3.1. Growth Mechanism: Anodization Curves with Different Organic Solvents
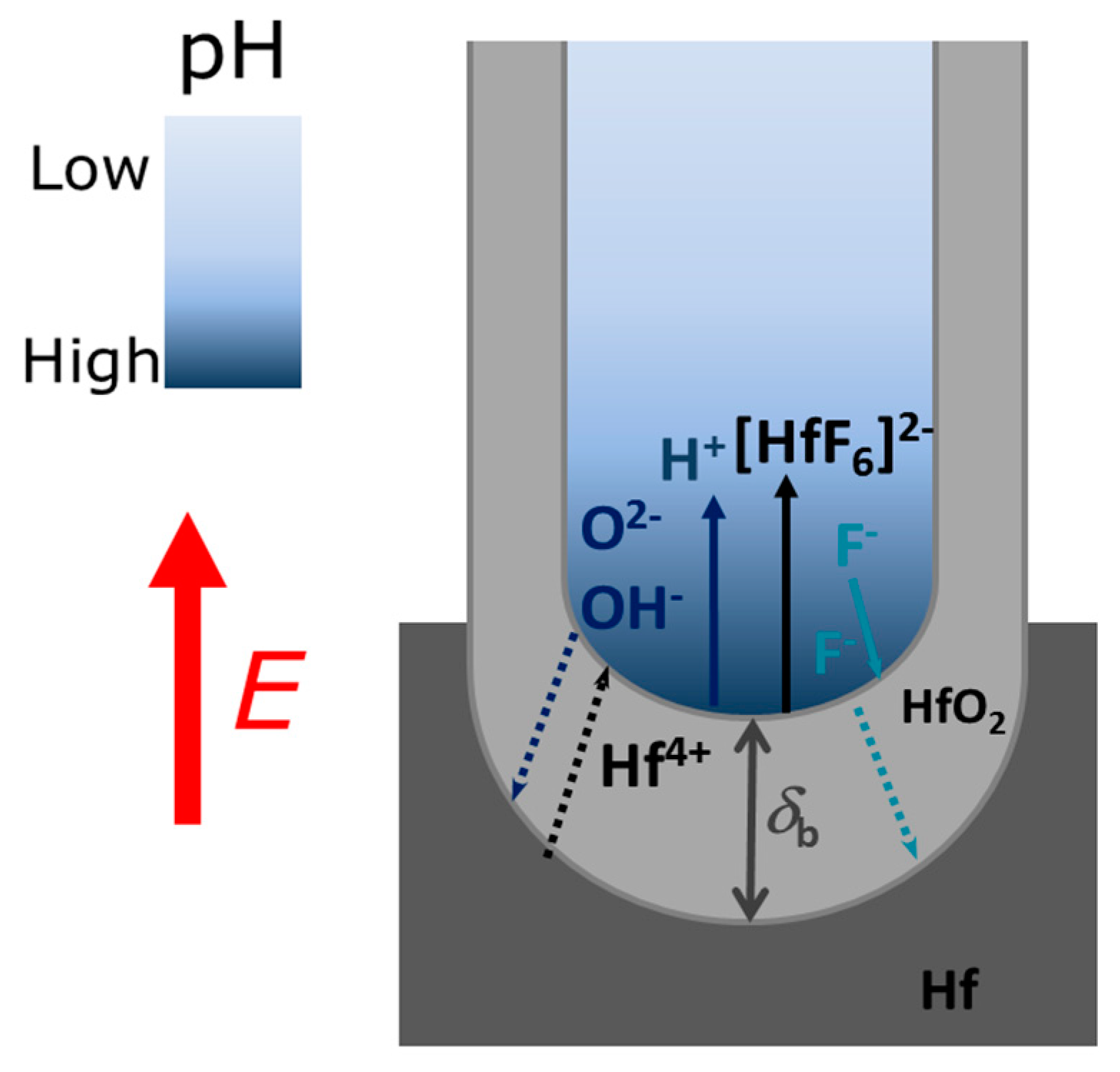
The main mechanisms responsible for the formation of NTs by an Hf anodization processes are: (i) The electric field-assisted oxidation at the metal/oxide interface, forming an HfO
2 continuous layer; (ii) the field-assisted dissolution of the oxide layer (at the oxide/electrolyte interface); and (iii) the chemical dissolution of the oxide by F
− ions at the metal/oxide and electrolyte/oxide interfaces (
Figure 1). The electrochemical equations for HfO
2 formation are:
and
for the oxidation (1) and dissolution (2) reactions, respectively (
Figure 1). The reactions occurring at the anode are oxidation of the metal, that releases Hf
4+ ions and electrons:
whereas, in the electrolyte one has the dissociation of water:
Differently from the Al metal anodization case [
25,
26], where a steady-state condition is achieved (oxidation rate is balanced with the dissolution rate), the Hf anodization case (as that of Ti anodization) consists in a non-steady state anodization process with higher oxidation than dissolution rates [
24]. Such effect severely compromises the HfO
2 NTs length and growth [
24]. There are also additional chemical dissolution effects during the anodization that affect the oxidation/dissolution equilibrium and limit NTs growth.
