 127, 108–116. [Google Scholar] [CrossRef]
- Ruffato, G.; Garoli, D.; Cattarin, S.; Barison, S.; Natali, M.; Canton, P.; Benedetti, A. Microporous and Mesoporous Materials Patterned nanoporous-gold thin layers: Structure control and tailoring of plasmonic properties. Microporous Mesoporous Mater. 2012, 163, 153–159. [Google Scholar] [CrossRef]
- Kamiuchi, N.; Sun, K.; Aso, R.; Tane, M.; Tamaoka, T.; Yoshida, H.; Takeda, S. Self-Activated surface dynamics in gold catalysts under reaction environments. Nat. Commun. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Calandrini, E.; Giovannini, G.; Garoli, D. 3D nanoporous antennas as a platform for high sensitivity IR plasmonic sensing. Opt. Express 2019, 27, 25912. [Google Scholar] [CrossRef]
- Jia, F.; Yu, C.; Ai, Z.; Zhang, L. Fabrication of Nanoporous Gold Film Electrodes with Ultrahigh Surface Area and Electrochemical Activity. Chem. Mater. 2007, 19, 3648–3653. [Google Scholar] [CrossRef]
- Rebbecchi, T.A.; Chen, Y. Template-based fabrication of nanoporous metals. J. Mater. Res. 2018, 33, 2–15. [Google Scholar] [CrossRef]
- Patel, J.; Radhakrishnan, L.; Zhao, B.; Uppalapati, B.; Daniels, R.C.; Ward, K.R.; Collinson, M.M. Electrochemical Properties of Nanostructured Porous Gold Electrodes in Biofouling Solutions. Anal. Chem. 2013, 85, 11610–11618. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lee, W.; Huang, Z.; Scholz, R. Fabrication and characterization of a flow-through nanoporous gold nanowire / AAO composite membrane. Nanotechnology 2008, 19, 335604. [Google Scholar] [CrossRef]
- Beets, N.; Stuckner, J.; Murayama, M.; Farkas, D. Fracture in nanoporous gold: An integrated computational and experimental study. Acta Mater. 2020, 185, 257–270. [Google Scholar] [CrossRef]
- Mattarozzi, L.; Cattarin, S.; Comisso, N.; Guerriero, P.; Musiani, M.; Verlato, E. Electrochimica Acta Preparation of porous nanostructured Ag electrodes for sensitive electrochemical detection of hydrogen peroxide. Electrochim. Acta 2016, 198, 296–303. [Google Scholar] [CrossRef]
- Rouya, E.; Cattarin, S.; Reed, M.L.; Kelly, R.G.; Zangari, G.; Energetica, L.; Uniti, C.S. Electrochemical Characterization of the Surface Area of Nanoporous Gold Films. J. Electrochem. 2012, 159, K97. [Google Scholar] [CrossRef]
- Nishio, K.; Masuda, H. Anodization of Gold in Oxalate Solution To Form a Nanoporous Black film. Angew. Chem. Int. Ed. Engl. 2011, 50, 1603–1607. [Google Scholar] [CrossRef]
- Simion, M.; Kleps, I.; Neghina, T.; Angelescu, A.; Miu, M.; Bragaru, A.; Danila, M.; Condac, E.; Costache, M.; Savu, L. Nanoporous silicon matrix used as biomaterial. J. Alloys Compd. 2007, 434–435, 830–832. [Google Scholar] [CrossRef]
- Ruffato, G.; Romanato, F.; Garoli, D.; Cattarin, S. Nanoporous gold plasmonic structures for sensing applications. Optics Express 2011, 19, 13164–13170. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. HISTORICAL commentary NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Asoh, H.; Uehara, A.; Ono, S. Fabrication of porous silica with three-dimensional periodicity by Si anodization. Jpn. J. Appl. Phys. Part 2 Lett. 2004, 43, 43–46. [Google Scholar] [CrossRef]