The evaluation of the mechanisms that lead to the formation and growth of self-ordered HfO
2 nanoporous/nanotubes, can be studied using current density [
j(
t)] curves (
Figure 2) [
23,
24,
25,
26,
27]. The evolution of the HfO
2 barrier layer thickness (
δb) at the bottom of the NTs (
Figure 1) was also estimated from the
j(
t) curves (
Figure 3) [
27]. According to the high-field conduction theory [
28], the current density (
j) is related to the voltage (
V) drop across the barrier layer as follows:
where
α and
β are electrolyte and material-dependent constants and the (
V/
δb) ratio is the effective electric-field across
δb [
27,
28]. From Equation (6) we obtain,
during the anodization [
24,
27]. It was considered the material constants,
α = 2.4 × 10
−9 mA·cm
−2 and
β = 27.98 nm·V
−1 (at room temperature), determined previously for TiO
2 [
27], and due to the physical similarities between these oxides here are also considered.
Figure 3 shows the evolutions of
δb along the anodization time calculated from the Equation (7).
j(
t) curves for all the samples (EG, FA, NMF and DMSO;
Figure 2 and corresponding inset) present the transient anodization characteristic of the successful formation of nanopore/nanotube arrays (as in the Al or Ti cases [
23,
24,
25,
26,
27]). After applying 60 V, a continuous HfO
2 layer is rapidly formed that leads to a resistance increase [rapid
j decrease (inset of
Figure 2) and
δb increase (
Figure 3)]. The following slight
j decrease marks the initiation of pore nucleation, likely on the surface valley-type irregularities where the electric field enhances the oxide dissolution and promotes hole formation (i.e., the dissolution promoted by F
− ions in favorable spots of the HfO
2 surface) [
21,
24]. Consequently, the HfO
2 layer thickness starts to increase, while the pores/tubes formation accelerates. This is evidenced by the increase of
j until a maximum is reached. A barrier layer, with thickness δ
b, forms at the pores/tubes bottom (
Figure 1 and
Figure 3). Afterwards, the emerging porous structure will mechanically adjust and compete with each other in a self-organization process.
The differences between
j(
t) transient periods of each sample (inset of
Figure 2) clearly reveals the decisive importance of the electrolyte solvent in promoting effective nucleation spots. In fact, comparing the
j(
t) transient period of each sample, we can observe three main aspects: (i) The lower
j(
t) values; (ii) the earlier emergence of NT nucleation; and (iii) the smaller nucleation time are attributed to samples FA, NMF, EG and DMSO, respectively.
As the anodization process evolves,
j(
t) of samples FA and EG present similar trends with the typical
j(
t)-decay of Ti anodization in fluoride-based electrolytes with EG [
23,
24,
25,
26,
27]. This behavior arises from the non-equilibrium in the oxidation/dissolution processes, being the HfO
2 dissolution lower than its formation, resulting in a slow decay of
j(
t) during the anodization. As a result, a progressive increase of δ
b of the NTs’ occurs, as shown in
Figure 3. Consequently, the ionic migration path along the oxide barrier [
27,
29] significantly extends, inhibiting the transport of F
−, Hf
4+ and O
2− ions across δ
b (Hf
4+ and O
2− for oxidation, F
− for dissolution) which subsequently limits a further NTs growth (
Figure 1). Additionally, chemical effects, such as local pH decrease, occur throughout the anodization leading to the chemical dissolution of the NTs wall preferentially at the NTs tops (V-shape NTs) [
17,
18,
27,
29,
30]. The NMF
j(
t) curve presents a large decay up to 8 min, similar to EG and FA, but then an overall constant
j(
t) emerges, although with some singularities during the anodization.
On the other hand, in the case of the DMSO, the
j(
t) curve remains approximately constant throughout the anodization, indicating a more optimized anodization for NTS growth, with balanced oxidation/dissolution processes that lead to a constant oxide growth rate (similar with Al nanoporous anodization were no limit in length is imposed [
25,
26]). Additionally, the DMSO
j(
t) transient curve shows an extended nucleation period of time (indication of low-rate pore nucleation) [
24] with fairly smaller
j(
t) values over such region [indicating a thinner δ
b (
Figure 3)] when comparing with other samples (FA or EG) [
24,
27].