- Parkhutik, V.; Curiel-Esparza, J.; Millan, M.C.; Albella, J. Spontaneous layering of porous silicon layers formed at high current densities. Phys. Status Solidi Appl. Mater. Sci. 2005, 202, 1576–1580. [Google Scholar] [CrossRef]
- Zhang, X.G. Electrochemistry of Silicon and Its Oxide; Springer Science & Business Media: Heidelberg/Berlin, Germany, 2007. [Google Scholar]
- Mohr, F. The chemistry of gold-fluoro compounds: A continuing challenge for gold chemists. Gold Bull. 2004, 37, 164–169. [Google Scholar] [CrossRef]
- Fang, C.; Ellis, A.V.; Voelcker, N.H. Electrochemically prepared porous silver and its application in surface-enhanced Raman scattering. J. Electroanal. Chem. 2011, 659, 151–160. [Google Scholar] [CrossRef]
- Lang, X.; Qian, L.; Guan, P.; Zi, J.; Chen, M. Localized surface plasmon resonance of nanoporous gold. Appl. Phys. Lett. 2011, 98, 2009–2012. [Google Scholar] [CrossRef]
- Romanitan, C.; Varasteanu, P.; Mihalache, I.; Culita, D.; Somacescu, S.; Pascu, R.; Tanasa, E.; Eremia, S.A.V.; Boldeiu, A.; Simion, M.; et al. High-performance solid state supercapacitors assembling graphene interconnected networks in porous silicon electrode by electrochemical methods using 2,6-dihydroxynaphthalen. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Bowen, K.; Tanner, B. High Resolution X-ray Diffractometer and Topography; Taylor & Francis: London, UK, 1998; ISBN 0850667585. [Google Scholar]
- Daillant, J.; Gibaud, A. X-Ray and Neutron Reflectivity: Principles and Applications; Springer: Berlin/Heidelberg, Germany, 1999; ISBN 3540661956. [Google Scholar]
- Birkholz, M.; Birkholz, M.; Fewster, P.F.; Genzel, C. Thin Film Analysis by X-Ray Scattering; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 9783527310524. [Google Scholar]
- Miu, M.; Danila, M.; Kleps, I.; Bragaru, A.; Simion, M. Nanostructure and internal strain distribution in porous silicon. J. Nanosci. Nanotechnol. 2011, 11, 9136–9142. [Google Scholar] [CrossRef]
- Xiao, X.; Si, P.; Magner, E. An overview of dealloyed nanoporous gold in bioelectrochemistry. Bioelectrochemistry 2016, 109, 117–126. [Google Scholar] [CrossRef]
- Jeyabharathi, C.; Ahrens, P.; Hasse, U.; Scholz, F. Identification of low-index crystal planes of polycrystalline gold on the basis of electrochemical oxide layer formation. J. Solid State Electrochem. 2016, 20, 3025–3031. [Google Scholar] [CrossRef]
- Valenti, G.; Scarabino, S.; Goudeau, B.; Lesch, A.; Jović, M.; Villani, E.; Sentic, M.; Rapino, S.; Arbault, S.; Paolucci, F.; et al. Single Cell Electrochemiluminescence Imaging: From the Proof-of-Concept to Disposable Device-Based Analysis. J. Am. Chem. Soc. 2017, 139, 16830–16837. [Google Scholar] [CrossRef] [PubMed]
- Bisquert, J.; Garcia-Belmonte, G.; Bueno, P.; Longo, E.; Bulhões, L.O.S. Impedance of constant phase element (CPE)-blocked diffusion in film electrodes. J. Electroanal. Chem. 1998, 452, 229–234. [Google Scholar] [CrossRef]
- Francis, N.J.; Knospe, C.R. Fabrication and Characterization of Nanoporous Gold Electrodes for Sensor Applications. Adv. Eng. Mater. 2019, 21, 1–13. [Google Scholar] [CrossRef]
- Bae, J.H.; Yu, Y.; Mirkin, M.V. Scanning Electrochemical Microscopy Study of Electron-Transfer Kinetics and Catalysis at Nanoporous Electrodes. J. Phys. Chem. C 2016, 120, 20651–20658. [Google Scholar] [CrossRef]
- Bard, A.J.; Fan, F.R.F.; Kwak, J.; Lev, O. Scanning Electrochemical Microscopy. Introduction and Principles. Anal. Chem. 1989, 61, 132–138. [Google Scholar] [CrossRef]
- Haensch, M.; Balboa, L.; Graf, M.; Silva Olaya, A.R.; Weissmüller, J.; Wittstock, G. Mass Transport in Porous Electrodes Studied by Scanning Electrochemical Microscopy: Example of Nanoporous Gold. ChemElectroChem 2019, 6, 3160–3166. [Google Scholar] [CrossRef]
- Seker, E.; Berdichevsky, Y.; Begley, M.R. The fabrication of low-impedance nanoporous gold multiple-electrode arrays for neural electrophysiology studies. Nanotechnology 2010, 21, 125504. [Google Scholar] [CrossRef]
- Hu, K.; Lan, D.; Li, X.; Zhang, S. Electrochemical DNA Biosensor Based on Nanoporous Gold Electrode and Multifunctional Encoded DNA-Au Bio Bar Codes. Anal. Chem. 2008, 80, 9124–9130. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | (%) | ||||
|---|---|---|---|---|---|
| NPG200 | 12.5 ± 0.2 | 35.2 ± 1 | 26 ± 3.15 | 2.34 | 1.92 |
| NPG400 | 9.67 ± 0.2 | 49.9 ± 1 | 46 ± 1.08 | 2.38 | 1.86 |
| NPG600 | 9.37 ± 0.1 | 51.5 ± 0.05 | 56 ± 1.32 | 2.39 | 1.76 |
| NPG800 | 8.79 ± 0.1 | 54.5 ± 0.05 | 63 ± 1.6 | 2.27 | 1.71 |
| NPG1000 | 6.29 ± 0.1 | 67.4 ± 0.05 | 66 ± 1.81 | 2.27 | 1.69 |
| Sample | Rg (nm) | dcryst. (nm) | Sn (m2/g) | ||
|---|---|---|---|---|---|
| NPG200 | 20.3 | 57.4 | 122.75 | 0.87 | 3.67 ± 0.18 |
| NPG400 | 22.1 | 62.5 | 59.83 | 0.61 | 9.44 ± 0.18 |
| NPG600 | 18.0 | 50.9 | 68.37 | 1.15 | 15.77 ± 0.14 |
| NPG800 | 16.9 | 47.8 | 63.27 | 1.12 | 17.70 ± 0.12 |
| NPG1000 | 16.2 | 45.8 | 67.81 | 1.20 | 17.96 ± 0.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varasteanu, P.; Romanitan, C.; Bujor, A.; Tutunaru, O.; Craciun, G.; Mihalache, I.; Radoi, A.; Kusko, M. From Chip Size to Wafer-Scale Nanoporous Gold Reliable Fabrication Using Low Currents Electrochemical Etching. Nanomaterials 2020, 10, 2321. https://doi.org/10.3390/nano10112321
Varasteanu P, Romanitan C, Bujor A, Tutunaru O, Craciun G, Mihalache I, Radoi A, Kusko M. From Chip Size to Wafer-Scale Nanoporous Gold Reliable Fabrication Using Low Currents Electrochemical Etching. Nanomaterials. 2020; 10(11):2321. https://doi.org/10.3390/nano10112321
Chicago/Turabian StyleVarasteanu, Pericle, Cosmin Romanitan, Alexandru Bujor, Oana Tutunaru, Gabriel Craciun, Iuliana Mihalache, Antonio Radoi, and Mihaela Kusko. 2020. "From Chip Size to Wafer-Scale Nanoporous Gold Reliable Fabrication Using Low Currents Electrochemical Etching" Nanomaterials 10, no. 11: 2321. https://doi.org/10.3390/nano10112321
APA StyleVarasteanu, P., Romanitan, C., Bujor, A., Tutunaru, O., Craciun, G., Mihalache, I., Radoi, A., & Kusko, M. (2020). From Chip Size to Wafer-Scale Nanoporous Gold Reliable Fabrication Using Low Currents Electrochemical Etching. Nanomaterials, 10(11), 2321. https://doi.org/10.3390/nano10112321