Additionally,
Figure 3 shows that by changing the electrolyte medium, we obtain different final δ
b. Furthermore, for each sample, the capacitance density (C) at the oxide barrier was also calculated from the
j(
t) curves [
Supporting Information (SI) - Figure S3 and Table S1]. At the end of the anodization (1 h), we can extract the final δ
b and C for each sample (
SI - Table S1). It shows that the FA/EG samples led to thicker δ
b (and lower C) and DMSO/NMF led to thinner δ
b (and higher C).
Moreover, we perform additional anodizations with the same conditions as for the samples EG, FA, NMF and DMSO during 17 h (
SI - Figure S1). In this case,
j(
t) of the sample NMF rapidly decays after 444 min. This corresponds that a complete conversion on the Hf foil into HfO
2 has occurred at this time (444 min), as observed by SEM cross-section images (
SI - Figure S2).
3.2. Growth Rate with Different Organic Solvents
Figure 4 shows the charge curves
Q(
t) obtained from the integration of the
j(
t) data. The
Q(
t) curves describe the growth rate along the anodization [
24]. Until the first 13 min both
Q(
t) slopes of NMF and FA samples are higher than those of the DMSO and EG. Although the NMF sample presents a higher charge over time for the entire anodization period (comparing to the rest of the samples),
Q(
t) of the DMSO sample overcomes that of the FA sample at the end of the anodization period (close to 50 min). Higher
Q(
t) curve indicates higher charge transfer, leading to a higher growth rate. The charge transferred during the anodization process can then be related to the solvent characteristics and
Q(
t) values. Additionally, the
Q(
t) curves present different slope’s trend: Whereas, in NMF and DMSO samples the slope is fairly linear, providing an almost constant AHO growth rate, that is not the case for the EG and FA samples, where
Q(
t) has a non-linear slope and presents two distinct growth rate regimes. After 11 min and 13 min of anodization time, for EG and FA, respectively, the growth rate slows down. With these electrolytes, the δ
b increases during the anodization, leading to a constant
Q(
t) over time [and significantly lower final
Q(
t) values]. As discussed before, the δ
b increase is related to the unbalanced oxidation-dissolution rate reactions, being the HfO
2 dissolution smaller than its formation, ultimately limiting the NTs growth and length [
24,
27]. δ
b(
t) curves (
Figure 3) of NMF/DMSO samples present thinner δ
b, while EG/FA samples shows thicker δ
b. One can observe the similar
Q(t) trend of the two groups of samples EG/FA and NMF/DMSO. EG and FA presents the transition of two regimes at 11 min and 13 min, respectively. After these anodization times, δ
b greatly increases (
Figure 3) and the growth rate slows down. On the other hand, NMF/DMSO samples show a
Q(
t) linear slope, corresponding to a constant δ
b over time (
Figure 3). At the end, EG/FA samples presents thicker δ
b than NMF/DMSM samples.
3.3. Morphology and Layer Thickness
Figure 5,
Figure 6,
Figure 7 and
Figure 8 shows SEM cross-section, and top view images of the AHO templates for all samples after 1 h of anodization. Comparing the different samples, one sees that the electrolyte solvent has a critical impact on the AHO morphology, growth rate and layer thickness. From the top view images, we can see that FA leads to a self-ordered nanoporous template (
Figure 5), while EG and DMSO lead to highly self-ordered NT arrays with hexagonal closely packed distribution (
Figure 6 and
Figure 7, respectively). For the NMF samples (
Figure 8), instead of homogeneous NTs or nanoporous structures, we obtained different morphologies, ranging from
Figure 8a a continuous oxide layer,
Figure 8b nanoporous,
Figure 8c nanoneedles,
Figure 8d nanoflakes or
Figure 8e agglomerated nanowires.
EDS analyses showed (
SI-Figure S5) that the anodic as-grown nanoporous (FA) nanotubes (DMSO/EG) presents significant amounts of F, which is typical of anodic HfO
2 or TiO
2 structures [
14,
18]. Literature associates the F presence with the formation of hafnium oxyfluoride in the AHO. For the sample NMF the same the F presence was obtained (
SI - Figure S5d). Additionally, in NMF sample we perform separated EDS analyses for bulk oxide (
SI - Figure S5e: Z1 area), and surface top nanostructures (flakes/needles;
SI - Figure S5f: Z2 area) identifying the same chemical elements in both areas and are in accordance with the other anodic hafnium oxide samples.
The pore diameter (
Dp), and interpore distance (
Dint) geometrical parameters were extracted from the SEM image (100 pores analyzed) for the EG, FA and DMSO samples as shown in
Table 1. The average
Dp and
Dint (and standard deviation SD) were estimated from the histogram of the size distribution, which were then fitted to a normal distribution (
Figure 9).
Usually, the regularity of the geometrical patterns in self-ordered nanoporous/nanotubes of anodic TiO
2, Al
2O
3 or HfO
2 is analyzed by a typical parameter—the porosity (
P). For the well-defined hexagonal porous structures,
P of the anodic oxide layer is given by the equation proposed by Nielsch et al.:
where
r is the pore radius (
r = D
p/2) [
31]. For the hexagonal self-ordered nanoporous Al
2O
3, or TiO
2 NT arrays the obtained porosities are close to 10% (10% porosity rule) when mild anodizations are implemented (low anodization potentials). This rule assumes that a perfect hexagonal structure shows a
P of 10% and deviations from these values results in the imperfect ordering of the structures. Qiu et al. presented a study of porosity for anodic HfO
2 NTs (electrolyte with ethylene glycol as solvent) and obtained porosity values of 10% when the anodizations were performed within the range of 10 to 40 V [
14]. In this work,
P was calculated for the samples FA, EG and DMSO, as shown in
Table 1. For the FA and DMSO samples, porosities closer to 10% (of ~9.3 and 14.2%, respectively) were obtained, consistent with the 10% rule. However, for the EG sample
P clearly deviates from the 10% rule (
P~18.1%).
The resulting AHO layer thicknesses are shown in
Table 2. The EG, FA and DMSO samples have a mean AHO layer thickness (
L) of approximately 8.0; 23.6 and 37.3 µm, respectively. On the other hand, the NFM sample shows a rapid AHO growth rate with
L = 94.8 µm, much larger than the other samples. Notice that while previously discussing the
Q(
t) curves (
Figure 3), the higher final
Q(
t) was indicative of thicker
L:
L(NMF) >
L(DMSO) >
L(FA) >
L(EG), as obtained [
24].
3.4. Electrolyte Solvent as the Driven Factor behind AHO Morphology, Porosity and Growth
In this study, there are two relevant parameters in the electrolyte solvent: The viscosity (η) and dielectric constant (κ) (
Table 2). The
Stokes-Einstein equation relates the diffusion constant (
D) of a macroscopic particle of radius
r, undergoing a Brownian motion, to the viscosity η of the fluid in which it is immersed [
32]. Thus, at a constant temperature, the individual ions [O
2−] or [F
−] diffusion constant will be inversely dependent on solvent η, limiting both oxidation and dissolution rates. On the other hand, a high-κ solvent draws a higher electrolyte capacitance (for a constant potential) inducing the formation of more charges at the oxide layer, thus, improving the extraction of the Hf
4+ ions and ultimately leading to a high oxidation rate [
17,
18]. Therefore, κ will be intimately related to the rate of the oxidation processes [Equation (1)] at the oxide/metal interface (higher κ, higher oxidation rate).
Figure 10a,b shows the analyses of the features
Dp,
Dint and
P as a function of the solvent physical parameters, η and κ.
Dp and
Dint decrease as η increases (
Figure 10a). The individual ions [O
2−] or [F
−] diffusion constant will be inversely dependent on solvent η (according to Stokes-Einstein equation), limiting both oxidation and dissolution processes rates. Increasing η, the diffusion of [O
2−] or [F
−] will decrease, leading to smaller
Dp (and
Dint) (
Figure 10a) and higher
P (
Figure 10b) (deviating from the optimized 10% rule for optimized self-ordered regularity). On the other hand, by decreasing κ [or the solvent conductivity (σ)
SI: Table S1],
P increases, also leading to deviations of 10% rule).
Figure 11a,b displays the counterplots (color-maps) of the AHO
L and
P as a function of the solvent physical parameters η and κ. We used the parameters for each sample, (η, κ,
L) from
Table 2 and (η, κ,
P) from
Table 1 and
Table 2, to perform a numerical estimation of 20 new data points by the interpolation method of cubic Spline. By this interpolating method, we are able to create an estimation of new values of
L and
P with certain conditions of the electrolyte (varying η and κ). The obtained (η, κ,
L) and (η, κ,
P) arrays were then plotted in 3D counterplot in
Figure 11a,b, respectively. One can observe that thicker AHO is obtained for high κ and low η values. These anodization conditions led to extremely fast oxidation rates as in the case of the NFM sample and demonstrated by the
Q(
t) curve (
Figure 4). The NFM sample shows a faulty structure without self-ordered nanoporous or tubes [only small areas revealed a nanoporous structure;
Figure 7c]. Indeed, much faster oxidation than field-enhanced dissolution occurred during the anodization, being the process out of the steady-state anodization conditions, which is mandatory for nanoporous/tubes upraise. We believe that, at the initial anodization stages, already nanoporous/NTs formation occurred [see initial
j(
t) transient in
Figure 2 and SEM image in
Figure 8c], but the NMF solvent high-κ (
Table 2) led to a much faster Hf
4+ extraction, and thus, leaving no time to maintain the nanostructures self-organization regime, i.e., to have a proper dissolution rate that would counter-balance the high oxidation rate. Additionally, the NMF
j(
t) singularities observed during the anodization (
Figure 2) can be related to the different nanostructures morphologies obtained (
Figure 8).
Figure 11b shows the counterplot of
P as a function of the solvent parameters η and κ. With this analysis, we establish a range of
P tunability. We can observe that porosities closer to the 10% rule can be obtained for higher κ and lower η. However, it is also observed that the porosity clearly deviates from the 10% rule for lower values of κ and higher η. Notice that moderate values of
P (closer to 10%) can be obtained with higher η values, but κ has to be at the higher value range. Contrarily to what is usually presented in literature, that
P depends exclusively on anodization parameters such, voltage, (
Dp,
Dint) or water content, we demonstrate for the first time that the porosity also critically depends on physical properties of the solvent (η, κ). These new results bring the possibility to mix the solvents in order to tune the anodic oxides with a perfect hexagonal arrangement.
The anodization conditions mandatory for the self-ordered nanoporous/tubes morphology to arise are obtained decreasing κ, either with low or high η. The FA solvent has a relatively lower κ, but a slightly higher η than those of NMF. This seems to be enough to establish the necessary conditions of a more equilibrated oxidation/dissolution balance for the formation of structures with self-organization (nanoporous structure;
Figure 5). Additionally, under these conditions, moderate
L is obtained, as shown in the counterplot map (
Figure 11a). On the other hand, the NT structure arises when decreasing even more κ, either in low or high η regimes, although with thinner or thicker thicknesses, respectively. Both DMSO and EG samples showed an NT structure, but the DMSO sample presented a higher
L. Both own a similar κ value, albeit much smaller than the one from the previously discussed samples. EG much higher viscosity, making F
− ions more difficult to be replaced by new ones at the NTs bottom. As a result, oxidation is faster than dissolution, leading to a
j(
t) decrease during the anodization, indicating the progressive increase of δ
b. Thus, the ionic migration path along the oxide barrier [
29] significantly extends, inhibiting the transport of F
−, Hf
4+ and O
2− ions across δ
b (Hf
4+ and O
2− for oxidation, F
− for dissolution) which subsequently limits a further NT growth (
Figure 1). On the other hand, from the dissolution reaction [Equation (2)] one can see that the failure of F
− leads to H
+ excess, and thus, to additional chemical dissolution effects that also result in limited NTs growth. DMSO showed a perfect balance between oxidation and dissolution,
j(
t) constant during the anodization, just as in the Al anodization case [
23,
25,
26]. The increased NT-array length when using a DMSO electrolyte can also be attributed to the controlled chemical dissolution process effect. Thus, the route to successfully obtain long NT arrays is to minimize the pH decrease promoted by H
+ additional etching. The DMSO aprotic photophilic solvent accepts an H
+ ion from NH
4F and reduces its activity, decreasing the chemical etching. Thus, allowing the DMSO NTs to grow deep into the hafnium foil without any significant loss at the tube tops. The presence of DMSO modifies the space charge region in the pores, thereby also avoiding the lateral etching and leading to a steady-state pore growth and low chemical etching of the NT walls.
As expected, the same conclusions were obtained for the counterplots with the solvent conductivity (σ) instead of using κ (
SI - Figure S6).
In summary, the electrolyte solvent affects the morphology and length of anodic HfO
2 (
Figure 12). Different nanostructures with different shapes of morphologies are obtained by changing the electrolyte solvent physical characteristics. For instance, we can tailor the anodic oxide morphology from NTs (EG and DMSO) to nanoporous (NPs; FA) to a thick oxide layer (NMF), by increasing the electrolyte κ, since the oxidation rate is higher (
Figure 11a (easier Hf
4+ extraction). Additionally, with the κ increase the porosity decreases (
Figure 11a). On the other hand, increasing η, the
L severally decreases (
Figure 11b), since the dissolution rate decreases because the ionic diffusion is limited. Overall, an accurate balance between the electrolyte solvents’ κ and η is needed to obtain the desired morphology, porosity and length.

 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}